

Abstract
Supplementation with members of the early-life microbiota or ‘probiotics’ is becoming increasingly popular to attempt to beneficially manipulate the preterm gut microbiota. We performed a large longitudinal study comprising two preterm groups; 101 orally supplemented with Bifidobacterium and Lactobacillus (Bif/Lacto) and 133 non-supplemented (Control) matched by age, sex, birth-mode, and diet. 16S rRNA metataxonomic profiling on stool samples (n = 592) indicated a predominance of Bifidobacterium, and a reduction of pathobionts in the Bif/Lacto group. Metabolic phenotyping found a parallel increase in fecal acetate and lactate in the Bif/Lacto group compared to the Control group, which positively correlated with Bifidobacterium abundance consistent with the ability of the supplemented Bifidobacterium strain to metabolize human milk oligosaccharides and reduced gut pH. This study demonstrates that microbiota supplementation can modify the preterm microbiome and the gastrointestinal environment to more closely resemble that of a full-term infant.
Introduction
Premature birth, defined as infants born <37 weeks gestation, accounts for 1 in 9 births globally1. Preterm infants are often born via Caesarean-section, have an underdeveloped immune system, receive numerous courses of antibiotics, and reside in neonatal intensive care units (NICUs); all of which disrupt the establishment of the early life gut microbiota2,3. This altered gut microbial ecosystem has been linked to an increased risk of serious morbidity during the NICU stay, including necrotizing enterocolitis (NEC)4 and late onset sepsis (LOS)5, and later life health problems such as asthma and eczema6,7.
Abnormal patterns of bacterial colonization are common in the preterm infant gut which is dominated by potentially pathogenic bacteria (i.e. pathobionts) such as Staphylococcus, Klebsiella, Escherichia, and Clostridium. These infants are also characterized by a low abundance or absence of beneficial Bifidobacterium and Lactobacillus, that are characteristic of the full-term infant gut. Thus, interventions to ‘normalize’ the preterm gut microbiota are an attractive proposition to improve health and prevent disease in preterm infants.
Oral administration of commensal infant bacteria via ‘probiotic8’ supplementation is one approach to encourage gut colonization of early life dominant microbiota members. Importantly, supplementation of preterm infants with Bifidobacterium or Lactobacillus has been reported to protect against NEC9,10 and LOS11. While large scale clinical studies have demonstrated the potential of probiotics to reduce NEC incidence, this has rarely been accompanied with in-depth longitudinal profiling to determine the impact of this type of supplementation on gut microbiota composition and metabolome of preterm infants. Thus, we compared longitudinal samples from two cohorts of preterm infants; 101 orally supplemented with a combination of Bifidobacterium and Lactobacillus and 133 non-supplemented infants. Cohorts were matched by gestational age, sex, delivery method, sample collection time, and diet across four tertiary-level NICUs. 16S rRNA gene profiling was used to determine the fecal bacterial composition (n = 592) and paired 1H nuclear magnetic resonance (NMR) spectroscopy was used to measure the metabolic content of the fecal samples, this included metabolites of microbial, host, and maternal origin (n = 157). To evaluate colonization potential whole genome sequencing was used to compare supplemented strains to isolates obtained from preterm infants, alongside in vitro studies to define underlying mechanisms governing preterm microbiota profiles.
Results
Study design
Fecal samples were collected from NICU-resident premature infants receiving a daily oral supplementation containing Bifidobacterium bifidum and Lactobacillus acidophilus (Bif/Lacto group), and from a group of similar aged premature infants (Control group) from three other NICUs that did not offer routine supplementation. This study design avoided cross-contamination amongst the study groups, which has been reported previously in other probiotic studies12,13 where study groups reside within the same NICU. Samples were collected corresponding to four time points at 0-9, 10-29, 30-49, and 50-99 days of age from birth (Fig. 1a).

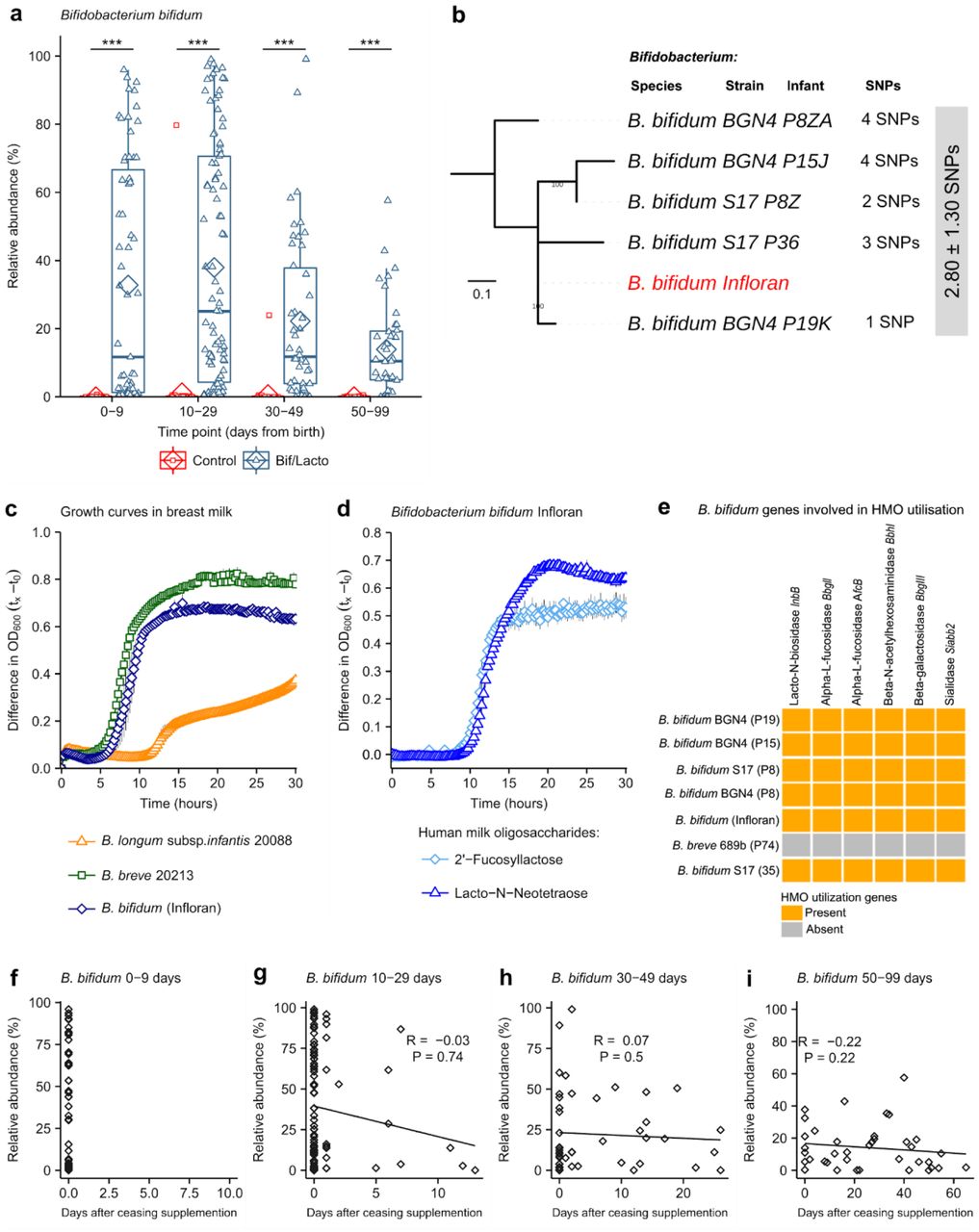
