

Abstract
Filopodia are dynamic membrane protrusions driven by polymerization of an actin filament core, mediated by formin molecules at the filopodia tips. Filopodia can adhere to the extracellular matrix and experience both external and cell generated pulling forces. The role of such forces in filopodia adhesion is however insufficiently understood. Here, we induced sustained growth of filopodia by applying pulling force to their tips via attached fibronectin-coated beads trapped by optical tweezers. Strikingly, pharmacological inhibition or knockdown of myosin IIA, which localized to the base of filopodia, resulted in weakening of filopodia adherence strength. Inhibition of formins, which caused detachment of actin filaments from formin molecules, produced similar effect. Thus, myosin IIA-generated centripetal force transmitted to the filopodia tips through interactions between formins and actin filaments are required for filopodia adhesion. Force-dependent adhesion led to preferential attachment of filopodia to rigid versus fluid substrates, which may underlie cell orientation and polarization.
INTRODUCTION
Filopodia are ubiquitous cell extensions involved in cell motility, exploration of the microenvironment and adhesion 1, 2. These finger-like membrane protrusions help cells to determine the direction of movement 3, establish contacts with other cells 4, 5 and capture inert particles or living objects (bacteria), which cells subsequently engulf 6–9. Filopodia are involved in numerous processes of embryonic development, as well as in cell migration in adult organisms. Moreover, augmented filopodia activity is a hallmark of tumor cells, which use them in the processes of invasion and metastasis1.
The main element of filopodia is the actin core, which consists of parallel actin filaments with barbed ends oriented towards the filopodium tip, and pointed ends toward the cell body 1, 2, 10. Actin filaments are connected to each other by several types of crosslinking proteins 11–14 The filopodia grow via actin polymerization at the tip, in a process driven by formin family proteins such as mDia2 15–17, 18, FMNL2 & 3 19–21, as well as by actin elongation protein Ena/VASP 15, 22, 23, 24. In addition to proteins that crosslink and polymerize actin, filopodia also contain actin based molecular motors, such as myosin X, localized to the tips of the filopodia 25. Although the function of myosin X is unclear, it is known to be required for filopodia growth, and its overexpression promotes filopodia formation 26, 27.
Adhesion of the filopodia to the extracellular matrix (ECM) is mediated by the integrin family of receptors (e.g. αvβ3) 25, 28, which are localized to the tip area. One possible function of myosin X is the delivery of integrins to this location 25. In addition to integrins, filopodia tips have been shown to contain other proteins involved in integrin mediated adhesion, such as talin 29 and RIAM 30. Several studies suggest that typical cell matrix adhesions, known as focal adhesions, could in some cases originate from filopodia 31, 32. Thus, filopodia could be considered as primary minimal cell matrix adhesion structures.
The hallmark of integrin mediated adhesions of focal adhesion type is their mechanosensitivity 33–35. They grow in response to pulling forces applied to them, either by the actomyosin cytoskeleton, or exogenously by micromanipulations, and may play a role in matrix rigidity sensing. Indeed, correlation between focal adhesion size and matrix rigidity is well-documented 36–38. Filopodia also may participate in matrix rigidity sensing. For example, it was demonstrated that cell durotaxis, a preferential cell movement along a gradient of substrate rigidity is mediated by filopodia 39. However, force dependence of filopodia adhesion has not yet been explored.
In the present study, we monitored filopodia adhesion and growth under conditions of pulling with a constant rate. We have demonstrated that adhesion of filopodia to the ECM strongly depends on myosin II activity and found myosin II filaments localized to the base regions of filopodia. Moreover, formin family protein activity at the filopodia tips is also required for filopodia adhesions, most probably through a role in the transmission of force through the actin core, from the filopodium base to the filopodium tip. Thus, filopodia are elementary units demonstrating adhesion-dependent mechanosensitivity.
RESULTS
Dynamics of filopodia induced by expression of myosin X in HeLa-JW cells
Transfection of HeLa-JW cells with either GFP-myosin X or mApple-myosin X resulted in a strong enhancement of filopodia formation in agreement with previous studies 40. During filopodia movement, myosin X was concentrated at the filopodia tips, forming characteristic patches sometimes also called “puncta” or “comet tails” (fig. S1A, movie S1). Here, we focused on filopodia originating from stable cell edges and extending along the fibronectin-coated substrate. These filopodia demonstrated periods of persistent growth, with an average velocity of 67 ± 6 nm/s (mean ± SEM, n = 89) interrupted by pauses and periods of shrinking with an average velocity of 28 ± 3 nm/s (mean ± SEM, n = 100). This behavior is consistent with previously published results 41. In addition to myosin X, the filopodia tips were also enriched in several other proteins such as mDia2, VASP and talin (fig. S1B and D).
To observe the dynamics of filopodia adhesion and protrusion under controlled experimental conditions, we monitored the growth of filopodia that were adhered to fibronectin-coated beads trapped by optical tweezers (see supplementary information for more details). First, 2μm diameter fibronectin-coated polystyrene beads were placed onto filopodia tips by the optical tweezers. After 20-30 s, which is required for the initial attachment of the bead to the filopodium, the movement of microscope piezo stage in the direction from the tip to base of filopodium was initiated (Fig. 1, movies S2, S3, S9A). The force exerted by filopodium on the bead was monitored by measuring the bead displacement from the center of the trap (Δx). In order to preserve the structural integrity of the filopodia, the velocity of the stage movement was set to approximately 10-20nm/s, which is slower than the average velocity of spontaneous filopodia growth. With this setup we observed sustained filopodia growth for more than 10 mins, during which time the tdTomato-Ftractin labelled actin core remained intact (Fig. 1B and 3A, movie S2). Pulling-induced filopodia growth was depended on integrin-mediated adhesion of filopodia tips to fibronectin-coated beads. When the beads were coated with concanavalin A instead of fibronectin, application of force never induced the growth of filopodia actin cores. Instead, pulling via concanavalin A-coated bead resulted either in detachment of filopodia tips from the beads (18%), or withdrawal of the bead from the trap (27%), or, in majority of cases (55%), in the formation of membrane tethers, n = 11 (movie S4).

(A) Experimental setup used to observe force-induced filopodia growth. Optical tweezers were used to trap fibronectin-coated microbeads attached to filopodia tips of HeLa-JW cells. (B) Confocal images of a typical cell expressing GFP-myosin X and tdTomato-Ftractin with an attached bead, taken immediately after starting of stage movement (top) and in the course of sustained growth (bottom). Note that both myosin X and actin remain at the filopodium tip during growth. See also movies S2-3. Scale bar, 5μm. (C) Top panel: A kymograph showing the dynamics of myosin X and actin in the filopodium shown in (B). This kymograph is composed from two parts smoothly combined next to each other: line was drawn through basis region (left part), and - proximal region of the filopodium (right). Middle panel: Filopodium growth in relation to the coordinate system of the microscope stage. The origin of the coordinate system corresponds to the bead position in the center of the laser trap at the initial time point. The coordinate of the bead is changing due to the uniform movement of the stage, and fluctuations of the bead position inside the trap. Lower panel: Forces experienced by the bead. Note the discrete peak force values corresponding to the moments of filopodia growth cessation (seen in the middle panel) as marked with dotted lines. Inset: The distribution of peak force values, based on the pooled measurements of 21 peaks from 6 beads.

