

Abstract
Under the current standard of care, individuals with HIV take three antiretroviral drugs simultaneously. Triple-drug combination therapies limit HIV drug resistance evolution, because viruses resistant to a subset of the cocktail are suppressed by the remainder of the drugs and should not complete replication and spread. Despite this, reanalysis of HIV genetic data shortly after triple drug therapies became available (1990s and 2000s) reveals ongoing drug resistance in patients on three-drug therapies. In disagreement with expected patterns of evolution in three-drug therapy-treated HIV populations, resistance usually evolves one mutation at a time in a semi-predictable order. We argue here that these surprising observations can be explained using a model that divides the human body into compartments (for example, the gut, lymph nodes and brain). If one drug reaches a compartment that the other two drugs cannot, this creates a single-drug compartment that can select for single-drug resistant viruses. Such viruses can potentially become resistant to additional drugs, if they migrate to another compartment where a second drug is present, and so on. In addition to a compartment model, for some drug combinations, an alternative model of time-heterogeneity due to short half-lives combined with sub-optimal adherence could also explain the observations. We discuss how these lessons from HIV drug resistance evolution may be useful for other systems.
Introduction
In the 1980s and 90s, many individuals with HIV died after their virus became resistant to all available treatments. Triple-drug therapies, introduced in 1995, were expected to prevent the evolution of drug resistance and subsequent treatment failure. While these therapies saved many lives (Hogg et al., 1999; Walensky et al., 2006), drug resistance evolution continued well into the 2000s. Indeed, we argue that what actually happened in the years following the introduction of triple-drug cocktails represents an enduring mystery, one that has been in plain sight this whole time. Resistance rates did fall, but each year, a substantial number of patients on triple-drug therapy continued to develop drug resistance (Lee et al., 2014; Rocheleau et al., 2018).
The assumption behind using three-drug therapies was that only a triple mutant would allow the virus to replicate in its presence. But does this assumption reflect how resistance actually emerged in patients - three mutations at once? This HIV mystery only deepened when resistance rates continued to fall well into the mid-2010s, even though the number of drugs making up the drug cocktails often remained the same, raising a second question: How did we ultimately curb drug resistance in HIV with tripledrug treatments, without understanding how resistance was evolving in the first place? Although this unappreciated mystery of HIV’s resistance to tripledrug cocktails is largely a historical one, we believe that solving it could help us address the problem of drug resistance better in the present.
We argue here that the mystery of drug resistance evolution in patients on triple-drug therapy can be explained using a model that divides the human body into compartments (for example, the gut, lymph nodes and brain). If one drug is able to reach a compartment that the other two drugs can’t, this creates a single-drug compartment. Virus in the singledrug compartment can become resistant to the drug it encounters there; this virus can then potentially become resistant to a second drug, if it migrates to another compartment where a second drug is present, and so on. In addition to the compartment model, an alternative explanation of time varying drug levels rooted in short half-life drugs and sub-optimal adherence could also explain the observations for some therapies.
To illustrate these arguments, we present data on drug resistance from the mid-1990s through the first decade of the 2000s and then explore how those data fit the predictions of the compartment model. Ultimately, by using the compartment model to answer key questions about drug resistance in HIV, we believe it will be possible to extract valuable lessons that the medical community can use to continue fighting drug resistance across systems.
Even as HIV drug resistance remains a critical problem in many parts of the world without access to extensive second and third-line therapies (Gupta et al., 2012), the topic is also highly relevant for research on other systems. For example in cancer treatment, where new studies are showing the importance of spatial structure (Raab et al., 2016; Shi et al., 2014; de Bruin et al., 2013; Heindl et al., 2015; Carmona-Fontaine et al., 2013) and the availability of new targeted drugs is making combination therapy ever more feasible (e.g., Carter et al. (2016); Lopez and Banerji (2017); Abramson and Arteaga (2011)).
How can resistance to multiple HIV drugs evolve one mutation at a time?
Today, we treat HIV with multiple drugs simultaneously. The rationale is that even if a virus acquires a single drug resistance mutation, that virus will not be able to spread within the body, because it will still be suppressed by one or more other drugs. A virus should need to acquire multiple resistance mutations before it is able to replicate in the presence of multiple drugs. How a fully resistant pathogen could emerge has been examined in modeling studies focused on HIV and other pathogens treated with multiple drugs simultaneously (Lipsitch and Levin, 1998; Rosenbloom et al., 2012; Kim et al., 2014; Moreno-Gamez et al., 2015). In the case of HIV, it is often claimed that three drugs are necessary to inhibit resistance evolution, because double drug-resistant mutants are expected to pre-exist within large viral populations (Ribeiro et al., 1998).