a, Study outline and sample collections times. Infloran supplementation was given until 34 weeks old, with the exception of very low birth weight infants (<1500 g) who received it until discharge. Control group was not given supplementation. b, Infant fecal microbiota similarity at 0-9 days and c, at 10-29 days shown using NMDS (Non-metric multidimensional scaling) analysis clustered with a Bray-Curtis dissimilarity. Arrows indicate bacterial genera driving the separation of points on the NMDS plots. d, Heatmaps showing the ten genera with highest proportional abundance at 0-9 days and e, at 10-29 days of age. Heatmap rows were clustered by total microbiota similarity using Bray-Curtis dissimilarity and the columns clustered by genera that occur more often together. Side bar plots show the proportional abundance of Bifidobacterium in each sample.
Supplementation with early life microbiota members influences preterm gut microbiota composition
The preterm gut is typically dominated by pathobionts such as Enterobacter, Escherichia, and Klebsiella14. We sought to determine whether preterm infants supplemented with Bifidobacterium and Lactobacillus, bacterial species associated with a healthy term infant gut, showed a modified preterm microbiota profile. Fecal metataxonomic bacterial composition was determined by 16S rRNA gene sequencing. Genus level clustering of samples using non-metric multidimensional scaling (NMDS) indicated clear variation in the microbiota profiles between Bif/Lacto supplemented infants and Controls (Fig. 1b-c; Supp Fig. 1a-b). The microbiota composition of Bif/Lacto and Control sample differed significantly at each of the four time points (PERMANOVA, P < 0.01). The clustering of the Bif/Lacto group was driven by the genus Bifidobacterium, while the genera driving the clustering of the Control group included Staphylococcus, Escherichia, and Klebsiella (Fig. 1b-c; Supp Fig 1a-b). Notably, while hospital NICUs may differ in their ‘environmental’ microbiota in ways that may influence infant colonization, NMDS and PERMANOVA tests showed no differences in the microbiota composition between infant samples from the three Control hospital NICUs (Supp Fig. 2a-d).
We also examined the ten most abundant genera by relative abundance at each time point using Bray-Curtis dissimilarity (Fig. 1d-e; Supp Fig. 1c-d), showing infant samples clustered into six main groups based on a single dominant bacterial genus; Bifidobacterium, Escherichia, Enterococcus, Klebsiella, Staphylococcus, or Streptococcus. These data indicate that the introduction of Bifidobacterium promote changes in the composition of the preterm gut microbiota.
Oral Bif/Lacto supplementation influences bacterial genus abundance and bacterial diversity
We sought to further define the genus composition based on relative abundance and diversity measures underlying these changes in microbiota composition. Bifidobacterium dominated the microbiota of the Bif/Lacto group with high relative abundance at all time points compared to the Control group (Fig. 2a-c). This indicated ‘efficient’ colonization by the supplemented strain, or ecosystem re-modeling to encourage colonization of other Bifidobacterium spp. Surprisingly, Lactobacillus was only detected in a minority of infants, but with a higher relative abundance in Bif/Lacto infants compared to the Control group at all time points (Fig. 2d), which may indicate a more transient and limited colonization potential for this strain. The relative abundance of bacteria such as Klebsiella, Escherichia, and Enterobacter was lower in Bif/Lacto infants compared to Control infants at earlier time points 0-29 days of age (Fig. 2e-g), with Klebsiella still lower at 30-99 days of age (Fig. 2e). Staphylococcus was initially abundant in both groups, but rapidly decreased as the infants aged (Fig. 2a-b; Supp Fig. 6f)15. Species level analysis of the 16S rRNA data matched the Staphylococcus present to S. epidermidis and S. haemolyticus, bacterial residents on the skin, indicating that these originate from initial colonization of skin-associated bacteria (Supplementary Fig. 6g-h). Indeed, the skin-associated commensal Cutibacterium was also found in higher relative abundance in Control infants at 10-29 days of age (Fig. 2h). These data suggest that the oral supplementation impacts the microbial colonization patterns, displacing other potentially pathogenic bacteria more typical of the preterm gut.

a-b, Bubble plots show the mean group abundance of the common bacterial genera at each time point in the Control group and the Bif/Lacto group. c, Bifidobacterium abundance at each time point. Relative abundance of d, Lactobacillus, e, Klebsiella, f, Escherichia, g, Enterobacter, h, Cutibacterium, and i, Clostridium. Individual points highlight individual infant samples, diamonds indicate the group mean, box plots show group median and interquartile range. Asterisks represent p values: ***P < 0.001.