(A) Visualization of mApple-myosin X (shown in red), RLC-GFP (green) and mTagBFP-Lifeact (blue) in HeLa-JW cell. (B) Zoomed images of bipolar myosin IIA filaments at the bases of filopodia. Upper panel: myosin IIA and myosin X were labeled as indicated above in HeLa-JW cell. Lower panel: Cos-7 cell expressing mApple-myosin X (red) and GFP-myosin IIA heavy chain (green). Arrows indicate myosin II mini-filaments. (C)-(H) Images of Cos-7 cells expressing myosin X (red) and myosin IIA or IIB (green) and their mutants: (C) GFP-myosin X. (D) mApple-myosin X and GFP-myosin IIA heavy chain. (E) GFP-myosin X ΔFERM. (F) GFP-myosin X ΔFERM and mCherry-myosin IIA heavy chain. (G) mApple-myosin X and GFP-myosin IIB heavy chain. (H) mApple-myosin X and GFP-myosin IIA N93K. Note the presence of myosin IIA filaments for A, B, D, F and H and the absence of myosin IIB at the filopodia bases. See also movies S8A-G, which correspond to images A, C-H, respectively. Scale bars, 2μm. (I) Survival fraction of the filopodia cohort in Cos-7 cells with the above constructs. (J) Lifetimes of filopodia in cells transfected with different constructs of myosin II and myosin X. To calculate the average filopodia lifetime, experimentally measured survival fraction of cell filopodia was fitted to the exponential decay function: survival fraction = e−t/λ, where t is time and λ is the average filopodia lifetime. Calculated lifetime values are (mean±SD) 151.0±5.1 (GFP-myosin X only, n=72, 3 cells), 537.2±19.8 (n=30, 3 cells), 152.4±1.7 (n=81, 3 cells), 513.0±16.9 (n=29, 3 cells), 194.7±2.6 (n=53, 3 cells), and 229.8±4.7 (n=70, 4 cells) seconds (in order corresponding to images C-H). All images and data for analysis were collected using structural illumination microscopy (SIM) except (C), which was obtained by spinning disk confocal microscopy (SDCM).
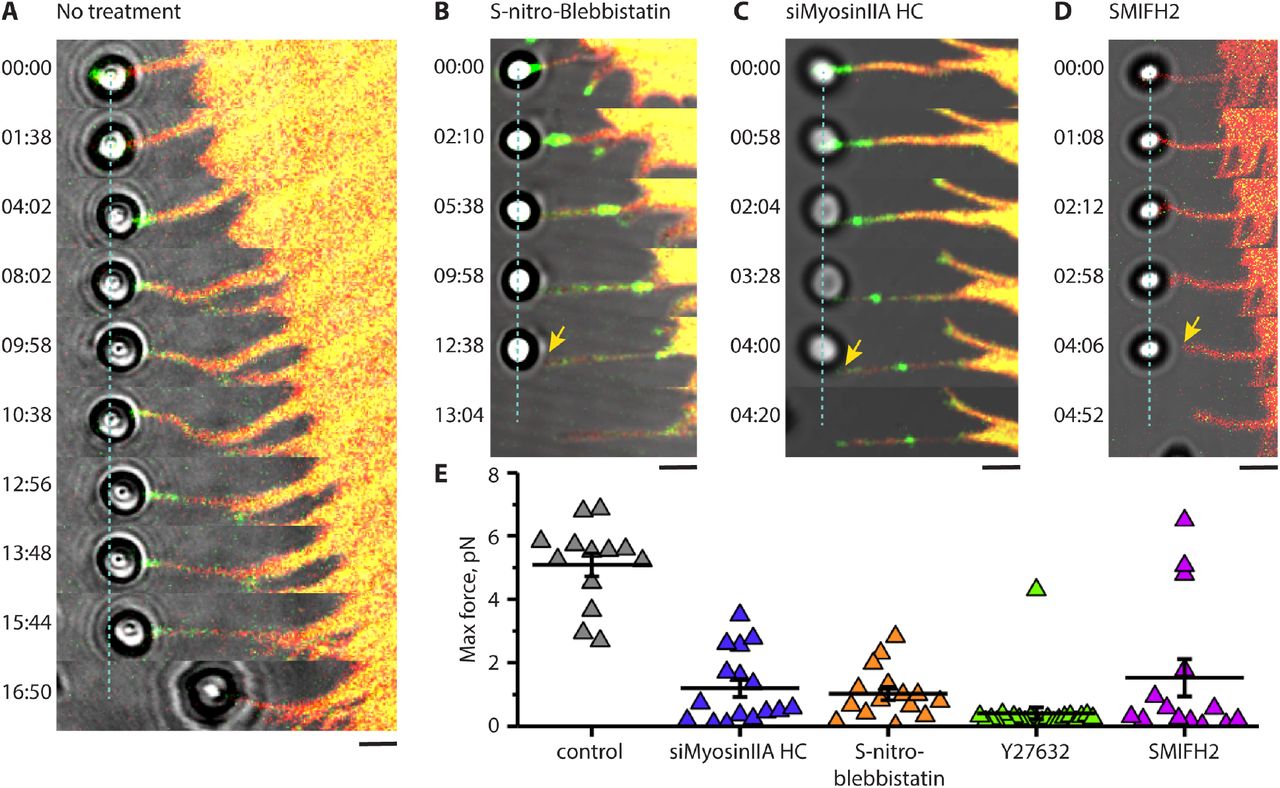
(A) Filopodium growth upon application of pulling force in HeLa-JW cells. The deflection of the bead from its initial position at the center of the laser trap (dashed line) is proportional to the forces exerted by the filopodium (see Fig. 1, movie S6A). At 16:50 min the filopodium retracted and pulled the bead out of the trap. (B-D) Filopodia in cells with suppressed myosin II or formin activity cannot maintain sustained adhesion to the bead and do not produce forces sufficient for noticeable bead deflection during the stage movement. Cells treated with 20μM of S-nitro-blebbistatin for 10-20 min (B), transfected with myosin IIA siRNA (C), or treated for with 40μM of the formin inhibitor SMIFH2 for 1 hour (D) are shown. GFP-myosin X and tdTomato-Ftractin are shown in green and red, respectively. See also movies S6B-D. The adhesions of the beads to filopodia were broken at 6 min 45 s, 3 min 50s and 2 min after starting the stage movement for S-nitro-blebbistatin-treated, myosin IIA knocked down, and SMIFH2-treated cells, respectively. Scale bars, 2μm. (E) Peak values of the forces exerted by filopodia on the beads during the stage movement in control cells (no treatment) and in cells transfected with myosin IIA siRNA, or treated with S-nitro-blebbistatin, Y27632 (30μM, 10-20 min), or SMIFH2. Mean values (horizontal lines) and SEMs (error bars) are indicated. The mean±SEM of the maximal forces exerted by control filopodia (5.1±0.4pN, n = 13) was significantly higher than those in myosin IIA knockdown, as well as S-nitro-blebbistatin-, Y27632-, and SMIFH2-treated cells (1.2±0.3, n = 16; 1.0±0.2, n = 15; 0.4±0.2, n = 22; and 1.5±0.6pN, n = 14, respectively) (p<0.0001 for control vs all treatment cases).
