

ABSTRACT
The Gram-negative opportunistic pathogen Pseudomonas aeruginosa has distinct genetic programs that favor either acute or chronic virulence gene expression. Acute virulence is associated with twitching and swimming motility, expression of a type III secretion system (T3SS), and the absence of alginate, Psl, or Pel polysaccharide production. Traits associated with chronic infection include growth as a biofilm, reduced motility, and expression of a type VI secretion system (T6SS). The Rsm post-transcriptional regulatory system plays an important role in the inverse control of phenotypes associated with acute and chronic virulence. RsmA and RsmF are RNA-binding proteins that interact with target mRNAs to control gene expression at the post-transcriptional level. Previous work found that RsmA activity is controlled by at least three small, non-coding regulatory RNAs (RsmW, RsmY, and RsmZ). In this study, we took an in-silico approach to identify additional sRNAs that might function in the sequestration of RsmA and/or RsmF and identified RsmV, a 192 nt transcript with four predicted RsmA/RsmF consensus binding sites. RsmV is capable of sequestering RsmA and RsmF in vivo to activate translation of tssA1, a component of the T6SS, and to inhibit T3SS gene expression. Each of the predicted RsmA/RsmF consensus binding sites contribute to RsmV activity. Electrophoretic mobility shifts assays show that RsmF binds RsmV with >10-fold higher affinity than RsmY and RsmZ. Gene expression studies revealed that the temporal expression pattern of RsmV differs from RsmW, RsmY, and RsmZ. These findings suggest that each sRNA may play distinct roles in controlling RsmA and RsmF activity.
IMPORTANCE The role of RsmF in post-transcriptional control of gene expression remains enigmatic. While numerous rsmA-dependent phenotypes are more pronounced in an rsmAF double mutant, deletion of rsmF alone has only modest effects. Understanding mechanisms that control RsmF activity will provide insight into additional roles for RsmF. In the current study we identify RsmV as an sRNA that controls RsmA and RsmF activity, and show that RsmV, RsmW, RsmY, and RsmZ are differentially expressed during growth.
INTRODUCTION
Pseudomonas aeruginosa is a Gram-negative opportunistic pathogen that can cause acute infections in the immunocompromised and chronic infections in individuals with cystic fibrosis (CF) (1, 2). Acute P. aeruginosa maladies include skin and soft tissue infections, ventilator associated pneumonia (VAP), and urinary tract infections. P. aeruginosa isolated from acute infections are typically motile, non-mucoid, and toxigenic. Acute infections by multi-drug resistant P. aeruginosa are difficult to resolve and can progress to sepsis resulting in a high rate of morbidity and mortality (3). Chronic P. aeruginosa infections are most common in CF patients and result from a variety of mutations in the CFTR ion channel that result in dehydrated and thickened mucus, and physiochemical changes in the airway surface fluid that result in a clearance defect (4). The persistence of P. aeruginosa in the CF airways is associated with adaptive changes including loss of motility, growth as a biofilm, mucoidy, and loss of some acute virulence functions (5, 6). The coordinate transition from an acute to a chronic infection phenotype is regulated by a variety of global regulatory networks including the Rsm system (7).
The Rsm system controls ~10% of the P. aeruginosa genome including the type III and type VI secretions, exopolysaccharides important for biofilm formation, and motility (8, 9). The Rsm system includes two small RNA-binding proteins (RsmA and RsmF/RsmN) and at least three small non-coding RNAs (RsmW, RsmY, and RsmZ) that function by sequestering RsmA and RsmF from mRNA targets. RsmA and RsmF are part of the CsrA family and regulate gene expression at the post-transcriptional level. RsmA and RsmF are 31% identical at the amino acid level and both rely on a conserved arginine residue for RNA-binding activity (13, 16). RsmA and RsmF directly interact with mRNA targets to positively or negatively alter translation efficiency and/or mRNA stability (8, 10, 11). The RsmA and RsmF bindings site on target mRNAs commonly overlap the ribosome binding site and consist of a conserved 5’- CANGGAYG sequence motif (where N is any nucleotide, the underlined GGA is 100% conserved, and Y is either a cytosine or uracil) that presents the GGA sequence in the loop portion of a stem-loop structure (12-15). While RsmA is able to bind mRNA targets with a single CANGGAYG sequence (12), RsmF differs in that high affinity binding is only observed with mRNAs targets possessing at least two CANGGAYG consensus binding sites (12). Although RsmA and RsmF share some targets in common, the full extent of overlap between the regulons is unknown (13, 16). The RsmF regulon, however, was recently determined using a pull-down method to identify 503 target RNAs (17).