However, this understanding does not fully capture the dynamics observed in intra-patient HIV populations. A large body of research shows that drug resistance has evolved in many patients on triple-drug cocktails, especially in the early years of these cocktails. For example, a 2006 study that followed 600 HIV-infected individuals over 3 years found a substantial risk of acquired drug resistance in every year of treatment, ranging from roughly 10% in year 1 to just over 3% in year 2 and 3 (Margot et al., 2006). Larger cohort studies have also documented a substantial risk of drug resistance among patients taking triple-drug therapies (Gill et al., 2010). Triple drug therapy does not always prevent the evolution of drug resistance.
Even more puzzling, evidence suggests that drug resistance mutations accumulate one at a time in HIV, even when a treatment contains multiple drugs (Pennings et al., 2014; Feder et al., 2016; Williams and Pennings, 2019). Such step-wise evolution should be impossible, according to the conventional wisdom. Why wouldn’t the other drugs keep a single-resistance mutant in check? In addition, step-wise mutation also happens in patients treated with just two drugs (Picard et al., 2001), which is even more surprising, as double drug-resistant mutants are likely already present in the body (Ribeiro et al., 1998). Therefore, the conventional reasoning that a fully drug-resistant virus must appear before resistant virus can successfully replicate in the body appears to be an oversimplification.
In the sections that follow, we will take a closer look at how exactly resistance to multiple drugs evolves in patients with HIV on triple-drug therapies, using the patterns that emerge across different types of regimens to learn about the evolutionary processes responsible.
Order of resistance mutations on protease inhibitor-based treatments is sequential and predictable
Until 1995, all HIV drugs were nucleoside analog reverse transcriptase inhibitors (NRTIs). In 1995, the FDA approved the first protease inhibitor (PI), saquinavir (Baker, 1995). In the industrialized world, treatments based on a PI and two NRTIs dramatically reduced mortality and morbidity due to HIV (Palella Jr et al., 1998). Yet, these treatments were far from evolution-proof.
For example, in a 2004 study with 653 participants, half (n=327) were randomized to treatment with lamivudine (3TC) + stavudine (D4T) + nelfinanvir (NFV), where NFV is the PI and 3TC and D4T are the NRTIs (Kempf et al., 2004). Within the first 2 years of therapy, drug resistance evolved in 79 of the 327 patients (24%). Among these patients, 35 acquired 3TC resistance (11%), 35 acquired 3TC resistance and NFV resistance (11%), and 8 acquired resistance to all three drugs (3TC, NFV and D4T, 2%). One patient acquired 3TC and D4T resistance, but not NFV resistance (0.1%). There were no patients with only D4T or only NFV resistance. These data strongly suggest that on 3TC+D4T+NFV treatment, drugs fail in a particular order: 3TC resistance arises first, then NFV and D4T resistance (see Figure 1A).

A. Resistance to the drug lamivudine (3TC) emerges first among patients with HIV taking the triple-drug cocktail nelfinanvir (NFV) + 3TC + stavudine (D4T) (n=327). NFV is a protease inhibitor and D4T and 3TC are nucleoside reverse transcriptase inhibitors. Source: Kempf et al. (2004). B. HIV populations treated with first-line protease inhibitor (PI)-based combination therapies possessing a single drug resistance mutation are most frequently resistant to 3TC or FTC via M184VI. Each bar represents a group of patients treated with a given PI-based combination therapy, and each sub-bar represents a patient with a single drug resistance mutation. Sub-bars are colored according to the identity of the single resistance mutation (PI drug resistance: yellow, M184VI conferring resistance to 3TC/FTC: dark blue, NRTI drug resistance: light blue). Sample sizes for each treatment are given by the therapy names. Data source: Feder et al. (2016). Abbreviations: abacavir, ABC; indinavir, IDV; lamivudine, 3TC; lopinavir, LPV; nelfinanvir, NFV; stavudine, D4T; saquinavir, SQV; zidovudine, AZT.
To determine if the trend of a predictable mutation order held for other PI-based combination therapies, we analyzed a large data set (around 7000 patients) encompassing many different treatments (assembled by the Stanford HIV drug resistance database, Rhee et al. (2003); Feder et al. (2016)) to identify the resistance profile of patients with just one drug resistance mutation. In this data set, a mutation conferring resistance to the NRTIs almost always occurred first when PI-based treatments were used; specifically, we observed the mutation M184V or M184I (shown in dark blue in Figure 1B), which confers resistance to 3TC, in around 90% of the cases.
These findings indicate that in PI-based combination therapies, drug resistance mutations occur in stepwise, predictable order.
Order of resistance mutations on non-nucleoside reverse transcriptase inhibitors-based three-drug treatments is also sequential and predictable
To determine whether the pattern of sequential and predictable resistance mutations was a general one in HIV, we investigate resistance to an additional class of drugs, the non-nucleoside reverse transcriptase inhibitors (NNRTIs). In 1996, the first NNRTI, nevirapine (NVP), was approved (Bowersox, 1996). NNRTIs, or “non-nukes,” had a different mode of action than other reverse transcriptase inhibitors then available, and triple reverse transcriptase-inhibitor therapy became an alternative to the existing PI-based treatments. In the 2000s and 2010s, NNRTI-based therapy became a very common HIV treatment (Rocheleau et al., 2018), but like all previous treatments, the NNRTI-based treatments were not evolution-proof.