During the first 3 mins after stage movement commenced, the exerted force approached the maximal value of 3-5 pN. However, it then dropped to the 1.5-2pN range, and remained at this level for a further 1-3 mins, after which it rapidly increased again (Fig. 1C). In a typical experiment, we detected 2-4 such peaks with a mean peak force value of 3pN alternating with the 1-3 min periods of lower force (1.5-2pN).
The pattern of force dependent filopodia elongation described above was typical for myosin X-induced filopodia, but not for filopodia induced by constitutively active Cdc42 (Q61L). Cdc42-induced filopodia attached to laser-trapped fibronectin-coated beads did not grow upon stage movement and eventually pulled the beads out of the trap (movie S5). In this study, we exclusively focused on the filopodia-induced by myosin X overexpression.
Immediately after attachment of the bead to the filopodium tip, the myosin X patch, or a significant portion that pinched off the main myosin X mass, started to move centripetally with an approximate velocity of 31 ± 5 nm/s (mean ± SEM, n = 42). Co-expression of myosin X with VASP in HeLa-JW cells revealed that the retrogradly moving myosin X patches were always colocalized with the patches of VASP (fig. S1B middle panel). Moreover, centripetal movement of myosin X patches in cells transfected with photoactivatable β-actin and myosin X proceeded with the same velocity as the movement of a photoactivated actin spots in the same filopodia (fig. S1C, movie S7). However, despite the retrograde movement of a large part of myosin X, it did not entirely disappear from the filopodium tip and the original amount was fully restored after several minutes (Fig. 1C, kymograph), even though detachment and subsequent centripetal movement of myosin X portions from the filopodium tip were occasionally observed throughout the entire period of force-induced filopodium growth (movie S3).
Involvement of myosin II in filopodia dynamics
Expression of GFP labeled myosin light chain in HeLa-JW cells showed that myosin II does not localize to the filopodia tips or shafts, but is often located at the proximal ends of the filopodia (Fig. 2A and B top, movie S8A). Structure illumination microscopy (SIM) revealed few myosin II mini-filaments on either side of the filopodium base.
Localization of exogenous myosin IIA at the filopodia bases was especially prominent in Cos-7 cell model. In should be noted that Cos-7 cells express myosin IIB and IIC, but do not contain myosin IIA 42, 43. Upon transfection with myosin X, the wild type Cos-7 cells form filopodia with a short life-time that apparently do not adhere to the fibronectin-coated substrate (Fig. 2C, movie S8B). Co-transfection of these cells with GFP-myosin IIA heavy chain resulted in formation of numerous bipolar myosin IIA filaments frequently localized to the bases of filopodia (Fig. 2B bottom, 2D, movie S8C). Majority of filopodia in such cells were associated with myosin IIA filaments during the period of observation. Indeed, the analysis of the histories of 25 filopodia from the movie S8C revealed that 17 of them (68%) had one or more myosin IIA doublets at their bases during the movie duration (4 min 5 s). Myosin X-containing filopodia in Cos-7 cells co-transfected with myosin IIA were somewhat longer than those in wild type Cos-7 lacking myosin IIA (fig. S2). Moreover, expression of myosin IIA significantly increased the lifetime of myosin X-induced filopodia (Fig. 2J) as it can be inferred from the analysis of survival fraction of the filopodia cohort (Fig. 2I).
In agreement with previous studies, formation of filopodia in Cos-7 cells can be also induced by expression of truncated myosin X lacking FERM domain (Fig. 2E, movie S8D) 44 Co-expression of myosin IIA significantly enhanced the lifetime of the filopodia induced by this truncated myosin X (Fig. 2I and J, movie S8E) showing that binding of myosin X to integrin via FERM domain 25 is not required for myosin IIA-driven enhancement of filopodia lifetime. In contrast, expression of GFP-myosin IIB heavy chain in Cos-7 cells did not affect the filopodia lifetime (Fig 2I, J). Consistently, myosin IIB was not localized to the filopodia base in these cells (Fig. 2G, movie S8F).
Mutant myosin IIA N93K has reduced myosin ATPase and motor activity, but preserves the ability to bind to actin filaments 45, 46. GFP fusion construct of this mutant forms filaments localized to the bases of myosin X-induced filopodia in Cos-7 cells, similarly to wild type myosin IIA (Fig. 2H). However, unlike the wild type, the mutant myosin IIA N93K only slightly enhanced the lifetime of filopodia (Fig. 2I, J). Thus, motor activity of myosin IIA is critically important for the effect of myosin IIA on filopodia life time.
We further studied how the presence and activity of myosin IIA affects unconstrained and force-induced filopodia growth. The function of myosin II was suppressed in HeLa-JW cells in three separate experiments: through the inhibition of ROCK by Y27632, by siRNA mediated knockdown of myosin IIA heavy chain (MYH9), and through the inhibition of myosin II ATPase activity by light-insensitive S-nitro-blebbistatin. Inhibition of ROCK blocks myosin II regulatory light chain (RLC) phosphorylation, which interferes with myosin II filament assembly 47-50. As a result, HeLa-JWcells treated with 30 μM of Y27632 essentially lose their myosin II filaments in less than half an hour. siRNA knockdown of MYH9 also resulted in a loss of most of the myosin II filaments (fig. S3). Inhibition of myosin II ATPase activity by S-nitro-blebbistatin did not disrupt myosin II filaments 50, although this treatment did result in profound changes to the organization of the actomyosin cytoskeleton, including a loss of stress fibers. While treatment with either S-nitro-blebbistatin or Y27632 did not change the average filopodia length, myosin IIA knockdown resulted in moderate but statistically significant shortening of filopodia, (fig. S2), which is consistent with the results on filopodia length in wild type and myosin IIA expressing Cos-7 cells mentioned above (fig. S2). Myosin X-positive comet tails persisted at the tips of filopodia in both HeLa-JW and Cos-7cells irrespectively to inhibition or lack of myosin IIA.