The RNA-binding activity of RsmA is controlled by the small non-coding RNAs RsmW, RsmY and RsmZ (13, 18). RsmW, RsmY, and RsmZ each have multiple CANGGAYG binding sites that allow for sequestration of RsmA from target mRNAs (19). It is unclear whether RsmW, RsmY, and RsmZ are the only sRNAs that function in the sequestration of RsmA or whether RsmW, RsmY, or RsmZ are the primary sRNAs that function in sequestration of RsmF. The affinity of RsmF for RsmY and RsmZ is 10-fold lower than RsmA. In this study, we sought to identify additional sRNAs that regulate RsmA and RsmF activity, and identified RsmV, a 192 nt transcript that has four CANGGAYG sequences presented in stem-loop structures. We demonstrate that RsmV is able to sequester RsmA and RsmF in vivo, that full RsmV activity is dependent upon each of the four CANGGAYG sequences, and that RsmV demonstrates a temporal expression pattern that is distinct from RsmW, RsmY, and RsmZ. We propose a model wherein each sRNA plays differential and distinct roles in control of the Rsm system.
RESULTS
Identification of RsmV as a sequestering RNA for RsmA and RsmF
We took an in-silico approach to identify candidate sRNAs that might control RsmA and/or RsmF activity in vivo. A prior SELEX study concluded that optimal RNA-binding activity by RsmF requires RNA targets with at least two GGA sequences presented in the loop portion of stem-loop structures. Transcriptome studies have identified ~500 potential sRNAs in P. aeruginosa (28, 29). The secondary structure of each sRNA was predicted using mFold and then examined for the presence of ≥2 GGA sequences presented in stem-loop structures. One sRNA candidate had six GGA sequences (Fig. 1A). Each GGA sequence demonstrated a ≥60% match to the full RsmA/RsmF consensus binding site (CAnGGAyG) (Fig. 1B). Four of the GGA sequences (designated sites 2, 3, 5 and 6) are predicted by mFold to be presented in stem-loop structures (Fig. 1A). The gene encoding the sRNA is located in the intergenic region between mucE and apqZ, and has been designated rsmV (Fig. 1C). A search of the Pseudomonas Genome Database indicates that the rsmV sequence is highly conserved in >100 sequenced P. aeruginosa genomes and is absent from the genomes of other Pseudomonads.

(A) Predicted mFold structure of RsmV. P. aeruginosa RsmV secondary structure determined by mFold modeling. Each of the six GGA sequences are highlighted in red and numbered by order of appearance from the 5’ end of the sequence. (B) Alignment of each GGA site to the full RsmA/RsmF consensus binding site. The GGA sites are 100% conserved (red) and other conserved portions of the consensus are highlighted in blue. (C) The genome context of rsmV, located between mucE and aqpZ. The positions of the primer used to generate cDNA for the experiment in Fig. 2B are labeled 1- 4. Primers used to generate PCR products in Fig. 2B are labeled Fprimer and Rprimer.
The RNAseq study that identified rsmV concluded that the RNA is 192 nt long (29). To verify the rsmV transcription start site, cDNA was generated using a primer within the gene (Fig. 2A). The cDNA was then used in a PCR reaction with primers positioned just upstream of and at the predicted start site. Whereas the primer positioned at the start site generated the expected product, the primer located just upstream did not generate a product. This finding is consistent with the rsmV transcription start site identified in the Wurtzel RNAseq study (29). There is no identifiable transcriptional terminator downstream of rsmV. To verify the 3’ boundary, cDNA was generated from total cellular RNA with primers positioned at the predicted 3’ end of rsmV and at several downstream positions as shown in Fig. 1C. The resulting cDNAs were then used as templates in PCR reactions with primers (Fprimer and Rprimer) positioned within the gene (Fig. 1C). The cDNA primer positioned within the aqpZ coding region (primer 4) did not yield a product. The cDNA primers positioned upstream of aqpZ (primers 1, 2, and 3) all yielded products, and the strongest product was observed with cDNA generated at the predicted 3’ terminus of rsmV using primer 2 (Fig. 2B). The weaker PCR products produced from cDNA generated with primers 2 and 3 may represent transcriptional read through.

(A) RNA purified from wt cells was used to generate cDNA using the indicated 3’ primer. The cDNA was then used in PCR reactions with the same 3’ primer and 5’ primers positioned just upstream of or at the predicted start of rsmV transcription. Genomic DNA (gDNA) served as a positive control. (B) Verification of the rsmV termination site. cDNA was generated using primers 1-4 as shown in Fig. 1C. The cDNA was then used in PCR reactions with the indicated primer sets in Fig. 1C. Genomic DNA (gDNA) served as a positive control.