In a study published in 2009 (Hoffmann et al., 2009), Hoffman and colleagues followed South African patients treated with one of two common NNRTI-based regimens: zidovudine (AZT)+ 3TC + efavirenz (EFV) (95% of patients) or AZT + 3TC + nevirapine (NVP) (5% of patients). We focused on patients taking the AZT+3TC+EFV regimen, where EFV is the NNRTI and AZT and 3TC are NRTIs. In this study, 68 patients who experienced virologic failure had their virus genotyped. At the first genotyping following virologic failure, 24 patients had no resistance mutations at all (treatment failure was likely due to lack of adherence), 19 patients had resistance to EFV, 20 patients had resistance to both EFV and 3TC, and 3 patients had resistance to all three drugs (EFV, 3TC, and AZT). Just 2 patients had resistance to 3TC only. No patients had resistance to AZT, AZT+3TC, or AZT+EFV. These data suggest that among patients on this NNRTI-based treatment, HIV evolved resistance in a particular order: resistance to EFV arose first, followed by resistance to 3TC and then AZT (see Figure 2A).

A. Resistance to the drug efavirenz (EFV) emerges first among patients with HIV taking the triple-drug cocktail EFV + lamivudine (3TC) + zidovudine (AZT) (n=68). EFV is a non-nucleoside reverse transcriptase inhibitor and AZT and 3TC are nucleoside reverse transcriptase inhibitors. Source: Hoffmann et al. (2009). B. HIV populations treated with first-line non-nucleoside reverse transcriptase inhibitor (NNRTI)-based combination therapies possessing a single drug resistance mutation are most frequently resistant to the NNRTI. Each bar represents a group of patients treated with a given NNRTI-based combination therapy, and each sub-bar represents a patient with a single drug resistance mutation. Sub-bars are colored according to the identity of the single resistance mutation (NNRTI resistance: red, M184VI conferring resistance to 3TC/FTC: dark blue, NRTI resistance: light blue). Sample sizes for each treatment are given by the therapy names. Data source: Feder et al. (2016). Abbreviations: abacavir, ABC; efavirenz, EFV; emtricitabine, FTC; lamivudine, 3TC; nevirapine, NVP; stavudine, D4T; tenofovir, TDF; zidovudine, AZT.
This example suggests that when someone is treated with AZT+3TC+EFV, NNRTI resistance (i.e., resistance to EFV) usually evolves first. Consistent with this prediction, in the Stanford HIV Drug Resistance Database dataset (Rhee et al., 2003; Feder et al., 2016), resistance to the NNRTI evolved first in about 80% of cases in patients taking an NNRTI-based regimen (red in Figure 2B), whereas 3TC resistance occurred first in only about 15% of cases (dark blue in Figure 2). Note the stark contrast with PI-based treatments, in which 3TC resistance almost always occurred first. Resistance to the other NR-TIs in NNRTI-based therapies, such as AZT, abacavir (ABC), D4T, and tenofovir (TDF) (light blue in Figure 2B), rarely occurred first. For some of the NNRTI-based therapies, the pattern was quite extreme. For example, for patients on EFV + TDF + emtricitabine (FTC), NNRTI resistance evolved first in each of the 31 cases in the Stanford data set.
These findings from a different class of drugs confirm that our model of drug resistance evolution in HIV must be able to explain why resistance evolution is (1) ongoing (2) stepwise and (3) predictable.
Surprises from the data presented
Collectively, the data we have presented yield three main surprises: (1) Drug resistance evolution was quite common in the 1990s and 2000s, even when treatment was with three drugs; (2) resistance evolves in a sequential manner (first one mutation, then a second, then a third, rather than two or three mutations at the same time); (3) the order of mutations is predictable; for example, on PI-based combination therapy, 3TC mutations almost always occur first. The predictability of the order of mutations cannot be explained by different mutational target sizes or mutation rates. For example, the product of the mutation rate and target size is two times higher for PI resistance than for 3TC resistance (1.7 10−4 for PIs such as NFV and LPV, vs 8 10−5 for 3TC, Supplemental Methods), yet 3TC resistance occurs first in about 90% of the cases (Figure 1B). The 3TC mutation M184V is also very costly for the virus, which should make it unlikely to be present as standing genetic variation. How, then, can we explain these patterns?