Despite the morphological integrity of filopodia being preserved in myosin II inhibited or depleted cells, adhesion of filopodia to the ECM was significantly impaired. While in control HeLa-JW cells application of pulling force via fibronectin-coated bead induced sustained growth of attached filopodia accompanied by the development of up to ∼ 5 pN force, in the cells with impaired myosin II activity the filopodia detached earlier, after developing rather small forces (Fig. 3B-C, E, movies S9 B-C). This suggests filopodia are unable to establish a proper adhesion contact in the absence of active myosin IIA. We also examined the immediate effect of Y27632 during the force-induced sustained growth of filopodia. Shortly after the drug was added to the experimental chamber, filopodia indeed detached from the bead (fig. S4, movie S10).
Interaction between actin filaments and formins is required for filopodia adhesion and myosin X localization
In myosin X-induced filopodia, the formin mDia2 is localized to the filopodia tips, and overlaps with myosin X patches (fig. S1B, upper panels). Small molecular inhibitor of formin homology domain 2 (SMIFH2) 51 was used to investigate the role of formins in attachment of filopodia to fibronectin-coated beads. We found that in SMIFH2 (40μM, 1hour) treated cells, adhesion of filopodia to the beads was impaired in a similar way to the adhesion of filopodia in myosin II inhibited/depleted cells. The duration of contact between the filopodia and bead was significantly shorter, and the maximal force exerted by filopodia to the bead was significantly weaker than in control cells (Fig. 3D-E, movie S9D).
While the number of filopodia in cells treated with SMIFH2 remained the same as in control cells and their mean length decreased only slightly (Fig. 4A), only about 25% of these filopodia preserved myosin X comet tails at their tips 1-2 hours following SMIFH2 addition (fig. S5; filopodia in 14 cells were scored in 2 independent experiments) despite originally being induced by over-expression of myosin X. We found that SMIFH2 induced rapid disintegration of the comet tails into myosin X patches, which rapidly moved centripetally towards the cell body (Fig. 4B, movie S12). Although such movement was occasionally observed in control cells (above), it was much more prominent in cells treated with SMIFH2, and led to the gradual disappearance of myosin X from the filopodia tips. Of note, the movement of myosin X patches in SMIFH2 treated cells occurred together with the movement of its partner VASP 52, another protein associated with barbed ends of actin filaments (fig. S6, movie S6).

(A) The average length of unconstrained filopodia (left) in control HeLa-JW cells expressing GFP-myosin X (mean±SEM) was 4.1±0.1μm (n = 1710 in 34 cells), which exceeded that of SMIFH2 treated cells 3.2±0.1μm (n = 1645 in 31 cells), while the numbers of filopodia (right) per micron of cell boundary did not differ significantly (mean±SEM): 0.36±0.01 (n = 34 cells) and 0.39±0.02 (n = 31 cells), respectively. The mean values are indicated by horizontal black lines; the error bars correspond to SEMs. (B) Upper panel: Disintegration of the myosin X comet tail following a 2 hours exposure to 20μM SMIFH2. Numerous myosin X patches are seen in the filopodia shaft. Lower panel: A kymograph showing fast centripetal movement of the patches boxed in the upper panel towards the cell body (red arrowheads, see also movie S12). Intervals of slow centripetal movements are indicated by yellow arrowheads. (C) Addition of Y27632 treatment stops the movement of myosin X patches in SMIFH2 treated cells. The same filopodium is shown before SMIFH2 treatment (upper panel), 15 min after the addition of 20μM SMIFH2 (middle panel) and 15 min after subsequent addition of 30μM Y27632 (lower panel). Myosin X patches are shown in the left images (see also movies S13A-C), and kymographs representing the movement of the patches in the boxed area - in the images on the right. All images and data for analysis were obtained with SDCM. Scale bars, 5μm.
The velocity of retrograde movement of myosin X patches in filopodia of cells treated with SMIFH2 was 84 ± 22 nm/s (mean ± SEM, n = 45) vs 31 ± 5 nm/s in control cells (see the first section of Results). Such movement might be a result of the detachment of myosin X-bearing actin filaments from the filopodia tips. Once free, their subsequent retrograde movement is driven by myosin II located at the bases of the filopodia. Indeed, incubation of SMIFH2 treated cells with Y27632 efficiently stopped the retrograde movement of the myosin X positive patches, reducing their average velocity to 0.66±0.14 nm/s (mean ± SEM, n = 17), see Fig. 4C and movies S13A-C.
We also studied the immediate effect of SMIFH2 during the force-induced sustained growth of filopodia. After the drug was added, myosin X retrograde movement, cessation of the polymerization of the actin core, and the drop of the pulling force generated by a filopodium were observed (movie S11). The fibronectin-coated bead, however, remained associated with the filopodium tip via the membrane tether (movie S11). This suggests that addition of SMIFH2 induced disruption of the link between actin core and the integrin adhesion receptors, which still remain associated with the membrane tether at the filopodium tip.
To prove that SMIFH2 treatment can detach actin filaments from formin located at the filopodia tips, we performed in vitro experiments where the actin filaments were growing from immobilized formin mDia1 construct (FH1FH2DAD) in the absence or presence of SMIFH2. Following treatment with 100μM SMIFH2, a rapid decrease in the fraction of filaments remaining associated with immobilized formins under conditions of mild shear flow was observed (fig. S7). Thus, SMIFH2 treatment disrupted physical contacts between formin molecules and actin filaments. Therefore, SMIFH2-induced rapid centripetal movement of myosin X is driven by myosin II mediated pulling of actin filaments detached from the filopodia tips.
Effect of myosin II and formin inhibition on the growth of unconstrained filopodia
In addition to the studies of filopodia growing in response to pulling forces, we examined the effects of myosin II and formin inhibition on the dynamics of free, unconstrained filopodia (Fig. 5 inset). We found that knockdown of myosin IIA and cell treatment with Y27632 or S-nitro-blebbistatin efficiently blocked growth and retraction of unconstrained filopodia, resulting in suppression of filopodia dynamics. In untreated myosin X-expressing cells, the fraction of filopodia in the “pause” state (with the growth rate between −15 and +15nm/s) was 13% (n = 194). At the same time, fractions of the “pausing” filopodia were 90% (n = 41), 80% (n = 83) and 55% (n = 42) for myosin IIA knockdown, S-nitro-blebbistatin-treated and Y27632-treated cells, respectively (Fig. 5). Similarly, the fraction of “pausing” filopodia in cells treated with the formin inhibitor SMIFH2 was 75% (n = 44) (Fig. 5)

A graph showing the distribution of growth/retraction velocities of unconstrained HeLa-JW cells filopodia for control, myosin II siRNA knockdown, S-nitro-blebbistatin, Y27632 and SMIFH2 treatment, observed in the same experiments as those assessing the fibronectin-coated bead attachment to filopodia. n represents the number of processed filopodia (with number of cells in parenthesis). (Inset) A cell expressing GFP-myosin X and tdTomato-Ftractin, which is representative of those used in experiments assessing filopodia growth. The filopodium attached to the laser trapped fibronectin-coated bead is indicated by the red arrowhead. Such filopodia were excluded from the score. Scale bar, 5μm.