RsmV interacts with and controls RsmA and RsmF activity
The presence of four predicted GGA sequences in stem-loop structures is consistent with RsmV serving as a sequestering sRNA for RsmA and/or RsmF. To test this prediction we measured binding using EMSA experiments. Full length RsmV was synthesized in vitro, radiolabeled at the 5’ end, and incubated with purified RsmAHis or RsmFHis prior to electrophoresis on non-denaturing gels. RsmAHis formed high affinity binding products with RsmV (Keq 14 nM) and two distinct binding complexes were evident (Fig. 3A). Those products could reflect binding of multiple RsmAHis dimers or differential interactions with multiple sites on the RsmV probe. RsmF also bound the RsmV probe with high affinity (Keq 2 nM), but only a single binding complex was detected (Fig. 3A).
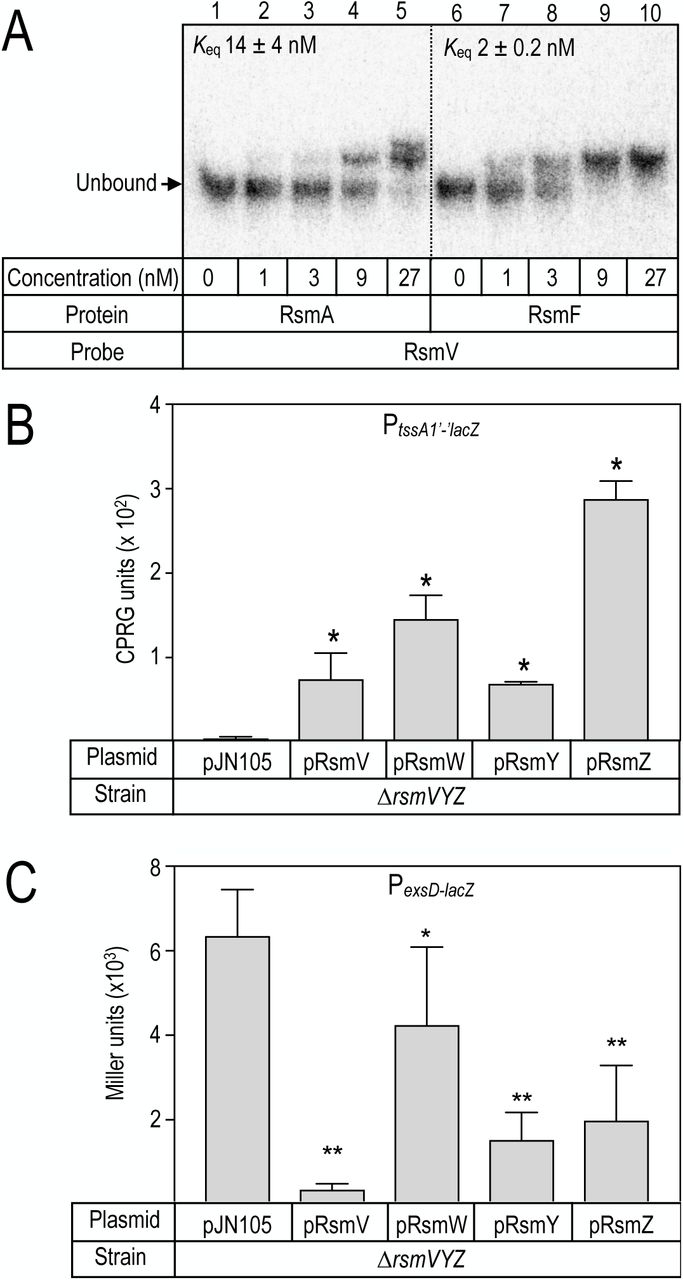
(A) RsmV was radiolabeled and used in electrophoretic mobility shift assays with purified RsmA (lanes 2-5) and RsmF (lanes 7-10) at the indicated concentrations. The position of the unbound RsmV probe is indicated. (B-C) Effect of RsmV on tssA1’-‘lacZ translational reporter (B) and PexsD-lacZ transcriptional reporter (C) activities. Strains consisting of a ∆rsmVYZ mutant transformed with either a vector control (pJN105) or the indicated sRNA expression plasmids were cultured in the presence of 0.4% arabinose to induce expression of the respective RNAs and assayed for ß-galactosidase activity. The reported values represent the average of at least three experiments with the standard error indicated. * P-value <0.05 relative to the vector control.