When examining diversity measures (Shannon and Inverse Simpson diversity), values were initially higher at 0-9 days in Bif/Lacto compared to Control infants (Supp Fig. 3b-c), although the abundance of Bifidobacterium was not correlated with the number of bacterial genera detected (Supp Fig. 3d). At later time-points (i.e. 30-99 days of age, Fig Supp Fig. 3b-c), the diversity values of the Bif/Lacto were lower than the Control group, which may correlate with the increasing Bifidobacterium abundance above 50% (Supp Fig. 3e-f, h-i). These data suggest there is a diversity ‘tipping-point’ in response to dominance of Bifidobacterium as the major early life member.
External factors including gestational age, birth weight, and antibiotics negatively affect Bifidobacterium abundance in Bif/Lacto infants
Previous studies have indicated that factors, such as gestational age and antibiotics16, significantly influence the developing early life gut microbiota; with preterm infants representing an infant cohort overexposed to potential microbiome-modulating factors. Focusing on Bifidobacterium as the dominant bacteria in the Bif/Lacto group, we noted that infants with a birth weight ≥1000 grams showed higher relative abundance of Bifidobacterium at 0-29 days (Fig. 3a). This was also the case at 10-29 days of age in infants born at a gestational age ≥28 weeks. (Fig. 3b). Furthermore, birth weight and gestational age were closely correlated (Fig. 3c) and correlated inversely with length of NICU stay (Supp Fig. 4e-f), indicating that the underdeveloped preterm gut may not represent an optimal niche for Bifidobacterium colonization. Higher Bifidobacterium proportions in Control infants with birth weights ≥1000 g compared to those of <1000 g supports this hypothesis (Supp Fig. 4a).

a, Bifidobacterium abundance between very-low birth weight (<1000 g) and low birth weight (>1000 g) b, Bifidobacterium abundance between infants with very low gestational age (<28 weeks) and low gestational age (≥28 weeks). c, Birth weight in grams correlated with gestational age in weeks. d, Bifidobacterium abundance in infants receiving antibiotics at the time of sample collection. e, Bifidobacterium abundance in infants delivered by caesarean and vaginal birth. f, Bifidobacterium abundance in infants still receiving or no-longer receiving supplementation. g-i Correlation between Bifidobacterium abundance and days after ceasing receiving supplementation. Asterisks represent p values: *P < 0.05, **P < 0.01, ***P < 0.001.
Preterm infants receive numerous antibiotics over the course of their NICU stay. Within the Bif/Lacto infants Bifidobacterium abundance was lower in infants currently being treated with antibiotics at all time points compared to those not receiving antibiotics, indicating antibiotic susceptibility of this genus (Fig. 3d). In contrast, the relative abundance of Staphylococcus, Klebsiella, and Escherichia was unchanged in infants receiving antibiotics, suggesting these were resistant to antibiotic treatment (Supp Fig. 4i-k).
Emergency Caesarean sections for maternal or fetal indications account for a large number of preterm births, and previous studies have indicated that Caesarean-section delivery can directly interrupt the transfer of maternal microbes (e.g. Bifidobacterium) to infants17. Surprisingly, there was no significant difference in the relative abundance of Bifidobacterium within the Bif/Lacto group in infants born by vaginal or cesarean birth (Fig. 3e). Gestational age, current antibiotic treatment, and delivery method did not significantly alter Bifidobacterium proportions in Control infants, however the low abundance of this bacteria in this cohort make robust statistical analysis difficult (Supp Fig. 4b-d).
In Bif/Lacto infants, supplementation ceased when infants reached a post-conceptual age of 34 weeks. However, no reduction was observed in the relative abundance of Bifidobacterium in samples collected from these infants after oral supplementation had ceased (Fig. 3f), with proportions maintained for up to 60 days (Fig. 3g-i). This indicates potentially longer-term colonization of the supplemented strain in these infants.
Diet is proposed to be one of the major factors modulating the early life microbiota, with significantly differences between formula and breast-fed infants18. Unusually, almost all infants recruited to this study were fed either their own mothers’ breast milk, or their mothers’ breast milk and donor breast milk in combination, or breast milk supplemented with preterm cows’ milk-based formula. Only a very small number of infants were exclusively formula fed (i.e. 4 out of 234), which may explain our findings that the relative abundance of Bifidobacterium was unaffected by diet within both the Bif/Lacto or Control infant cohorts (Supp Fig. 4g-h).
Bif/Lacto infants shown distinct changes in species composition over time including prolonged colonization of the Bifidobacterium bifidum Infloran strain, which correlates with human breast milk metabolism and routine supplementation
Our data so far indicated a dominance of Bifidobacterium in the Bif/Lacto cohort, and poor colonization ability of Lactobacillus in the preterm gut. To probe this with greater resolution, we compared the abundance of species present within these two genera. B. bifidum was highly abundant in Bif/Lacto infants, while only being abundant in 2/133 Control infants (Fig. 4a). Bifidobacterium breve was also more abundant in the Bif/Lacto supplemented group (Supp Fig. 5a), with Bifidobacterium longum present in a small number of infants from both groups (Supp Fig. 5b). B. bifidum relative abundance declined with increasing infant age (Fig. 4a), with concurrent increases in B. breve (Supp Fig. 5a). As B. breve coexisted with, rather than replaced (Supp Fig. 5i) B. bifidum this suggests close species interactions; potentially via metabolite cross-feeding. In contrast, while the Lactobacillus acidophilus Infloran strain (for genome analysis see Supp Fig. 6d-e) was more prevalent in Bif/Lacto infants (Supp Fig. 5d), abundance decreased to zero within days after cessation of supplementation (Supp Fig. 5e-h), indicating inefficient colonization of this species.