In this study, we have shown that filopodia adhesion to the ECM is a force dependent process. This conclusion is based on experiments in which sustained growth of filopodia was maintained by the application of pulling force at the interface between a fibronectin-coated bead, and the tip of a filopodia. With this setup, inhibition of myosin II filament formation or myosin II ATPase activity resulted in suppression of filopodia adhesion to fibronectin-coated bead. In should be noted that our experiments were performed on filopodia induced by over-expressing myosin X and, therefore, our conclusions are, strictly speaking, only valid for this class of filopodia. However, myosin X has been shown to be a universal component of filopodia 26, so employment of such an experimental system does not restrict the generality of our finding.
Since myosin II is located at the bases of filopodia (Fig. 2), a question requiring further clarification is how the pulling force is transmitted to the filopodia tips. Our data are consistent with the idea that this force is transmitted by the filaments of the actin core attached to formin molecules and the filopodia tips. We have shown that formin inhibition by SMIFH2 suppresses filopodia adhesion to the beads in the same manner as inhibition of myosin II. Moreover, we demonstrated that SMIFH2 treatment led to a rapid, myosin II-dependent, movement of actin filament associated proteins, myosin X and VASP, from the filopodia tips towards the cell body. Since centripetal movements of myosin X patches proceed together with the movements of photoactivated actin spots, we interpret the acceleration of myosin X/VASP movement upon SMIFH2 treatment as an evidence of actin filament detachment from formins at the filopodia tips. Indeed, in vitro experiments demonstrated that addition of SMIFH2 to actin filaments growing from the immobilized formins under a condition of moderate flow results in the detachment of actin filaments from the formin molecules. Together, these experiments suggest that myosin II inhibition, or inhibition of the formin-mediated association between actin filaments and the filopodia tips, make filopodia unable to form stable adhesions with fibronectin-coated beads. This in turn prevents them from growing upon force application.
To check whether adhesion and growth of filopodia require the pulling force, we compared behavior of unconstrained filopodia on rigid substrate with that on fluid supported lipid bilayer (SLB) where the traction forces cannot develop 53. To this end, we created a composite substrate on which rigid surface was covered by orderly patterned small islands (D = 3μm) of SLB. Both rigid and fluid areas were coated with integrin ligand, RGD peptide, with the same density (fig. S8). We found that dynamics of filopodia extended over rigid regions of this substrate was similar to that of filopodia growing on rigid fibronectin-coated substrate used in rest of experiments. At the same time, filopodia that encountered the SLB islands could not attach properly and as a result spent over such substrate significantly shorter time than over rigid area of the same geometry (fig. S9A, B, movie S14). Accordingly, the average density of filopodia tips remaining inside the SLB islands during period of observation (> 10min) was lower than that on the rigid substrate (fig. S9C). Thus, not only inhibition of myosin II or formin, but also micro-environmental conditions under which filopodia tips do not develop traction force, prevent proper adhesion of filopodia.
DISCUSSION
In the present study, we have demonstrated that adhesion of myosin X-induced filopodia to extracellular matrix depends on the forces generated by associated myosin IIA filaments. Our interest to filopodia induced by myosin X is in part justified by the fact that this protein is overexpressed in many types of cancer cells and adhesion of myosin X containing filopodia to the matrix may play an important role in the cancer cells invasion and metastasis. 54
The structured illumination microscopy (SIM) revealed the presence of individual myosin IIA filaments at the base of filopodia. The force generated by one bipolar myosin IIA filament (consisting of about 30 individual myosin molecules 55, 15 at each side) can be estimated based on the stall force for individual myosin IIA molecule (3.4pN according 56) and duty ratio (5-11% according 57) as 2.6-5.6pN. This value is consistent with pulling forces generated by individual filopodia as measured in our experiments. In the presence of formin inhibitor SMIFH2 detaching the filaments of the actin core from formin molecules at the filopodia tips, the filaments retract centripetally towards the cell body as can be inferred from the analysis of movements of myosin X and VASP clusters associated with these filaments. Inhibition of myosin IIA filament formation by Y-27632 suppressed such retrograde movement inside the filopodia. The forces generated by myosin IIA filaments appear to be sufficient to overcome the actin filament crosslinking inside the core and generate retrograde filament movement. Altogether, these data suggest that myosin IIA-generated forces are transmitted to filopodia tips via actin cores associated with formin molecules at the tips.
Dependence of filopodia adhesion on myosin IIA-generated forces was revealed in two types of experimental systems. First, we studied the effects of application of pulling force on the filopodia. In control HeLa-JW cells, filopodia attached to optically trapped fibronectin-coated beads were growing upon slow movement of the microscope stage and developed periodic 2-5pN forces. Myosin IIA-depletion or cell treatment with inhibitors of myosin II assembly (Y-27632) or motor activity (blebbistatin) led to significant decrease of the forces exerted by filopodia and subsequent detachment of filopodia from the trapped beads. Thus, myosin IIA-generated forces are required for the adhesion of filopodia to the beads and for force-induced filopodia growth.
Second, the function of myosin IIA in the regulation of filopodia lifetime in Cos-7 cells was studied. In Cos-7 cells lacking myosin IIA, myosin X overexpression induces only unstable short living filopodia. Their lifetime, however, can be significantly increased upon expression of exogenous myosin IIA. Such increase of the lifetime can be explained by stabilization of adhesion of these filopodia to the substrate. In contrast, myosin IIB overexpression does not increase the filopodia lifetime and does not localize to filopodia bases. Importantly, even though myosin IIA mutant with compromised motor activity (myosin IIA N93K) still localizes to filopodia bases, it only slightly enhances the filopodia lifetime. Thus, the motor function of myosin IIA rather than its actin crosslinking function 45 is important for stabilization of filopodia adhesion.
Force-driven growth of filopodia attached to the bead is an integrin-dependent process and was not observed in experiments with integrin-independent adhesion of filopodia to beads coated by concanavalin A. At the same time, association of integrin with myosin X FERM domain was dispensable for the myosin IIA-driven augmentation of the filopodia lifetime. This suggests that potential link between integrin and actin filaments via myosin X is not critically important for filopodia adhesion stabilization.
A major linker between integrin and actin filaments, talin, has been detected at the filopodia tips in this and other publications 30. Previously, it was established that force-driven unfolding of talin facilitates interaction of talin with another adhesion complex component, vinculin, resulting in reinforcement of the association between talin and actin filaments 58–61. The question whether this mechanism is applicable to filopodia adhesion reinforcement requires additional studies. While vinculin enrichment was detected in shafts but not tips of filopodia 62, a RIAM protein that competes with vinculin for talin-binding and localizes to filopodia tips 30, 63 may be involved in force dependent filopodia adhesion. RIAM binds Ena/VASP and profilin64 and could recruit these actin polymerization-promoting proteins to the filopodia tips.