With evidence that both RsmA and RsmF interact with RsmV we next examined whether RsmV can sequester RsmA/RsmF in vivo. RsmA and RsmF have inverse effects on expression of the type VI (T6SS) and type III (T3SS) secretion systems (13). We used the previously described PtssA1’-‘lacZ translational reporter as surrogate for regulatory control of T6SS (13) and PexsD-lacZ transcriptional reporter as marker for the T3SS (30). RsmA/RsmF directly bind the tssA1 leader region to inhibit translation (12, 13) and positively regulate T3SS gene expression through a mechanism that remains to be defined (13). In a mutant lacking rsmV, rsmY, and rsmZ, RsmA/RsmF availability is high resulting in repression of PtssA1’-‘lacZ reporter activity and high levels of PexsD-lacZ reporter activity (Fig. 3B-C). Plasmid expressed RsmV resulted in significant activation of PtssA1’-‘lacZ reporter activity and inhibition of the PexsD-lacZ reporter. Both of these findings are consistent with RsmV serving a role in RsmA/RsmF sequestration. When compared to the previously identified sequestering RNAs RsmW, RsmY, and RsmZ (13, 31), RsmV demonstrated activity comparable to RsmY when using the PtssA1’-‘lacZ reporter (Fig. 3B) and had the strongest inhibitory activity for the PexsD-lacZ reporter (Fig. 3C).
To determine whether RsmV preferentially sequesters either RsmA or RsmF, the PtssA1’- ‘lacZ translational reporter was introduced into ∆rsmAVYZ and ∆rsmFVYZ mutant backgrounds. RsmA is more active than RsmF resulting in stronger repression of PtssA1’-‘lacZ reporter activity in the ∆rsmFVYZ mutant when compared to the ∆rsmAVYZ background for strains carrying the vector control (pJN105) (Fig. 4A vs 4B). In the ∆rsmFVYZ mutant, where repression of PtssA1’-‘lacZ activity is attributable to RsmA, RsmV demonstrated relatively weak suppressive activity when compared to RsmW, RsmY, and RsmZ (Fig. 4A). A similar picture emerged when using the ∆rsmAVYZ background to examine RsmF sequestration in that RsmV demonstrated the weakest suppressive activity (Fig. 4B). Thus, RsmV is capable of sequestering both RsmA and RsmF and appears to lack strong preference for one vs the other under the conditions tested.

(A-B) Either ∆rsmAVYZ (A) or ∆rsmFVYZ (B) quadruple mutants carrying the Plac-tssA1’-‘lacZ translational reporter was transformed with either a vector control (pJN105) or RsmV, RsmW, RsmY and RsmZ expression plasmids. The resulting strains were cultured in the presence of 0.4% arabinose to induce expression of the respective RNAs and assayed for ß-galactosidase activity. Reported values represent the average of at least three experiments with the standard error indicated. * P-value <0.05 relative to the vector control.
Contribution of GGA sites 2, 3, 5 and 6 to RsmV activity
The RsmV primary sequence contains six GGA sequences, four of which (GGA2, 3, 5, and 6) may be presented in the loop portions of stem-loop structures (Fig. 1A). To determine which GGA sites are important for RsmV regulatory activity each of the GGA sequences in stem-loop structures was changed to CCU. The activity of each mutant RNA was tested using the PtssA1’-‘lacZ translational and PexsD- lacZ transcriptional reporters. The GGA4 and GGA6 mutant RNAs demonstrated a significant loss of regulatory activity for the PtssA1’-‘lacZ translational reporter when compared to wt RsmV (Fig. 5A). In contrast, each of the GGA sites was required for full regulatory control of the PexsD-lacZ transcriptional reporter (Fig. 5B). The most likely explanation for the differential requirement for the GGA2 and GGA5 sites is that the PexsD-lacZ reporter is more sensitive to changes in RsmA availability relative to the PtssA1’-‘lacZ reporter.

The PA103 ∆rsmVYZ mutant carrying the (A) Plac tssA1’-‘lacZ translational reporter or (B) PexsD-lacZ transcriptional reporter were transformed with either a vector control (pJN105) or the indicated RsmV expression plasmids. The resulting strains were cultured in the presence of 0.4% arabinose to induce expression of the respective RNAs and assayed for ß-galactosidase activity. Reported values represent the average of at least three experiments with the standard error indicated. * P-value <0.05 relative to the vector control. (C-D) EMSA experiments with wt RsmV and the indicated mutant radiolabeled probes. 40 nM RsmA (C) or RsmF (D) were incubated with the indicated probes, subjected to non-denaturing gel electrophoresis, and phosphorimaging. The positions of the unbound probes are indicated.