a, Bifidobacterium bifidum abundance in Bif/Lacto and Control group infants. b, Mid-point rooted maximum-likelihood tree based on 12 SNPs called via reference-based approach (strain Infloran as the reference genome) from 5 Bifidobacterium bifidum genomes. Grey box denotes pairwise SNP distance between these 6 genomes. Data: mean ± S.D. c, Growth curves of B. bifidum Infloran, B. breve 20213, and B. longum subsp. infantis 20088, in whole human milk. d, Growth curves B. bifidum infloran in human milk oligosaccharides (HMO) Lacto-N-tetraose and 2-fucosyllactose e, Heat map representing B. bifidum genes involved in utilisation of human milk oligosaccharides. f-i, Correlation between B.bifidum abundance and days after ceasing receiving supplementation. Asterisks represent p values: ***P < 0.001.
Previous research studies have shown that colonization of the gut by probiotic bacteria may vary depending on the strains used, mode of administration, dose, and inclusion of prebiotics19. To confirm putative bifidobacterial supplemented strain colonization (as described above), we performed whole genome sequencing-based analysis to compare the B. bifidum Infloran strain to nine Bifidobacterium isolates cultured from fecal samples from seven Bif/Lacto supplemented infants. Core-genome single nucleotide polymorphisms (SNPs) analysis indicated the five B. bifidum isolates were identical at 0 SNP difference (based on 87 core genes; Suppl. Fig. 6a). Reference-based genome-mapping of whole genome sequences of five B. bifidum genomes to B. bifidum Infloran strain (as reference genome, Fig 4b) indicated a near-identical similarity (mean pairwise SNPs: 2.80 ± 1.30), strongly suggesting they belong to the same bacterial strain (i.e. Infloran). Average nucleotide identity (ANI) analysis also supported these findings (100.00% nucleotide identity, Suppl. Fig 6b). These data support the elevated B. bifidum relative abundances in our 16S rRNA metataxonomic data (Fig. 4a), including after supplementation had finished in Bif/Lacto infants, indicating longer-term colonization of this strain (Fig. 4f-i).
Bifidobacterium represents a dominant genus in the full-term healthy breast-fed infant selectively fed by complex oligosaccharides (i.e. human milk oligosaccharides (HMOs)) within breast milk. However, the ability of Bifidobacterium to digest HMOs varies between species and strains of this genus20. Thus, we analyzed B. bifidum genomes (our 5 isolates and Infloran strain) for the presence of genes involved in HMO utilization; all B. bifidum isolates contained specific genes involved in HMO utilization (Fig. 4e), and mucin degradation genes which may aid gut colonization (Suppl. Fig. 6c). Notably, growth curves in whole breast milk (Fig. 4c), confirmed that the B. bifidum Infloran strain utilized whole breast milk. Further phenotypic analysis indicated this strain was able to metabolize specific HMOs; 2-fucosyllactose (2’-FL) and Lacto-N-Neotetraose (LnNT), corresponding to genes AfcA and BBgIII respectively, encoding for extracellular enzymes involved in their utilization (Fig. 4d). Therefore, the ability to digest breast milk and HMOs in these predominantly breast milk fed infants, is likely to be a key factor driving high rates of Bifidobacterium colonization.
Bacterial strains used as probiotics commonly lack antibiotic resistance genes. However, in the NICU environment, which has high antibiotic prescription practices, this may reduce the colonization of supplemented strains in the preterm gut. Analysis of the B. bifidum Infloran strain genome indicated the presence of only the intrinsic ileS gene (associated with mupirocin resistance, Supp Table 2a). Minimum antibiotic concentration testing confirmed sensitivity to commonly prescribed antibiotics in NICUs (Suppl Table 2b). These data are in agreement with the reduced relative abundance of Bifidobacterium in Bif/Lacto infants receiving antibiotics (Fig. 3d) However, by giving the supplement twice daily (up to 34 weeks post-conceptual age) this may have aided rapid re-establishment after antibiotic treatment and promoted relatively stable colonization.
Infants receiving oral supplementation show differences in metabolomic profiles and lower fecal pH
Microbial metabolites are key molecules involved in microbe-microbe and microbe-host interactions. To define the ‘functional’ impact of Bif/Lacto supplementation, 1H nuclear magnetic resonance (NMR) spectroscopy was used to characterize the metabolomes of a subset of paired fecal samples (75 from Bif/Lacto group, and 81 from Control group; all timepoints; N = 157). A principal component analysis (PCA) model (R2 = 53.6%) was built using these metabolic phenotypes and clear biochemical variation was observed between the Bif/Lacto and Control samples (Fig. 5a). Pairwise orthogonal projection to latent structures-discriminant analysis (OPLS-DA) models constructed for each time point confirmed these metabolic differences throughout the study period (p values < 0.01, Suppl Fig. 7a). A covariate-adjusted-PLS-DA (CA-PLS-DA) model comparing the fecal profiles at all sampling points and adjusted for sampling age showed that infants in the Bif/Lacto group excreted greater amounts of the short chain fatty acid (SCFA) acetate (Fig. 5c) and lower amounts of the sugars 2’-FL, 3-fucosyllactose (3’-FL), arabinose, and trehalose compared to those in the Control group (Fig. 5e-h). Compared directly, fecal lactate was also higher in Bif/Lacto infants compared to Control infants (Fig. 5d). Notably, these differences observed were maintained throughout the study period.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
a, Principal Component Analysis (PCA) scores plot comparing the fecal metabolic profiles of the Bif/Lacto and Control groups at all time points. b, Discriminatory metabolites that contribute to the covariate-adjusted projection to latent structures-discriminant analysis (CA-PLS-DA) model comparing the fecal metabolic profiles of the Bif/Lacto and Control infants adjusted for sampling age. Upper panel. Average 1H NMR spectrum from all samples indicating metabolites that are excreted in greater amounts by the Bif/Lacto infants (red) and those excreted in greater amounts by the control infants (blue). Lower panel. Manhattan plot showing P-values calculated for each variable in the multivariate model, corrected for multiple testing using the False Discovery Rate (allowing 5% false discoveries). Horizontal lines indicate cut-off values for the false discovery rate on the log10 scale. Blue points indicate metabolites significantly higher in the control feces and red points indicate those metabolites significantly higher in the Bif/Lacto feces. c, Relative acetate concentration. d, Relative lactate concentration. e, Relative 2’-fucosyllactose (2-FL) concentration. f, Relative 3’-fucosyllactose (3-FL) concentration. g, Relative arabinose concentration. h, Relative trehalose concentration. i, Spearman correlation heat map displaying main faecal metabolites (rows) versus the most abundant bacterial groups (columns). Red denotes positive correlation and blue denotes for negative correlation. j, Group faecal sample pH. Asterisks represent p values: *p < 0.05, **p < 0.01, ***p < 0.001. Details of metabolites data for each sample investigated and pH measurements can be found in Supplementary Tables 8 and 9.