Another mechanism of myosin II-dependence of filopodia adhesion and growth might involve formin-driven actin polymerization known to be a major factor in filopodia extension 15–17, 19–21. Recent studies demonstrate that formin-driven actin polymerization can be enhanced by pulling forces 65–68. Thus, myosin II-generated force transmitted via actin core to formins at the filopodium tip can stimulate actin polymerization, promoting filopodia growth. Polymerization of actin could also be important for recruitment of new adhesion components to the filopodia tips and adhesion reinforcement 69.
Filopodia adhesion is tightly associated with filopodia growth and shrinking. In our experiments, inhibition of myosin II and formins not only suppressed filopodia adhesion but also resulted in reduction of motility of filopodia along the substrate. During pulling-induced growth of bead-attached filopodia, periods of filopodia elongation alternate with periods of growth cessation accompanied by increase of the pulling force. Notably the periods of force development preceded the periods of filopodia elongation. Thus, force developed during growth cessation may trigger the subsequent filopodia growth. Similarly, the growth of unconstrained filopodia along rigid substrate can proceed via periods of attachment, development of force, and consequent filopodia elongation 41. Inhibition of force generation or transmission suppresses such dynamics. Interestingly, in filopodia of other types the attachment of filopodia tips to the substrate instead of further elongation led to formation of associated lamellipodia 70. It is not yet known, whether this switch is also mediated by myosin II-generated force.
Our finding that filopodia adhesion and growth are both is force-dependent explains how filopodia could respond differently to substrates of varying stiffness. On a stiff substrate, the force generated by myosin II and applied to the adhesion complex will develop faster than on a compliant substrate 71. Accordingly, filopodia adhesion should be more efficient on stiff substrates than on compliant substrates. Indeed, we showed that the contacts of filopodia with RGD ligands associated with fluid membrane bilayer were less stable than with the areas of rigid substrate covered with RGD of the same density. These considerations can explain involvement of filopodia in the phenomenon of durotaxis 39, a preferential cell movement towards stiffer substrates 72. This may provide a mechanism to rectify directional cell migration.
Orientation based on filopodia adhesion is characteristic for several cell types, in particular for nerve cells. The growth cones of most neurites produce numerous filopodia, and the adhesion of these filopodia can determine the direction of neurite growth 73, 74 Interestingly, the filopodia-mediated traction force in growth cones is myosin II-dependent 75 and application of external force can regulate the direction of growth cone advance 76. The results from these experiments can now be explained by preferential adhesion/growth of filopodia, which experience larger force. The mechanosensitivity of filopodia adhesion provides a mechanism of cell orientation that complements that mediated by focal adhesions. Focal adhesions are formed by cells attached to rigid two-dimensional substrates, whereas filopodia adhesion can be formed by cells embedded in three-dimensional fibrillar ECM network. Thus, further investigation of filopodia mechanosensitivity could shed a new light on a variety of processes related to tissue morphogenesis.
METHODS
Cell culture and transfection
Hela-JW, a subline of a HeLa cervical carcinoma cell line derived in the laboratory of J. Willams (Carnegie-Mellon University, USA) on the basis of better attachment to plastic dishes 77, was obtained from the laboratory of B. Geiger 78. Cos-7 (African green monkey fibroblast-like cell line, was obtain from ATCC (ATCC® CRL-1651™). The cells from both cell lines were grown in DMEM-Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum (FBS), 100 U ml−1 penicillin/streptomycin, 2 mM glutamine and 1 mM sodium pyruvate at 5% CO2 at 37 °C. HeLa-JW cells were transfected with DNA plasmids by electroporation (two pulses of 1005 V for 35 ms) using Neon transfection system (Thermofisher), while Cos-7 cells – by jetPRIME transfection reagent (Polyplus) according to manufacturer protocol. Expression vectors encoding the following fluorescent fusion proteins were used: GFP-myosin X and GFP-Myo10 ΔFERM 40, 44 (gift from R. Cheney, University of North Carolina Medical School, USA), reduced size myosin X construct (without 3’UTR) derived from GFP-myosin X was sub-cloned into mApple-C1 cloning vector backbone (M. Davidson collection, Florida State University, USA, kindly provided by P. Kanchanawong, MBI, NUS, Singapore), tdTomato–Ftractin 79 (gift from M. J. Schell, Uniformed Services University, Bethesda, Maryland, USA), mCherry-Utrophin 80, 81 was kindly provided by W. Bement (University of Wisconsin, Madison, USA), mTagBFP-Lifeact was obtained from M. Davidson collection, Florida State University, USA (Addgene), myosin regulatory light chain (RLC)-GFP82 (gift from W. Wolf and R. Chisholm, Northwestern University, Chicago, Illinois, USA), GFP-myosin IIA heavy chain was sub-cloned from pTRE-GFP-NMHCIIA (from R. Adelstein, NHLBI, NIH, USA) into pEGFP-C3 by MluI & HindIIIcloning sites (cloned by M. Tamada, M. Sheetz laboratory), GFP-myosin IIA N93K 49 was kindly provided by Dr. Vicente-Manzanares (Universidad Autónoma de Madrid, Madrin, Spain), GFP-myosin IIB, mCherry-VASP and mCherry-talin (M. Davidson collection, Florida State University, kindly provided by P. Kanchanawong, MBI, NUS, Singapore), full-length mDia2 was sub-cloned from GFP-C1-mDia2 83 (gift from S. Narumiya, Faculty of Medicine, Kyoto University, Japan) into mCherry-C1 vector(Clontech), PAmCherry-b-actin was kindly provided by V. Verkhusha (Albert Einstein College of Medicine, NY, USA). All cell culture and transfection materials were obtained from Life Technologies.
Live cell imaging and confocal microscopy
Following electroporation, cells were seeded at a density of 2 × 104 cells ml−1 in 2ml onto 35mm glass based dishes with 12 or 27 mm bottom base cover glass #1 in diameter (Iwaki, Japan) coated with 10 μg ml−1 fibronectin (Calbiochem) for 20 min. Cells were imaged in Leibovitz’s L-15 medium without Phenol Red containing 10% FBS at 5% CO2 at 37 °C. Snapshot or time-lapse images were acquired with a spinning-disc confocal system (PerkinElmer Ultraview VoX) based on an Olympus IX81 inverted microscope, equipped with a 100 × oil immersion objective (1.40 NA, UPlanSApo), an EMCCD camera (C9100-13, Hamamatsu Photonics), and Volocity control software (PerkinElmer).
We perform photoactivation experiments by activation of a defined region inside filopodia using blue diode laser (405 nm, 100mW). Photoactivation and livecell imaging were performed on CSU-W1 spinning disk confocal system on Nikon Eclipse Ti-E inverted microscope with Perfect Focus System, controlled by MetaMorph software (Molecular device) supplemented with a 100x oil 1.45 NA CFI Plan Apo Lambda oil immersion objective and sCMOS camera (Prime 95B, Photometrics).