The simplest interpretation of the reporter findings is that the mutant RNAs with altered activity have reduced capacity to sequester RsmA/RsmF. To test this prediction, binding assays were performed with radiolabeled RNA probes. RsmA bound each of the single GGA substitution mutants with affinities similar to or greater than wt RsmV (Fig. 5C, Table 1, Fig. S1). Whereas two distinct products are formed upon RsmA binding to the wt, GGA2, and GGA5 probes, only a single product was observed for the GGA3 and GGA6 probes. This is noteworthy as the mutant GGA3 and GGA6 RNAs also demonstrated a defect in activation of the PtssA1’-‘lacZ translational reporter (Fig. 5A). RsmF also bound each of the mutant probes with high affinity, with the exception of GGA6, which was significantly reduced (Fig. 5D, Table 1, Fig. S1). Given that RsmA and RsmF are homodimers with two RNA binding sites (one from each monomer), and that each mutant RNA still has three potential GGA interaction sites, high affinity binding to the mutant probes was not unexpected. We thus generated a probe bearing CCU substitutions at all four sites (Quad) and found that RsmA (Keq >27 nM) and RsmF (Keq >243 nM) were unable to bind (Fig. 5C-D, Table 1, Fig. S1) or exert regulatory control over the PtssA1’-‘lacZ translational and PexsD-lacZ transcriptional reporters. We conclude that the primary, if not exclusive, sites for RsmA/RsmF binding are GGA2, 3, 5, and 6.
RsmA and RsmF affinities for wt and mutant RsmV
Role of RsmV in vivo
Data presented thus far have relied upon plasmid-expressed RsmV, which may result in RNA levels that exceed the native level expressed by cells under physiologically relevant conditions. To address the effect of RsmV expressed at native levels on the output of the Rsm system we generated an in-frame rsmV deletion mutant (∆rsmV) and measured PexsD-lacZ reporter activity. When compared to wt cells, the ∆rsmV demonstrated a modest but significant increase in reporter activity (Fig. 6A). This increase in reporter activity is consistent with reduced sequestration of RsmA and/RsmF, both of which have a positive effect on T3SS gene expression. By comparison, PexsD-lacZ reporter activity is also elevated in an ∆rsmYZ double mutant. The higher level of reporter activity in the ∆rsmYZ mutant is consistent with the data presented in Fig. 3C showing that RsmY and RsmZ each have stronger effects on activation of PexsD-lacZ reporter activity relative to RsmV.

(A) Strain PA103 (wt), and the ∆rsmV and ∆rsmYZ mutants carrying the PexsD-lacZ transcriptional reporter were cultured under inducing conditions for T3SS gene expression and assayed for ß-galactosidase activity. * P-value <0.05 relative to wt. (B) A ∆rsmAF mutant transformed with either a vector control, or pRsmAHis or pRsmFHis expression vectors was cultured and subjected to rapid purification of pRsmAHis or pRsmFHis and bound RNAs. Select RNAs (as indicated) were quantified from the purified RNA pool by qRT-PCR and reported as fold change relative to the vector control. The coding sequences for lolB and rnpB were included as negative controls and tssA1 served as a positive control. Reported values represent the average of at least three replicates with the standard error reported. *P-value <0.05 when compared to expression of the wild type vector is indicated.
A second approach to test the relevance of RsmV in vivo involved precipitation experiments with histidine-tagged RsmA or RsmF. A ∆rsmAF double mutant transformed with either RsmAHis or RsmFHis expression plasmids was cultured to mid-log phase and then rapidly subjected to precipitation with Ni2+-agarose beads and isolation of bound RNA. The presence of specific RNAs was detected from the entire pool of bound RNAs by qRT-PCR. Positive controls were the known RsmA/RsmF targets RsmY, RsmZ, and the tssA1 leader region (13). Negative controls included two mRNAs (lolB and rnpB) that are not known targets of RsmA or RsmF, and Ni2+-agarose beads alone. Whereas no enrichment of the lolB or rnpB mRNAs was detected, there was significant enrichment of the tssA1 mRNA and the RsmV, RsmY, and RsmZ sRNAs by both RsmA and RsmF (Fig. 6B).