The relative abundance of Bifidobacterium was found to be significantly positively associated with fecal acetate and negatively associated with fecal 2’-FL, 3’-FL, arabinose, and trehalose (Fig 5i). Acetate and lactate are known metabolic by-products of Bifidobacterium21, while 2’-FL and, 3’-FL are common components of HMOs, with certain Bifidobacterium strains (including Infloran Fig 4e) able to selectively metabolize these breast milk components22. These results indicate that the higher relative abundance of Bifidobacterium in Bif/Lacto infants correlates with greater HMO metabolism, and with acetate and lactate generated as major end products.
To determine the impact of increased acetate and lactate on the infant gut environment, fecal pH was measured in a subset of infants (n = 74). At 0-9 days of age fecal pH was 5.5 (SD=0.7) in Bif/Lacto infants compared to pH 7.0 (SD=0.5) in Control infants. These differences in fecal pH remained throughout the study (Fig. 5j) and fecal pH was significantly negatively correlated with fecal acetate and lactate (Supp Fig. 7c-d) and the relative abundance of Bifidobacterium (Supp Fig. 7e). Comparing the relative bacterial abundance at species level, B. bifidum had a stronger negative correlation with fecal pH and positive correlation with fecal acetate and lactate (Supp Fig 8a-c) compared to B. breve, the other main Bifidobacterium species present (Supp Fig. 8d-f). Metabolomic analysis on bacterial culture supernatant confirmed the strong acetate producing ability of the supplemented strain B. bifidum (Supp Fig. 8g).
Discussion
Oral supplementation with B. bifidum and L. acidophilus in premature infants drives a fecal microbiota composition and environment more similar to a healthy full-term breast-fed infant. Crucially, we determined that certain clinical practices and relevant external factors may positively or negatively influence the ability of introduced strains, particularly Bifidobacterium, to successfully establish a niche within the preterm infant gut microbiome.
As diet is a major driver of microbiota diversity, particularly the strong relationship between breastmilk and Bifidobacterium abundance, we were careful to match our study cohorts. Although both groups of premature infants received high rates of breast milk, via maternal or donor milk, the low abundance of Bifidobacterium found in Control infants indicates breast milk consumption itself (without supplementation), was not sufficient to encourage high rates of ‘natural’ Bifidobacterium colonization. Maternal to infant transmission of Bifidobacterium occurs in term infants23,24, however the NICU environment and antibiotic treatment may limit establishment of parental Bifidobacterium, leaving infants susceptible to colonization by hospital-environmental bacteria25. For the Bif/Lacto group, the combination of supplementation of early life microbiota members and a known prebiotic food source i.e. breast milk and HMOs, likely aided colonization. Crucially, this symbiotic approach was successful because the ‘right’ bacterial strain was matched to the appropriate nutritional environment. In this case a B. bifidum strain with the genetic potential to metabolize HMOs, and phenotypically shown to use these early life dietary sources for growth. Interestingly, previous studies show B. bifidum secretes extracellular enzymes that facilitate cross-feeding of oligosaccharide degradation products among other Bifidobacterium species20. Furthermore, B. bifidum strains are known to breakdown mucin which may aid gut colonization26. This personalized microbiota supplementation strategy, including genomic and phenotypic analysis of the probiotic strain, tailored to the nutritional environment of the receiving gut, is an important consideration for future studies.
Extremely low birth weight infants (<1000 g) represented the most vulnerable cohort in this study, and presented less abundance of genus Bifidobacterium, potentially due to several factors including; lengthened antibiotic courses27, underdeveloped gut physiology (i.e. poorer gut motility, and thinner mucus layer28), and difficulties in establishing full enteral feeding29. Indeed, previous clinical studies have had difficulties evaluating the beneficial effects of supplementation in this at-risk cohort of preterm infants30,31. Notably, although extremely low birth weight Bif/Lacto infants had lower bifidobacterial abundance, supplementation in our study did enhance levels when compared to Control infants. Thus, from an intervention strategy perspective, daily and prolonged supplementation may contribute to faster (re)establishment of Bifidobacterium, which may also promote colonization resistance against exogenous or resident pathogens in this particularly fragile preterm cohort.
Surprisingly, Bifidobacterium abundance in supplemented infants was not affected by delivery mode (i.e. vaginal vs. caesarean-section). The early and frequent antibiotic treatment in preterm infants may create a ‘naïve niche’, which enables supplemented bifidobacterial strains to successful colonize. Indeed, routine oral supplementation in Bif/Lacto infants may help to ameliorate caesarean delivery-associated bacterial alterations32 in the preterm microbiome. Follow-up studies in this infant cohort could determine if this also contributes to reducing diseases associated with caesarean delivery or prematurity such as ashma33.
Rates of antibiotic prescription in preterms are remarkably high, ranging from 79% to 87% in extremely low birth neonates <1000 g34,35. Antibiotic treatment favors the establishment of antibiotic-resistant bacteria, while indirectly eradicates highly susceptible microbiota members such as Bifidobacterium. A recent research study in term infants correlated abundance of Bifidobacterium species with a reduction in antimicrobial resistance genes and transferable elements36. As Bif/Lacto infants were predominantly colonized by genus Bifidobacterium, this may have contributed to reduce the reservoir of potentially multidrug resistance pathogens (i.e. Staphylococcus, Escherichia, and Klebsiella), which were prevalent in Control infants.