For structured illumination microscopy (SIM), two types of equipment were used: (1) Live SR (Roper Scientific) module on Nikon Eclipse Ti-E inverted microscope (specifications of setup described above), (2) Nikon N-SIM microscope, based on a Nikon Ti-E inverted microscope with Perfect Focus System controlled by Nikon NIS-Elements AR software supplemented with a 100x oil immersion objective (1.40 NA, CFI Plan-ApochromatVC) and EMCCD camera (Andor Ixon DU-897). For life cell imaging the samples were mounted in a humidified cell culture chamber and maintained at 37 °C with 5% CO2. All SIM images with obtained with system (1) except images on Fig. 2A and B (upper panel), which were obtained with set up (2).
Transfection of siRNA and immunoblotting
Cells were seeded into a 35 mm dish on day 0 and transfected with 100μM of MYH9 ON-TARGET plus SMART pool siRNA (L-007668-00-0005, Dharmacon) using ScreenfectTMA (WAKO, Japan) on day 1. Control cells were transfected with scrambled control ON-TARGET plus Non-targeting pool siRNA (D-001810-10, Dharmacon). Transfection of plasmid GFP-myosin X and tdTomato-Ftractin was performed in the evening of the day 1 using Jet Prime transfection reagent (Polyplus) and cells were imaged on day 2. For assessment of myosin IIA heavy chain expression, transfected cells were lysed in RIPA buffer on day 2 (exactly 24 hours following siRNA transfection) and analyzed by Western blotting with primary rabbit antibodies to the myosin IIA tail domain (M8064, Sigma-Aldrich, dilution 1:1000); staining of α-tubulin with monoclonal DM1A antibody (T6199, Sigma-Aldrich, dilution 1:5000) was used as a loading control. HRP-conjugated anti-rabbit IgG (Bio-Rad, 1706515, dilution 5000) and anti-mouse IgG (A4416, Sigma-Aldrich, dilution 1:10000) were used as secondary antibodies, respectively.
Immunofluorescence antibody staining
Anti-myosin IIA tail domain (M8064, Sigma-Aldrich, dilution 1:800), and 405 Alexa-Fluor-conjugated secondary antibodies (A31556, Molecular Probes, dilution 1:200). Cells were pre-fixed by addition of warm 20% PFA (Tousimis) into medium (to make 8% solution) and subsequent 15 min incubation at room temperature. This was followed by fixation and permeabilization by 3.7% PFA, 0.2% glutaraldehyde and 0.25% Triton X-100 in PBS for 15 min. The fixed cells were then washed two times with PBS and blocked with 5% bovine serum albumin (BSA) for 30 min. The cells were then stained with primary antibodies overnight at 4 °C, and incubated with secondary antibodies for 1h at room temperature.
Drug treatment
For formin drug inhibition studies, cells were incubated with 20 or 40μM SMIFH2 (4401, TOCRIS, UK) in serum containing DMEM for 1-2h at 5% CO2, 37 °C. In in vitro experiments, SMIFH2 (340316-62-3, Sigma-Aldrich) was used at a concentration of 100μM (see below). For myosin II inhibition studies, 30μM or 50μM Rho-kinase (ROCK) inhibitor Y27632 (Y0503, Sigma-Aldrich) or 20μM S-nitro-blebbistatin (13013-10, Cayman Chemicals) was added for 10-20 min before the experiments or directly during observations. All inhibitors remained in the medium during the entire period of observation.
Optical tweezers and data acquisition
All experiments involving filopodia pulling were carried out on a Nikon A1R confocal microscope adapted for the use of laser tweezers. 2.19 μm diameter polystyrene beads (PC05N, Bangs Laboratories) were coated with fibronectin (341635, Calbiochem) according to a previous protocol 84 For bead trapping we used an infrared laser (λ = 1064nm, power 0.5-1W, YLM-5-LP-SC Ytterbium Fiber Laser, IPG photonics).
To determine the forces, F, applied to the bead by the optical trap, we measured the displacement of the bead from the trap center, Δx, and then knowing the stiffness of the trap, k, the force was calculated as: F = kΔx. The trap stiffness was calibrated using the equipartition method 85 by tracking the fluctuations of a bead trapped by optical tweezers, using an Andor Neo sCMOS camera, at 100fps. The displacement of the beads from the center of the optical trap in confocal microscopy observations was monitored using piA640-210gm camera (Basler) at 0.5-1fps and Metamorph software for tracking. The smallest detectable bead displacement was ∼5 nm, corresponding to the smallest force measured of ∼0.04 pN.
For laser trap experiments, HeLa-JW cells transfected by electroporation with GFP-myosin X and tdTomato-Ftractin, were seeded at a density of 2 × 104 cells ml−1 in 750μl onto chambered #1 borosilicate cover glasses (155383, Lab-Tek) coated with fibronectin (341635, Calbiochem) by incubation in 1ng ml−1 solution in PBS for 20min at 37.0 °C. A reduced concentration of fibronectin (compared to that used for regular cell observations) was used to prevent the beads sticking to the cover slip. Chambers with cells were mounted on P-545.3R7 stage equipped with E-545 Plnano piezo controller (Physik Instrumente), which was moved in order to generate the pulling force between filopodia and trapped beads. The velocity of the stage movement was maintained in a range 10-20 nm/s by PIMikroMove 2.15.0.0 software. The specimen were incubated in a custom-built microscope hood at 37.0 °C, 5% CO2 humidified environment. Simultaneously with pulling, the cells were imaged using lasers λ = 488nm and 561nm for excitation of GFP and tdTomato, respectively. Collected experimental data were processed by particle tracking algorithms of the Metamorph software.
In vitro assay for formin processivity in the presence of SMIFH2
We used a microfluidics based assay to assess formin processivity in the course of formin-driven actin polymerization. The method has been described in more detail elsewhere 66. Briefly, formin construct Snap-mDia1(FH1FH2DAD)-6xHisTag was specifically anchored to the bottom surface of a microchamber, using a biotinylated pentaHis antibody (Qiagen) and streptavidin to bind to a biotin-BSA-functionalized glass surface which was further passivated with BSA. The microfluidics PDMS chamber height was 60 μm. Immobilized formin constructs were allowed to nucleate actin filaments by exposing them for 30s to a 2 μM solution of 20% Alexa488-labeled at Lys328 actin 86 and 0.4μM profilin in a buffer containing 10 mM Tris-HCl (Euromedex) pH 7.8, 1 mM MgCl2 (Merck), 200 μM ATP (Roche), 50 mM KCl (VWR Chemicals) and supplemented with 5 mM DTT (Euromedex), 1 mM DABCO (1,4-diazabicyclo[2.2.2]octane) to reduce photobleaching. Recording of actin filament elongation was started after the buffer was changed to contain 1 μM unlabeled actin and 4 μM profilin, with or without 100 μM SMIFH2 (Sigma-Aldrich). The microfluidics flow was kept low enough to ensure that the viscous drag had no impact on filament detachment from formin. Images were acquired every 10 s on a Nikon Ti-E microscope using a 60x objective and a Hamamatsu Orca Flash 4.0 V2+ sCMOS camera. Actin was purified from rabbit muscle 87. Recombinant Profilin I and Snap-mDia1(FH1FH2DAD)-6xHisTag were expressed in E. Coli and purified 88.