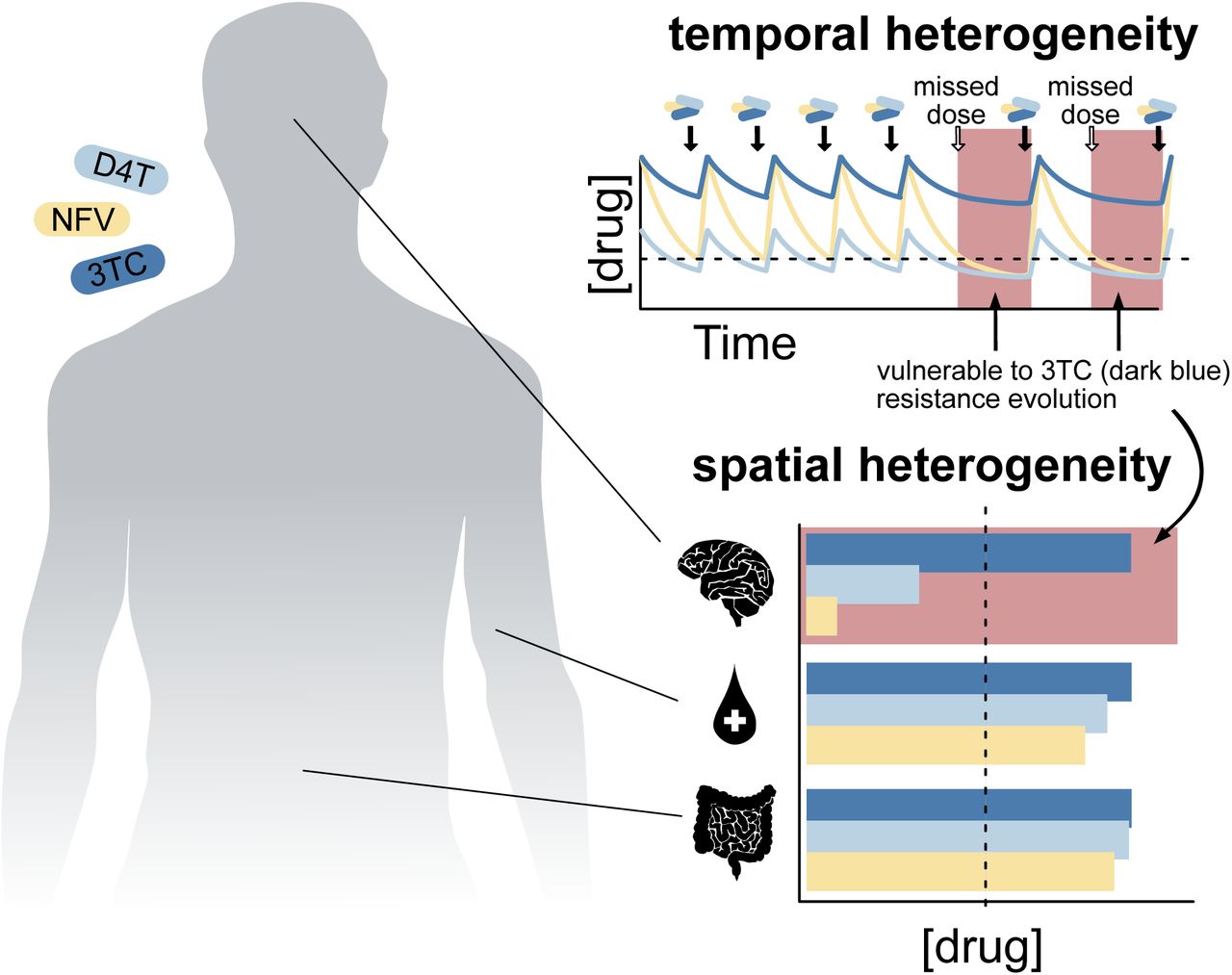
When three drugs are present at suppressive concentrations, resistance mutations should not spread in the body. Therefore, to explain the patterns of ongoing drug resistance evolution in HIV, we must invoke some heterogeneity in drug levels. This heterogeneity may manifest in space (incomplete penetration to all tissues) or in time (via differences in drug half lives). We discuss these two hypotheses below.
Lessons from a spatially-varying bodily compartment model
It is well known that HIV drugs do not penetrate throughout the body uniformly, and that penetration profiles differ among individual drugs and organs (Else et al., 2011; Letendre et al., 2008). Because penetration profiles differ between drugs, there may be parts of the body (e.g., brain, lymph nodes) that are only reached by one drug, even when someone is treated with three drugs (see Figure 3). These so-called single-drug compartments provide a space where a virus resistant to one drug can replicate. A viral particle in such a single-drug compartment with the right drug resistance mutation can establish an actively replicating viral population in the compartment. One of us (Pennings) and colleagues previously proposed a model (Moreno-Gamez et al., 2015) that showed that unequal penetration profiles can the evolution of drug resistance on combination therapies much more likely. Here, we show how that model fits the patterns described thus far.
{kind=link}
{kind=link}
{kind=link}
In patients treated with three drug therapies, drug levels may vary either in time (A.) or through space (B.). A. Drug concentrations decay at different rates over time, including to below suppressive concentration (dashed line). Missed doses combined with short half-lives may result in temporal monotherapy (red box), wherein two of the three drugs do not suppress the virus. B. Different drugs reach different compartments (here, the brain, the blood and the gut) above or below the level needed to suppress viral replication (dashed grey line). This results in spatial monotherapy (red box), whereby some compartments of the body (such as the brain) contain active concentrations of only a single drug, facilitating the evolution of resistance to that drug.