Previous studies have indicated that although preterm infants are particularly at risk of serious diseases with an infectious aetiology (e.g. NEC and late onset sepsis), probiotic supplementation can reduce incidence. However, as highlighted above there are some notable exceptions, which may relate to the differences in strain(s) chosen, infant diet or infant age. Notably, a recent clinical audit in the same NICU where the oral supplementation was given (i.e. Norfolk and Norwich University Hospital), indicated a >50% reduction in NEC rates and late-onset sepsis when comparing 5-year epochs before and after introducing probiotic supplementation, with no episodes of probiotic ‘sepsis’ indicated37. Whilst the processes leading to life-threatening conditions including NEC in premature infants are complex, overgrowth of potentially pathogenic bacteria is thought to be a key factor38. We show that supplemented premature infants have lower relative abundance of pathobionts including Klebsiella and Escherichia, which have previously been linked to NEC and late onset sepsis. This may be due to direct inhibition through compounds secreted by Bifidobacterium (e.g. bacteriocins39), competition for space and/or nutrients availability.
Changes in the gut environment were also indicated through our metabolomic analyses; highlighted by elevated abundance of acetate and lactate in feces from the Bif/Lacto Group, which are known to be primary metabolic end products of HMO degradation by Bifidobacterium40. Acetate and lactate have beneficial health effects enhancing defense functions in host epithelial cells41 and mucosal dendritic cells42. The lower fecal pH detected in the Bif/Lacto Group correlated with higher amounts of these acids and higher abundance of Bifidobacterium, which may also create an unfavorable environment for overgrowth of pathobionts43,44.
Alongside the key microbiological findings of this study, we have also provided context for further trials focusing on clinical practice in NICU and suggestions for future intervention studies in this at-risk infant population. A key strength relates to the size and scope of the study; representing one of the largest reported longitudinal studies in preterm infants, where study cohorts were matched by gestational age, sex, birth-mode, time points of sample collection, and diet, which are all factors that may significantly impact the microbiota, and thus conclusions obtained. The use of high-throughput sequencing and metabolomic phenotyping, linked to clinical metadata, complemented by phenotypic studies, has allowed us to provide a comprehensive basis for the beneficial impact of Bif/Lacto supplementation on the wider microbiota over time, providing a more mechanism-orientated approach to our data analysis. Providing maternal and donor breast milk are essential practices required for successful gut colonization by Bifidobacterium, which also contributes to the enhanced metabolic end-products such as acetate and lactate in the preterm gut. These products will play an important role in direct antagonism of potentially pathogenic microbes, and the maturation of immune cells in early life. This large-scale longitudinal multi-center-controlled study emphasizes the important role that targeted microbiota or probiotic supplementation plays in preterm infants, exerting protective and functional effects on preterm gut microbial communities.
Online Methods
Study design
Two distinct preterm groups were recruited for this study: 1) Bif/Lacto Group who routinely received oral Bifidobacterium and Lactobacillus supplementation (n = 101 infants), and 2) Control Group infants who did not receive supplementation (n = 133 infants). Infants in the Bif/Lacto group were prescribed daily oral supplementation of 109 colony forming units (CFU) of Bifidobacterium bifidum and 109 of Lactobacillus acidophilus (Infloran®, Desma Healthcare, Chiasso, Switzerland). This supplementation was given twice daily in a divided dose and commenced with the first enteral colostrum/milk feed (usually day 1 postnatal). Oral supplementation was normally administered until 34 weeks post-conceptual age, with the exception of very low birth weight infants (<1500 g) who received it until discharge. Half a capsule of Infloran (125 mg) was dissolved in 1 ml of expressed breastmilk and/or sterile water, and this dose was given twice daily (250mg/total/day) to the infant via nasogastric tube. Recruitment inclusion criteria included gestational age ≤ 34 weeks, and infants resident in the same NICU for study duration. Infants diagnosed with advanced stages of necrotizing enterocolitis or severe congenital abnormalities, were excluded from the study.
Preterm infants were recruited from four different NICUs across England, UK; Norfolk and Norwich University Hospital (NNUH) enrolled the Bif/Lacto group, and Rosie Hospital, Queen Charlotte’s and Chelsea Hospital, and St Mary’s Hospital recruited Control group infants. All NICUs had comparable health care practices including antibiotic and antifungal policies. Bif/Lacto vs. Control groups included similar sex ratios, delivery mode (i.e. Caesarean-section or vaginal delivery) and feeding modes (Suppl Table 1). Specifically, we controlled for diet by preferably selecting preterm infants who received their mother’s own breast milk or donor breast milk; majority were exclusively breastfed or received donor breast milk (78% in Bif/Lacto Group and 76% Control Group), mixed fed with a combination of breastmilk, formula or donor breast milk (20% Bif/Lacto Group and 20% Control Group), and exclusively formula fed (2% Bif/Lacto Group, and 4% Control Group).
Ethical approval for the study
Fecal collection from NNUH and Rosie Hospital was approved by the Faculty of Medical and Health Sciences Ethics Committee at the University of East Anglia (UEA), and followed protocols laid out by the UEA Biorepository (License no: 11208). Fecal collection for Queen Charlotte’s and Chelsea Hospital and St Mary’s Hospital was approved by West London Research Ethics Committee (REC) under the REC approval reference number 10/H0711/39. In all cases, doctors and nurses recruited infants after parents gave written consent.
Time points of sample collection for this study included 0-9 days, 10-29 days, 30-49 days, 50-99 days. Clinical data collected included gestational age, delivery mode, antibiotic courses received, and dietary information can be found in Supplementary Table 3.
DNA extraction of preterm stool samples
FastDNA Spin Kit for Soil (MP) was used to extract DNA from preterm feces following manufacturer instructions, with extended 3 min bead-beating. DNA concentration and quality were quantified using a Qubit® 2.0 fluorometer (Invitrogen).