Differential expression of RsmV, RsmW, RsmY, and RsmZ
The in vivo data demonstrate that RsmV, RsmW, RsmY, and RsmZ are each capable of sequestering RsmA and RsmF. We hypothesized that differential expression of the RNAs might allow cells to fine-tune the output of the Rsm system. To test for differential expression, RNA samples were collected from cells cultured to OD600 readings of 0.5 (early log phase), 1.0 (mid-log phase), 2.0, 5.0, and 7.0 (late stationary phase). The amount of each RNA detected by qRT-PCR at early log phase was normalized to 1.0 and the reported values for each subsequent time point are relative to those values (Fig. 7). Both RsmY and RsmZ showed a transient increase in expression at mid-log phase, followed by a decrease at OD600 readings of 2.0 and 5.0, and then a significant increase in late stationary phase (OD600 7.0) The expression pattern for RsmW was delayed until the OD600 reached 2.0 but demonstrated the highest fold changes in expression at OD600 2.0 and 5.0, and then approached the fold changes observed for RsmY and RsmZ at OD600 7.0. By contrast, RsmV demonstrated a slow but steady increase throughout the growth curve but was the least dynamic of the four RNAs. The observed differences in expression patterns are consistent with the hypothesis that the sRNAs may serve distinct roles in RsmA/RsmF sequestration based upon their timing of expression.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
RNA was isolated from wt cells harvested at the indicated A600 readings and used as template in RT-qPCR experiments with primers specific to rsmV, rsmW, rsmY, and rsmZ. The reported values for each RNA are relative to the measurement of the sample collected at A600 0.5. The data represent the average of at least three replicates.
One mechanism to account for the differential expression of RsmV, RsmW, RsmY, and RsmZ is by distinct transcription factors. Transcription of rsmY and rsmZ is controlled by the GacAS two component system (20). A previous study found that GacAS does not control rsmW transcription (31). To determine whether rsmV transcription is regulated by GacA, a PrsmV-lacZ transcriptional reporter was integrated at the ΦCTX phage attachment site of wt cells and a ∆gacA mutant. Whereas PrsmY-lacZ and PrsmZ-lacZ reporter activity demonstrate strong gacA- dependence, PrsmV-lacZ activity showed no difference between wildtype and the gacA mutant (Fig. S2).
DISCUSSION
The primary RsmA/RsmF sequestering RNAs in Pseudomonas aeruginosa are RsmY and RsmZ. In addition to RsmY/RsmZ, RsmW plays a smaller role in the sequestration of RsmA (31) and can also sequester RsmF (Fig. 2). RsmV represents a fourth RsmA/RsmF sequestering RNA in P. aeruginosa. RsmV shares sequence and structural characteristics with RsmY and RsmZ including multiple GGA motifs (6), four of which are likely presented in stem-loop structures. RsmY and RsmZ are also the primary sequestering RNAs in P. fluorescens (now P. protegens) (18). At least one additional sRNA, RsmX, also contributes to Rsm control in P. protegens (32). The involvement of multiple sequestering sRNAs in the control of CsrA/RsmA activity is common. CsrB is the primary CsrA-sequestering RNA in E. coli and contains 18 GGA motifs (33). Other E. coli sRNAs can also sequester CsrA including CsrC and McaS (34, 35). CsrC has a structure similar to CsrB but with fewer GGA motifs (34). McaS is an sRNA that basepairs with some mRNAs involved in curli and flagella synthesis and can also sequester CsrA via two GGA motifs (35). In addition to sRNAs, the 5’ untranslated region of mRNAs can also function in the sequestration of CrsA (36).
The relative activities of RsmV, RsmW, RsmY, and RsmZ were compared by expressing each sRNA from an arabinose-inducible expression vector. Plasmid expressed RsmV activated PtssA1’-‘lacZ reporter activity and inhibited PexsD-lacZ reporter activity (Fig. 3B-C). RsmV had activity comparable to RsmY for activation of the PtssA1’-‘lacZ reporter activity and the strongest effect on activation for PexsD-lacZ reporter activity. RsmA and RsmF both bind to RsmV with high affinity in vitro (Fig. 3A), and the affinity of RsmF for RsmV is at least 10-fold higher than for RsmY and RsmZ (12, 13). Although the affinity of RsmF for RsmV is higher in vitro, RsmV does not seem to show preferential activity towards RsmF over RsmA in vivo (Fig. 4). The reason for this is unclear but may reflect differences between the in vitro and in vivo binding conditions. A difference between in vitro and in vivo conditions was also evident when the RsmV GGA mutants were examined. Whereas each of single GGA substitution mutants demonstrated altered regulatory control of PexsD-lacZ and/or PtssA1’-‘lacZ reporter activity (Fig. 5A-B), the binding affinity of RsmA and RsmF was relatively unaffected by the single GGA substitutions (Table 1). A similar trend was observed in a previous mutagenesis study of RsmY and RsmZ wherein the in vivo activity did not strictly correlate with in vitro binding (37). It was speculated that other RNA binding proteins, such as Hfq, may prevent binding to suboptimal sites in vivo.