We start with an example of how the compartment model may work for one of the HIV treatments discussed above: 3TC+NFV+D4T. If someone is treated with 3TC, NFV, and D4T (see Figure 1A), 3TC may be present in the brain at high levels, because of its superior penetration, but the other two drugs are likely not (Letendre et al., 2008) - NFV because of its low penetration, and D4T because of its very short half-life. This difference in drug levels between 3TC and the other two drugs creates a compartment, the brain, in which a virus needs to evolve resistance to only one drug, 3TC. We have good reasons to believe that this compartment actually exists (Letendre et al., 2008; Browne et al., 1993), and this model explains why resistance to 3TC predictably arises before resistance to the other drugs.
The drug compartment model predicts a constant risk that a 3TC-resistant virus emerges (from the reservoir or from a drug sanctuary), invades the single-drug compartment, and creates a selfsustaining, replicating viral population in the brain. Because the HIV compartments in the human body are linked by migration, the resistant viral population in the brain may create enough viral particles to be noticed in a routine viral load measurement of the blood. In fact, if virus from blood plasma is sequenced, it may be almost 100% 3TC resistant.
Once resistance to 3TC is established in the brain, this viral population also constitutes a risk for two-drug resistance, because it needs only one additional mutation to become resistant to a second drug. If a compartment exists that contains only two drugs, such as 3TC and NFV (but not D4T), then the most likely next evolutionary step would be for the virus to acquire resistance to NFV and invade the two-drug compartment. While we do not have direct evidence for the existence of a 3TC/NFV compartment, because D4T is not an effective drug (Rosenbloom et al., 2012), most of the body may effectively be the 3TC/NFV compartment (see Fig 1A). Finally, if the patient remains on the same treatment, a viral particle may acquire the third mutation, making it resistant to all three drugs in the treatment (this is also seen in Fig 1A).
The model in which 3TC has a better penetration profile than the other two drugs explains all three of our surprising observations about drug resistance on triple-drug therapies: it explains how drug resistance can evolve, why it evolves by one mutation at a time, and that it evolves in a predictable order - namely in response to the best-penetrating drug first. Intuitively, one might think that resistance will first arise in response to the “worst” drug in a cocktail. However, in the compartment model, a desirable property –high penetration– actually makes 3TC vulnerable to resistance. Evidence shows that 3TC penetration is better than NFV penetration for both the central nervous system (CNS) (Letendre et al., 2008)) and the male genital compartment (Lambert-Niclot et al., 2011) and that NFV has generally poor penetration (Aweeka et al., 1999). As a result, counterintuitively, the best way to prevent evolution of 3TC resistance is not to target the shortcomings of 3TC, but to replace one or both of the other two drugs with agents that have better penetration profiles. Historically, introducing highly penetrant lopinavir/ritonavir in 2000 resulted in exactly this effect, as we will describe later in this paper.
Lessons from a temporally-varying model
Alternatively, if multiple drugs applied have different half-lives, this could also lead to only a single drug (the one with the longest half-life) being present and thus a scenario in which single drug resistance mutations could be selected. Therapies are normally dosed in such a way that all three drugs should be present at high enough concentrations at all times. However two factors complicate this: 1) some drugs have very short half-lives and 2) adherence varies among patients. If a patient is put on a complicated regimen requiring multiple drug doses per day and is not perfectly adherent to the treatment, this can lead to single-drug time periods.
We believe that this model with time-varying drug levels could explain the patterns we observe in treatments with unboosted PIs. We return to the example of 3TC+NFV+D4T treatment. First, it is important to know that D4T is not a very potent drug (Rosenbloom et al., 2012). Second, NFV is dosed twice a day and has a very short half-life. These two things together mean that if a patient misses a dose, 3TC may quickly be the only potent drug present, and a virus that is resistant to 3TC may be able to spread. A modeling study should be conducted to determine whether relative timescales of drug decay and viral replication could allow the spread of a 3TC resistant mutant during 3TC+NFV+D4T treatment with imperfect adherence (Rosenbloom et al., 2012).
While single-drug time periods plausibly explain the observed data when some component drugs are not totally suppressive and others have short halflives, such explanations cannot explain the failure under NNRTIs.
Unlike PIs, NNRTIs have particularly long halflives of days. Treatment interruptions of a few days are long enough for the other drugs to leave the system, but short enough for NNRTIs to remain present at a high level. However, several trials found no effect of interrupting NNRTI-based treatment for 2 days every week for 72 weeks (Reynolds et al., 2010) or 3 days a week for 48 weeks (De Truchis et al., 2017). Consistent with this, another clinical trial did not find an effect of “covering” the monotherapy tail of interrupted NNRTI-based treatment on the risk of drug resistance evolution (Fox et al., 2008).