16S rRNA gene sequencing: library preparation and bioinformatics analysis
16S rRNA region (V1-V2) primers were used for library construction (Suppl Table 4), with the following PCR conditions; cycle of 94°C 3 min and 25 cycles of 94°C for 45 s, 55°C for 15 s and 72°C for 30 s. Sequencing of the 16S rRNA gene libraries was performed using Illumina MiSeq platform with 300 bp paired end reads.
Raw reads were filtered through quality control using trim galore (version 0.4.3), minimum quality threshold of phred 33, and minimum read length of 60 bp. Reads that passed threshold were aligned against SILVA database (version: SILVA_132_SSURef_tax_silva) using BLASTN (ncbi-blast-2.2.25+; Max e-value 10e-3) separately for both pairs. After performing BLASTN alignment, all output files were imported and annotated using the paired-end protocol of MEGAN on default Lowest Common Ancestor (LCA) parameters.
R Studio version 1.1.463 and using the ggplot2 R package version 3.1.0 was used for the analysis of microbiota sequence data and generation of figures. The 16S rRNA bacterial sequence data was subsampled to an even depth of 20,000 read using phyloseq package version 1.24.2. Sample details with proportion of reads assigned to each bacterial genus can be found in Supplementary Table 6 and species in Supplementary Table 7. NMDS (Non-metric multidimensional scaling) plots were generated with a Bray-Curtis dissimilarity calculation in R Studio using with the vegan package version 2.5-4 using code adapted from Torondel et al. (2016)45. Permutational MANOVA in the Adonis function of the vegan R package version 2.5-4 was used to determine significant differences between NMDS community structure. Heatmaps were generated using the ComplexHeatmap package version 1.18.1 and clustered using a Bray-Curtis dissimilarity calculation. Genus number, Shannon diversity, and Inverse Simpson diversity were calculated using the vegan package version 2.5-4. Statistically significant differences in genus and species abundance were determined using a Kruskal Wallace test with Family-Wise Error Rate using to correct for multiple testing. R scripts are available at: https://github.com/dalbymj/BAMBI-Paper-Files.
Genomic DNA Extraction from bacterial isolates
We isolated the strains present in the oral supplementation (i.e. Bifidobacterium bifidum and Lactobacillus acidophilus) as well as additional Bifidobacterium isolates from infant samples. Overnight pure cultures in Brain Heart Infusion Broth (BHI) were harvested for phenol-chloroform DNA extraction. Bacterial pellets were resuspended in 2⍰ml 25% sucrose in 10⍰mM Tris and 1⍰mM EDTA at pH 8. Cells were subsequently lysed adding 50⍰μl 100⍰mg/ml lysozyme (Roche) and incubating at 37 °C for 1 h. 100⍰μl 201mg/ml Proteinase K (Roche), 30⍰μl 10⍰mg/ml RNase A (Roche), 400⍰μl 0.5⍰M EDTA (pH 8.0) and 250⍰μl 10% Sarkosyl NL30 (Fisher) was added into the lysed bacterial suspension, incubated 1 h on ice and left overnight at 50 °C. Next, washes of phenol-chloroform-isoamyl alcohol (PCIA, Sigma) using 15⍰ml gel-lock tubes (Qiagen), with E Buffer (10mM Tris pH 8 (Fisher Scientific, UK)) added to sample to a final volume of 5 ml, mixed with 5 ml of PCIA (Sigma) and centrifuged for 15 min at 4000 rpm. The CIA step was repeated three times, after which the final aqueous phase was transferred into sterile Corning™ 50 ml centrifuge tubes, and 2.5 volumes of ethanol (VWR Chemicals, USA) added, incubated for 15 min at −20 °C, and centrifuged 10 min at 4000 rpm and 4 °C. Finally, the pellet was washed twice with 10 ml of 70% ethanol and centrifuged at 4000 rpm for 10 min, dried overnight, and re-suspended in 300 μl of E Buffer.
Whole genome sequencing analysis: library preparation and bioinformatics analysis
DNA from pure cultures was sequenced at Wellcome Trust Sanger Institute using 96-plex Illumina HiSeq 2500 platform to generate 125⍰bp paired end reads as described previously46. Genome assembly was performed by the sequencing provider using the assembly pipeline described by Page et al. 201647. Next, genome assemblies were annotated using Prokka (version 1.12). We predicted the 16s rRNA gene from the whole genome data using barrnap (version 0.7) and compare it to with existing 16S rRNA sequences. Single nucleotide variants (SNPs) were identified using Snippy (version 4.0) by mapping assembled contigs to annotated reference Infloran B. bifidum strain to reconstruct SNP phylogeny of six B. bifidum strains48.
To construct a phylogeny of 10 Bifidobacterium strains, we used pangenome pipeline Roary (version 3.12.0) to build a core gene alignment (87 core genes, with options -e -n otherwise default), followed by snp-sites (version 2.3.3) to call SNPs (6,202 SNPs in total)47,49. We used the SNP site-alignments obtained from both reference-based and core-gene alignment approaches to infer Maximum Likelihood (ML) phylogenies using RAxML (version 8.2.10) with GTR+ nucleotide substitution model at 100 permutations conducted for bootstrap convergence test50. The ML tree reconstructed was with the highest likelihood out of 5 runs (option -N 5). Pairwise SNP distances were calculated and compared using snp-dists (version 0.2)51. Pairwise Average Nucleotide Identity (ANI) was computed and graphed using module pyani (version 0.2.7)52. Web tool iTOL was used to visualize and annotate ML trees53.
Determination of Minimal Inhibitory Concentration (MIC) of Bifidobacterium bifidum Infloran strain
The microdilution method was used to test Minimal Inhibitory Concentration (MIC) of the probiotic strains (B. bifidum) against routinely prescribed antibiotics; benzylpenicillin, gentamicin, and meropenem. Serial twofold dilutions of antibiotics in MRS medium (Difco) and 10 μL from fresh overnight culture were incubated for 24 h at 37 °C under anaerobic conditions. Cell density was monitored using a plate reader (BMG Labtech, UK) at 595 nm. MICs were determined as the lowest concentration of antibiotic inhibiting any bacterial growth, with tests performed in triplicate.