RsmV activity is clearly evident when expressed from a plasmid (Fig. 3). A role for RsmV when expressed at native levels from the chromosome was also detected. Deletion of rsmV resulted in a modest but significant increase in T3SS reporter activity (Fig. 6A) and co-purification experiments found that RsmV interacts with RsmA and RsmF (Fig. 6B). Both of these findings suggest that RsmV can compete with RsmY and RsmZ for RsmA/RsmF binding in wt cells (Fig. 6B). Unclear is whether conditions exist where rsmV transcription is elevated and might result in more pronounced phenotypes. Transcription of rsmY and rsmZ is directly controlled by the GacA/S two-component system, a highly conserved system in Gamma-proteobacteria (20, 38, 39). GacS is a sensor kinase whose activity is controlled by two orphan kinases, RetS and LadS (40-43). Additional regulators interact with and alter the effect of RetS on GacS (44, 45). SuhB regulates rsmY and rsmZ transcription indirectly by altering gacA levels (24). The phosphotransfer protein, HptB, regulates rsmY and rsmZ transcription when P. aeruginosa is grown on a surface (22, 23). Other regulators contribute to rsmY and rsmZ transcription through mechanisms that do not alter GacS/GacA activity. MvaT, a H-NS like protein, binds A+T rich regions of DNA and silences rsmZ transcription, while BswR, a transcriptional regulator, counteracts negative regulation of rsmZ by MvaT (20, 21). Recently, MgtE, a magnesium transporter, was shown to alter rsmY and rsmZ transcription, yet a mechanism of action is yet to be defined (25).
Neither rsmV nor rsmW are under positive transcriptional control of the GacAS system (31) (Fig. S2). GacA may repress rsmW transcription through an indirect mechanism (30). RsmW expression appears to be highest during stationary phase in minimal media, which may be more biologically representative of a biofilm (31). RsmW is encoded directly downstream of PA4570, a protein of unknown function. RsmW and PA4570 are likely co-transcribed and separated by an RNase cleavage event. Determining the transcriptional regulation of PA4570 may provide insight into rsmW transcriptional control. A search for potential promoters upstream of rsmV predicted binding sites for the transcriptional activators RhlR, AlgU, and FleQ. mRNA levels for rsmV, however, were unaffected in PA14 transposon mutants within each of those genes relative to wild type as measured by qRT-PCR (data not shown). Additional studies will be required to determine how rsmV and rsmW transcription is controlled, and if RsmV plays a larger role in regulating RsmA and/or RsmF activity under a different set of growth conditions.
RsmX, RsmY, and RsmZ in P. protogens are differentially expressed, thus contributing to a mechanism of fine-tuning RsmA and RsmE activity (32). Expression of P. protogens RsmX and RsmY occurs in parallel during exponential growth while RsmZ expression is delayed (32). This may allow cells to fine-tune expression of these sRNAs based on the environmental conditions. We propose a similar scenario for expression of RsmV, RsmW, RsmY, and RsmZ in P. aeruginosa. The differences in binding affinities for RsmA/RsmF, timing of gene expression, and expression levels of the sRNAs may provide a mechanism of fine-tuning the expression of genes under control of the Rsm system.
METHODS AND MATERIALS
Strain and plasmid construction
Routine cloning was performed with E. coli DH5α cultured in LB-Lennox medium with gentamycin (15 μg/ml) as required. P. aeruginosa strain PA103 and the ∆gacA, ∆rsmYZ mutants were reported previously (Table 1) (47). The in-frame ∆rsmV deletion mutant was constructed by allelic exchange. The upstream and downstream flanking regions (~800 bp) of rsmV were generated by PCR using primer pairs 118845409- 118845410 and 118845411-118845412. The PCR products were cloned into pEXG2 (48) and the resulting construct was mobilized into wild type PA103 and the ∆rsmYZ, ∆rsmAYZ, and ∆rsmFYZ mutant by conjugation. Merodiploids were resolved by sucrose counter-selection as previously described (49). The RsmV expression plasmid was constructed by positioning the rsmV transcription start site immediately downstream of the PBAD promoter start site using the Gibson assembly method (New England Biolabs). Briefly, the PBAD promoter region from pJN105 (primer pair 117830775-117830776) and rsmV (primer pair 118845423-118845424) were amplified by PCR and then assembled into the MluI and SacI digested pJN105 (50). pRsmV vectors bearing single GGA to CCT substitutions, or various combinations therefore, were assembled using the Gibson method from gene blocks listed in Table 2 and cloned into the NruI and PvuI sites of pJN105 as outlined in Table 3. The rsmV transcriptional reporter (primer pair 150592489-150592490) includes 500 nucleotides upstream of the rsmV transcription start site. The rsmV reporter was integrated into the CTX phage attachment site in WT and gacA strains.