On the other hand, long NNRTI treatment interruptions of several weeks, which allow for substantial viral population growth, do promote drug resistance evolution (Pennings, 2012). Population growth, rather than the single-drug time periods created by the long half-lives of NNRTIs, have outsize impact on the risk of drug resistance evolution.
As long as the viral population is suppressed ahead of the NNRTI combination treatment interruption, there is insufficient virus for evolutionary forces to operate efficiently over the relevant timescales of single-drug time periods.
Taken together, these two factors (heterogeneity in time or space) we believe reflect plausible mechanisms through which ongoing resistance evolution has occurred. Different drug failure mechanisms could explain the patterns of ongoing resistance in different types of drug combinations. Also, both mechanisms could be happening at once. When penetration of drugs into tissues is poor and drugs are declining at different rates, this could create a scenario where some time-spaces (as opposed to simply times or spaces) create the environment to select for single drug resistance. More work needs to be done to understand how these factors interact.
Below we discuss some difficulties of applying these models:
Issues with identifying and measuring compartments
In principle, the compartment model combined with a comprehensive understanding of where and when drugs penetrate should allow us to make predictions concerning the order in which drug resistance mutations occur. In practice, understanding where, when, and how certain drugs reach parts of the body is complicated (Vendel et al., 2019). Indeed, different studies have measured different relative penetrances of drugs into the tissue versus the plasma.
Systematic differences among people complicate our understanding of drug penetrance. For example, treatment with other drugs (e.g., TB drugs) may affect the half-lives and penetration of HIV drugs (López-Cortés et al., 2002). Genetic variation among patients may also mediate penetrance (see Apostolova et al. (2015)). For example, EFV concentrations are higher in people with certain mutations, and these mutations are more common in Black/African groups. Different adherence patterns could hypothetically make different mutational orderings more or less likely as well. A recent paper suggests that the combination of imperfect adherence with imperfect penetration makes sequential drug resistance evolution likely in an in vitro system (Lustig et al., 2019).
In addition, studies of drug penetrance may not be quantifying the correct factors to help us understand where drug resistance can emerge. First, the administered version of a drug may be at a different concentration than the active form (Dumond et al., 2008). Second, the intracellular concentration of a drug, rather than the tissue concentration, may mediate effectiveness, requiring that we understand not only whether drugs reach a tissue, but the rates at which they are transported into cells (Bazzoli et al., 2010; Dumond et al., 2008). Third, we may not be investigating penetrance in the correct compartments, or at the correct spatial scale. For example, there can be considerable heterogeneity of drug penetrance within a single lymph node or gut cross section (Thompson et al., 2015, 2019). Fourth, many studies report tissue drug concentrations relative to the blood plasma, without considering the actual tissue drug level and its inhibitory effect on the virus (Else et al., 2011). Even comparing tissue drug concentration to the IC50 does not capture the full dynamics of drug concentrations within the body (Shen et al., 2008).
In conclusion, predicting evolutionary escape in a spatio-temporally heterogeneous body on the basis of available drug penetrance data is not straightforward, even when data are consistent with the existence of single-drug compartments. An exciting possibility is that if drug penetration influences the order of mutations, we may be able to use data on the order of mutations to learn about relative drug penetration.
Could small fitness differences between mutations explain the mystery of ordered and predictable resistance mutations?
Here we explore an alternative explanation and why we believe this explanation cannot explain all features of the data. We ask if differences in the selective advantage conferred by different classes of mutations can cause ongoing stepwise and predictably-ordered evolution. For example, NNRTI mutations may occur first simply because they result in higher viral fitness than NRTI mutations (See Fig 2), or because they carry less of a fitness cost. A number of lines of evidence suggest that increased fitness in the face of three drugs cannot account for the emergence of drug-resistant viruses.
Consider the ability of a virus to replicate. If its absolute fitness value is 1, it will maintain its population size. If the absolute fitness value is above 1, the population will grow. If it is below 1, the population will shrink until extinct (Perelson et al., 1997). Note that HIV populations do not go extinct as the viral population is sustained latently as provirus in T cells and can reactivate at a later time. Note, however, fully suppressivetherapy halts continual rounds of viral replication (Kearney et al., 2014; Brodin et al., 2016; McManus et al., 2019).
In patients on triple-drug therapy, when there is no drug resistance, we typically see almost no wild-type (WT) virus in the blood because the absolute fitness of the wildtype virus is below 1. Even if a virus has a single resistance mutation, its absolute fitness will still be below 1 in the context of multidrug therapy, and its population size will shrink, although possibly more slowly than the the population without the virus. Although differences in the rate of population decline could lead to the apparent increase in frequency of a drug resistance mutation, these shrinking populations are not expected to impact the viral reservoir (Brodin et al., 2016) and will not lead to virological failure.