Gene search using BLAST
Genomes from B. bifidum Infloran strain and five other B. bifidum isolates were searched for genes involved in utilization of human milk oligosaccharides, and mucin degradation. Nucleotide sequences of genes of interest were extracted from National Centre of Biotechnology Information (NCBI). Supplementary table 5 summarizes genes analyzed and publication source. BLAST alignment (ncbi-blast-2.2.25) was performed using a filtering criteria of 80% coverage and 80% identity.
Breast milk and human milk oligosaccharides utilization study
Growth kinetics of the Infloran isolate B. bifidum and control type strains B. longum subsp. infantis DSM 20088 and B. breve DSM 20213 in breast milk and individual HMOs (LNnT or 2’FL) were performed. Isolates were grown overnight in RCM (Oxoid) then subcultured into modified MRS (Difco) with breast milk (pooled from multiple mothers, 1% w/v), or individual HMOs (2% w/v). Growth kinetics were measured every 15 minutes for 48 hours using a microplate spectrophotometer (Tecan Infinite F50).
Metabolomic profiling of preterm feces using one dimensional 1D-nuclear magnetic resonance spectroscopy (NMR) and 2Dnuclear magnetic resonance spectroscopy (NMR)
A subset of 157 paired fecal samples (75 from Bif/Lacto group, and 81 from Control group) were analyzed by standard one-dimensional (1D) 1H NMR spectroscopy using a Bruker 600 MHz spectrometer operating at 300 K. Feces (50 mg) were combined with 700 μl of phosphate buffer (pH 7.4; 100% D2O) containing 1 mmol/L of 3-trimethylsilyl-1-[2,2,3,3-2H4] propionate (TSP), and 10 zirconium beads (1 mm diameter) (BioSpec Products). Samples were homogenized using a Precellys bead beater (Bertin) with 2 cycles of 40 s at 6,500 Hz speed, centrifuged at 14,000 g for 10 min and the supernatant was transferred to NMR tubes. 1D NMR spectra were acquired for each sample using a nuclear overhauser effect pulse sequence for water suppression as described by Beckonert and colleagues54). Spectra were automatically phased and calibrated to the TSP reference using Topspin 3.6 (Bruker BioSpin). Spectra were imported into Matlab 9.4 (R2018a), redundant spectral regions (those arising from TSP and imperfect water suppression) were removed, and the spectral profiles were normalized using a probabilistic quotient method. Data analysis was performed using principal components analysis (PCA), orthogonal projection to latent structures discriminant analysis (OPLS-DA) and covariate-adjusted-projection to latent structures-discriminant analysis (CA-PLSDA) using in-house scripts. Pairwise OPLS-DA models (Bif/Lacto versus Control) were constructed for each sampling point and for all sampling points combined. Here, the complete spectral datapoints (metabolic profile) served as the predictors (X variables) and class membership (Bif/Lacto versus Control) served as the response (Y) variable. The predictive ability (Q2Y) of the models were calculated using a 7-fold cross-validation approach and the validity of the Q2Y values were assessed through permutation testing (100 permutations). A CA-PLS model was also built using the fecal profiles from all sampling points and the model was adjusted for sampling age. Additional two-dimensional (2D) 1H-1H NMR spectroscopy was performed on two selected fecal samples to assist with metabolite identification. Conventional 2D NMR spectra were acquired using homonuclear correlation spectroscopy (COSY) and heteronuclear single quantum coherence spectroscopy (HSQC) experiments with water suppression to assist with structural elucidation.
pH measurement of the fecal samples
The pH of a randomly selected subset of fecal samples used in the metabolomics analysis (39 samples from the Bif/Lacto Group, and 39 samples from the Control Group) was assessed. Fifty mg of fecal sample was added 1 ml of sterile water, vortexed and measured using a glass electrode pH meter (Martini Mi151).
Accession codes
The datasets supporting the conclusions of this article are available in the European Nucleotide Archive (http://www.ebi.ac.uk/ena repository) accession numbers PRJEB31653 (Illumina 16S MiSeq sequencing and Illumina HiSeq sequencing data).
Author’s contributions
Overall study design was conceived by LJH, with PC, JSK, MJD and CAG contributing to refinement. Sample preparation for sequencing was performed by CAG, JK, CL, LC, MK, KLL, AS and KS. Bioinformatics and related computational analyses were performed by SC, RK and SM. Metabolomic analysis was performed by CAG, FFR and JRS. Phenotypic assays were performed by CAG and ML. Statistical analyses and final figure preparation was performed by MJD with input from LJH, CAG, SC, RK, ML, and JRS. LJH, CAG, and MJD wrote the manuscript with important contributions to intellectual content from all authors.
Competing Interests
All authors have no competing interests to disclose.
Acknowledgements
This work was funded via a Wellcome Trust Investigator Award to LJH (100974/Z/13/Z) and support of the BBSRC Norwich Research Park Bioscience Doctoral Training Grant (BB/M011216/1, supervisor LJH, student CAG), Institute Strategic Programme (ISP) grant for Gut Health and Food Safety, BB/J004529/1 (LJH), and ISP grant for Gut Microbes and Health BB/R012490/1 and its constituent project(s), BBS/E/F/000PR10353 and BBS/E/F/000PR10355 to LJH. Work at St Mary’s was supported by a programme grant from the Winnicott Foundation to JSK, and the National Institute for Health Research (NIHR) Biomedical Research Centre based at Imperial Healthcare NHS Trust and Imperial College London. KS was funded by an NIHR Doctoral Research Fellowship [NIHR-DRF-2011-04-128]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We sincerely thank all clinical nurses at Norfolk and Norwich University Hospital (NNUH), Rosie Hospital, Queen Charlotte’s and Chelsea Hospital, and St Mary’s Hospital for collecting stool samples. We would like to give a special mention to research nurses Karen Few, Hayley Aylmer and Zoe McClure for obtaining consent from parents and collecting samples. We thank Wellcome Trust Sanger Institute for their sequencing support.
Footnotes
↵# Joint first author
Typo corrected on author (K Lloyd) surname
References