Filopodia density and length measurement; tracking filopodia tips
Cells labeled with GFP-myosin X, tdTomato-Ftractin or mCherry-Utrophin were imaged in 488nm (green) and 594nm (red) channels, simultaneously. Cell segmentation was performed using an in-house algorithm implemented as a Fiji macro. The segmented cell was analyzed using MATLAB software CellGeo 89 to estimate filopodia density and their length. First, each data set, which corresponded to 10 frames per movie taken at 1-2 fps at x100, was averaged over time to generate a smooth image for each channel. The averaged images from the green and red channels were then summed to produce an enhanced image for segmentation. To optimize the segmentation results, filopodia segmentation and cell body segmentation was conducted in two separate steps. For filopodia segmentation, a background subtraction procedure with a rolling ball of 20-pixel radius was applied first, followed by Triangle auto-thresholding (Fiji Auto Threshold v1.16.1). For cell body segmentation, Gray Morphology open operator with a circle of 5-pixel radius was used (Fiji Morphology) to remove filopodia before applying Li auto-thresholding (Fiji Auto Threshold v1.16.1). The final segmentation result was obtained by combining the masks from the above two steps. After segmentation, the BisectoGraph module of CellGeo 89 was used to partition each cell into the cell body and individual filopodia, based on the parameters called critical radius (roughly the half of maximal filopodia width) and minimum filopodia length. In our analysis, a critical radius of 0.7μm and a minimum filopodia length of 1.5μm were used. Note that, our definition of filopodia length slightly differed from that used in original paper by 89. Namely, in our analysis the filopodia length is defined to be the distance from the filopodia tip to the cell body boundary. The MATLAB code for computing this length was kindly provided by D. Tsygankov (Georgia Institute of Technology, Atlanta, GA, USA). For filopodia density quantification, the perimeter of the cell body was measured and the number of filopodia per unit of length (μm−1) was computed.
To track the filopodia tips, the Imaris 8.3.1 spots tracking procedure was used. The filopodia tips were identified using segmentation procedure described above followed by MATLAB binary morphology function bwmorph with ‘skel’ and ‘endpoints’ operations.
Unconstrained filopodia elongation and shrinking rates were obtained by linear regression fitting of the filopodia tips trajectories at the regions corresponding to the periods of filopodia persistent growth or shrinking, respectively.
Supported lipid bilayer micro patterns chamber and filopodia tips trajectory analysis on rigid and fluid substrates
The cleaned glass coverslips were coated with PLL-g-PEG-biotin (Susos) for two hours followed by UV etching using 3 μm circular shape arrays of chromium (Cr) photomask 90, which resulted in removal of PLL-g-PEG-biotin polymers on the etched circular regions. After multiple rinses with UHQ water, phospholipid vesicles were introduced to the surface for 5 min, allowing the self-assembly of lipid bilayer on the etched glass surfaces. Phospholipid vesicles were synthesized using the existing protocol 90. Specifically, 98 % of DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) was mixed with 2 % of biotin-DOGC (1,2-dioleoylsn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)-succinyl]) biotin lipids. The lipid solution was further diluted with Tris buffered saline (TBS, Sigma-Aldrich) at a ratio of 1:1 before introduction. The glass coverslips were assembled into the donut shape chamber at the UHQ water reservoir. The hybrid substrate was then incubated with 0.1 % bovine serum albumin (BSA, Sigma Aldrich) for two hours to reduce nonspecific protein absorption. Next, 1 μg/ml of Dylight-405 NeutrAvidin (Thermo Fisher) was introduced to the chamber for one hour, and then incubated with 1ug/ml of RGD-biotin (Peptides International) for one hour. Multiple rinses with UHQ were applied after each step. A fluidity test of RGD molecules at the surface of supported lipid bilayer (SLB) was performed by fluorescence recovery after photobleaching. The concentration of RGD on both SLB and PLL-g-PEG polymer was similar, based on the estimated fluorescence of Dylight-405 NeutrAvidin (fig. S8).
HeLa-JW cells expressing a myosinX-GFP chimera were introduced to the chamber, allowing cells to adhere to and spread on the RGD-coated hybrid substrate. Cells were visualized using a charge coupled EMCCD camera (Photometrics) coupled with total internal reflection microscopy (TIRF) with 100x objective (1.5 NA, Nikon). The chamber was maintained at 37°C during observation. The time-lapse video was recorded at varying intervals over a range of 3 – 5 s.
The GFP-Myosin X cluster at the filopodia tip was tracked by using a cross-correlation single particle tracking method 91. Obtained trajectories were analyzed using the “inpolygon” matlab function to find trajectory segments that crossed SLB circular islands and computer drawn islands in the center of the SLB pattern. The time intervals between the initial and end points of the segments were then used to estimate the average filopodia tip dwell time on the SLB and computer drawn circular islands. In addition, the ratio between the number of filopodia tip trajectories remaining inside rigid and fluid circles relatively to the total number of trajectories in the circles during the period of observation was calculated to characterize the filopodia adhesion preferences to rigid and fluid substrates (Fig. 6). In this analysis, we used only trajectories that spanned more than 5 frames.
Statistics and reproducibility
Prism (GraphPad 6.0 Software) was used for statistical analysis. Each exact n value is indicated in the corresponding figure or figure legend. The significance of the differences (P value) was calculated using the two-tailed unpaired Student’s t-test.
Acknowledgments
Encouraging and stimulating discussions with Drs. D. Bray (University of Cambrige, UK), M.M. Kozlov (Tel Aviv University, Israel) are much appreciated. We are grateful to Dr. T. Kachanawong (MBI, Singapore) for providing genetic constructs, Dr. D. Kovar (University of Chicago, IL, USA) for a sample of SMIFH2 inhibitor, and Dr. Tsygankov (Georgia Tech, USA) for providing code for filopodia length computation. We thank the Protein Cloning and Expression Core facility of the MBI for help with sub-cloning of mCherry-mDia2 and mApple-myosin X. We also thank Dr. F. Margadant and Lau Wai Han (MBI Microscopy Core facility) and Dr. V. Vyasnoff (MBI, Singapore) for their kind help with the optical tweezers setup. This research has been supported by the National Research Foundation Singapore, Ministry of Education of Singapore, Grant R714006006271 & R714019006271 (awarded to A.D.B.), Grant MOE2012T31001 (awarded to Y.J.) and BMRC Grant A*Star-JST 1514324022 (awarded to A.D.B.). As well we thank S. Wolf and A. Wang (Science Communication MBI, Singapore) for excellent editorial help.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.
- 8.
- 9.↵
- 10.↵
- 11.↵
- 12.
- 13.
- 14.↵
- 15.↵
- 16.
- 17.↵
- 18.↵
- 19.↵
- 20.
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.
- 35.↵
- 36.↵
- 37.
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.
- 60.
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}