In addition, our knowledge viral dynamics inside the body is inconsistent with the idea of a resistant mutant with higher fitness out-competing the wild-type virus already present. Typically, when virologic failure occurs (i.e., treatment fails because the virus has started replicating again), we observe either the WT virus replicating (when the patient has not been adherent to the treatment) or a drug-resistant virus replicating, but almost never both. When drug resistance is present, the majority of the virus sampled is typically drug resistant (see, e.g., Williams and Pennings (2019)). This suggests that rather than drugresistant viruses slowly replacing WT viruses, drugresistant viruses are instead replicating in a niche that was not previously occupied by any virus.
In short, though most explicit and implicit models of adapting populations include a mutant outcompeting the WT organism, this does not seem to be what is happening when drug resistance arises in HIV infection. Instead, it appears that the mutant drug-resistant virus is able replicate in a part of the body where (or during a time when) the WT virus cannot replicate, which is only possible if there is a space (or time) in which only one drug is present at a high level.
Spatial-temporal heterogeneity can also explain why acquired resistance rates have fallen since 1996
Having proposed the compartment model as an explanation for why acquired drug resistance rates were a significant problem at the beginning of the highly active antiretroviral therapy (HAART) era, in the mid 1990s, we would now like to propose the model as an explanation for a second phenomenon: the gradual fall in acquired drug resistance rates that occurred between 1996 and the mid-2010s. The nature of the treatments did not change over this period: NNRTI- and PI-based triple-drug therapies continued to be used almost exclusively (Tseng et al., 2015). However, we hypothesize that several subtle but key improvements in treatment either reduced overall viral replication or reduced the size of single-drug compartments in patients, limiting opportunities for drug resistance to evolve.
Above, we predicted that, according to the compartment model, the best way to curb resistance is to improve the penetrance of the “worst” drugs in a cocktail. In particular, for the combination 3TC+NFV+D4T, we predicted that replacing NFV or D4T with better-penetrating drugs would decrease the rate of resistance to 3TC and the rate of acquired resistance in general. As it happens, replacement of the low-penetration drug NFV is exactly what occurred historically. In 2000, the first generation of PIs (including NFV) was replaced by a second generation of PIs “boosted” by ritonavir (Tseng et al., 2015). These boosted PIs featured better penetration and longer half-lives (Capparelli et al., 2005) For example, ritonavir-boosted lopinavir (LPV/r) consistently reaches a level above the 50% inhibitory concentration in cerebrospinal fluid (CSF) (Capparelli et al., 2005), thereby likely shrinking the size or number of compartments in which 3TC exists alone. The effect of replacing NFV with LPV/r is clear in the clinical trial we described previously (Figure 1A and Kempf et al. (2004)). In the NFV arm of the study, 79 of 327 patients developed some type of drug resistance (24%) within 2 years, whereas in the LPV/r arm, only 19 of 326 did (6%) (Figure 1); this represents a reduction in treatment resistance of 76%. The four-fold lower rate of drug resistance evolution among patients on the LPV/r regimen suggests that the compartments of the body that were only reached by 3TC became four times smaller when physicians replaced NFV with LPV/r in their patients’ drug cocktails.
NRTI drugs also significantly improved over time. Before 1995, NRTIs were the only drug class we had for treating HIV. Now, in 2019, most people in highincome countries start treatment with regimens based on integrase inhibitors, but NRTIs are still a part of those regimens. While we argued earlier that the long half-lives of NNRTIs probably don’t contribute much to the risk of drug resistance evolution, drugs with very short half-lives likely pose a risk for resistance evolution to their companion drugs when they don’t reach high enough levels to suppress the virus. The initial NRTIs had short half-lives (AZT, 1987, 0.5-3 hrs (Retrovir FDA factsheet, Revised: 2008); didanosine (DDI), 1991, 1.5 hrs (Videx FDA Factsheet, 1999); D4T, 1994, 0.8-1.5 hrs (Stavudine FDA factsheet, 2008)). Newer NRTIs had longer half-lives (3TC, 1995, 5-7 hrs (Empivir FDA factsheet, Revised: 2017); TDF, 2001, 17 hrs (Viread FDA factsheet, 2012b); FTC, 2003, 10 hrs (Emtriva FDA factsheet, 2012a)). Other improvements followed. In 2004, the FDA approved Truvada, a pill that combined FTC and TDF, two of the long half-life NRTIs, which made it possible for patients to take a single NRTI pill once a day. Then, in 2006, the FDA approved Atripla, which combines FTC and TDF with efavirenz (EFV), an NNRTI with an even longer half-life (40 hours). Atripla was the first pill that contained all three drugs that traditionally make up a HAART regimen in a single pill that can be taken just once a day. Simpler and less frequent dosing makes it easier for patients to adhere to treatment regimens, and the longer half-lives of NRTIs mean that missing a dose becomes less problematic with regard to resistance. Combined, these improvements led to a 12-fold reduction of the risk of acquired HIV drug resistance per month from 1.7% in 1997 to 0.13% in 2008 (Gill et al., 2010).