β-Galactosidase Assays
PA103 strains were grown overnight at 37°C in LB containing 80 μg/ml gentamicin as required. The next day strains were diluted to an absorbance (A600) of 0.1 in tryptic soy broth (TSB) for measurement of tssA1’-‘lacZ reporter activity or TSB supplemented with 100 mM monosodium glutamate, and 1% glycerol for measurement of PexsD-lacZ reporter activity. Arabinose (0.4%) was also added to induce rsmV expression from the PBAD promoter. The cultures were incubated at 37°C and harvested when the A600 reached 1.0. β-galactosidase activity was assayed with the substrates ortho-nitrophenyl-galactopyranoside (ONPG) as previously described (51) or chlorophenol red-β-D-galactopyranoside (CPRG). CPRG activity was determined by measuring product formation at 578 nM and using an adaptation of the Miller equation: CPRG units = (A578/culture A600/time /culture vol [ml]) x 1000. CPRG and Miller units are reported as the average of at least three independent experiments with error bars representing the standard deviation (SD).
Electrophoretic mobility shift assays
DNA templates encoding wildtype rsmV or rsmV bearing point mutations within the GGA sequences were PCR amplified and used as templates for in vitro generation of RNA probes. RNA probes were end-labeled with [γ-32] ATP as previously described (13). Purified RsmA or RsmF, as described previously (13), were incubated with the RNA probes at the indicated concentrations in 1X binding buffer (10 mM Tris-HCl pH [7.5], 10 mM MgCl2, 100 mM KCl), 3.25 ng/μl total yeast tRNA (Life Technologies), 10 mM DTT, 5% (vol/vol) glycerol, 0.1 units RNAse Out (Life Technologies). Reactions were incubated at 37° C for 30 min, and then mixed with 2 μl of gel loading buffer II (Life Technologies) and immediately subjected to electrophoresis on 7.5 % (wt/vol) native polyacrylamide glycine gels (10 mM Tris-HCl pH[7.5], 380 mM glycine, 1 mM EDTA) at 4° C. Imaging was performed using an FLA-7000 phosphorimager (Fujifilm), and analyzed using MultiGuage v3.0 software.
RNA enrichment experiments
Strain PA14 ΔrsmAF carrying either an empty vector control, pRsmAHis6, or pRsmFHis6 was grown in TSB supplemented with 20 mM MgCl2, 5 mM EGTA, 15 μg/ml gentamicin, and 0.1% arabinose to mid log phase, chilled, and pelleted for immediate lysis. Cells were lysed under native conditions to retain protein structure (Qiagen QIAexpressionist manual native purification buffer recipe) supplemented with 2.5 mM ribonucleoside vanadyl complex (NEB) to inhibit RNAse activity, 1 mg/mL lysozyme, and 0.1% Triton X-100. Lysis was completed by freeze-thaw cycles. Lysates were treated with 10 uL RQ-1 RNase-free DNAse and cleared by centrifugation. An aliquot was removed from the cleared lysate for total RNA isolation and preserved in Trizol, and the remaining lysate was incubated with Ni-NTA agarose at 4°C for 1 hour under non-denaturing binding conditions. Ni-NTA agarose was then loaded into a column and washed 3 times with non-denaturing binding buffer containing 10 mM imidazole. Protein and associated RNAs were eluted in 4 fractions with 250 mM imidazole and 4 fractions with 500 mM imidazole. Protein-containing fractions from the RsmAHis6, or RsmFHis6 expressing strains, and an equivalent volume from the vector control strain, were treated with TRIzol (Thermofisher) and RNA was extracted according to the manufacturer’s protocol. RNA was treated with RQ-1 RNase-free DNase and concentrated using a RNA Clean and Concentrator kit (Zymo). First strand cDNA was synthesized using Superscript II (ThermoFisher) according to manufacturer’s protocol with Random Primer 9 (NEB). The copy number of the indicated genes was determined by qPCR using SYBR Green Master Mix (Bio-rad).
Statistical analyses
One-way ANOVA was performed using Prism 6.0 (GraphPad Software, Inc., La Jolla, CA).
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health, grant number AI097264 to MCW and TLY. KHS was supported by T32GM082729 and 5T32AI007511-19.
REFERENCES