Conclusion
HIV was once the poster child for the rapid evolution of drug resistance. While HIV drug resistance evolution remains an enduring problem in many parts of the world, it has become rare in high-income countries. In this paper, we answer some questions about how this transition happened, thereby expanding the lessons the medical world can take away from the example of HIV. Although multiple simultaneous drugs were necessary for the successful treatment of HIV in the 90s and 2000s, we show how these triple-drug therapies still allowed the evolution of drug resistance. In theory, the reasoning behind why combination therapy should work is correct, but in practice, single drug compartments may occur inside the body, allowing step-wise evolution of drug resistance.
One thing is clear: in numerous and diverse health problems (cancer, malaria, bacteria, viruses), drug resistance is an evolutionary problem. In some cases (cancer, HIV, hepatitis C virus), resistance evolves again and again within individual patients. In other cases (malaria, TB), resistance evolves de novo a limited number of times and then spreads from patient to patient. For antibiotic resistant bacteria other than Mycobacterium tuberculosis, the situation is even more complex, because plasmids, transposons, and individual resistance genes can be transferred within and between bacterial species. Mutation, recombination, and horizontal gene transfer create variants that selection can than work on.
Perhaps the most obvious lesson that we can learn from HIV is that, in all of these systems, the stories of how resistance evolves are unlikely to be simple. In particular, spatial or temporal heterogeneity, whether within the patient or at the level of a patient population, probably makes each of these cases much more complex than the common textbook explanations of resistance evolution would imply (as illustrated by recent work on spatial structure in the lungs of TB patients Strydom et al. (2019)). In addition, the environments in which these systems evolve are temporally heterogeneous because of drug half-lives, dosing schedules, and imperfect adherence. For these reasons, we believe that the fight against drug resistance can best proceed with evolutionary biologists on the team. While there is excellent work being done on the theory of drug resistance and experimental evolution is being investigated in lab settings, there is also tremendous opportunity and need for evolutionary biologists to work in the messy world of cohort and clinical trial data.
Population genetics has a rich ensemble of tools that can be put to use on complete genetic data, to understand how and why drug resistance evolves. For example, population genetic models of evolutionary rescue from standing genetic variation could be fit to explain treatment outcome differences among tumors of different volumes (Shiao et al., 2017). Studies of genetic variation among resistant and susceptible bacterial strains may be used to better understand the role of de novo evolution and transmission in drug resistance across medically relevant bacteria (Croucher et al., 2014).
In conclusion, the problem of drug resistance is a persistent one across many diseases. It only makes sense to learn from our rare successes, and the study of ever-more-effective HIV regimens constitutes just such a success. Although much remains to be learned about the evolution of drug resistance in HIV, what we do know reminds us to be skeptical of simple stories and enthusiastic about incremental improvements in drugs.
Methods
To determine the first mutation for a variety of therapies, we analyzed data taken from Feder et al. (2016) representing Sanger-sequenced HIV populations treated with a broad range of regimens between 1989 and 2013. Each patient was treated with exactly one regimen to select against patients with pre-existing resistance before the onset of therapy. Ambiguous underlying nucleotide calls (i.e., non-A/T/C/G calls) were interpreted as population polymorphisms among all possible resulting amino acids. For example, an AAS residue (AAC/AAG) was recorded as an asparagine/lysine polymorphism, but an AAY (AAC/AAT) was recorded as asparagine. When multiple sequences were available for the same patient at the same time point, polymorphisms were also recorded.
We then determined the number of drug resistance mutations by comparing sequences to the WHO list of surveillance drug resistance mutations (DRMs). DRMs were only counted if HIV sequences contained residues conferring resistance to the therapy with which they were treated. Two classes of DRMs were recorded: 1) polymorphic DRMs in which some calls supported the DRM and some supported a non-DRM and 2) non-polymorphic DRMs, in which all calls supported the DRM.
To determine the first DRM, we retained only patients with exactly one polymorphic DRM or exactly one non-polymorphic DRM and any number of polymorphic DRMs (under the assumption that fixed mutations occurred before polymorphic mutations). For patients with sequences taken at multiple time points, we retained the first sample meeting the conditions above.
Calculating target size and total mutation rate per drug
We based our calculations on the consensus subtype B sequence provided by the Los Alamos HIV Database (https://www.hiv.lanl.gov/content/index), the WHO list of resistance mutations, and mutation rates estimated in Abram et al. (2010). For each relevant amino acid position in protease and reverse transcriptase proteins, we determined all possible onestep mutations. For all one-step mutations, we determined whether they led to resistance to the relevant drugs according to the WHO list. We then summed the mutation probability for all possible one-step mutations that lead to resistance to a certain drug. The resulting total mutation probabilities ranged from 4 · 10−5 for TDF to 2 · 10−4 for LPV.
Acknowledgments
We thank Alison Hill, Jonathan Pritchard, Noah Rosenberg and Dmitri Petrov for valuable comments on earlier versions of this manuscript. We thank Chris Hoffmann for providing help interpreting the raw data from his 2009 CID paper.
References




