

ABSTRACT
Background Hemodynamic shear stress critically regulates endothelial activation and atherogenesis by affecting cytoskeletal dynamics and endothelial gene expression. The Nck adaptor proteins (Nck1 and Nck2) regulate cytoskeletal remodeling pathways and play redundant roles during development. While a cell permeable Nck-binding peptide reduces shear-induced inflammation, the roles of Nck1 and Nck2 in atherosclerosis remain unknown.
Methods and Results Herein, we show that Nck1 deficiency (siRNA/shRNA knockdown, genetic knockout), but not Nck2, decreases basal and shear stress-induced proinflammatory signaling (NF-κB phosphorylation and nuclear translocation) and ICAM-1/VCAM-1 expression. In contrast, neither Nck1 nor Nck2 were required for flow-induced Akt and ERK1/2 activation, and only Nck2 was required for laminar flow-induced cytoskeletal alignment. Using the partial carotid ligation model of disturbed flow, we found that Nck1 knockout mice showed significantly reduced proinflammatory gene expression and macrophage infiltration that was not further diminished upon Nck2 deletion. Consistent with these findings, Nck1 knockout mice showed significantly diminished diet-induced atherosclerosis, associated with reduced plasma cytokine levels and diminished macrophage content. To define the mechanisms of differential Nck1 and Nck2 signaling in endothelial activation, we performed domain swap experiments mixing SH2 and SH3 domains between Nck1 and Nck2. These Nck1/Nck2 chimeras define a critical role for the Nck1 SH2 domain (phosphotyrosine binding) but a redundant role for Nck1/2 SH3 domains (proline rich binding) in rescuing shear stress-induced endothelial activation in Nck1/2 DKO cells. Using domain point mutations, we confirmed the vital role for Nck1’s SH2 domain and identify the first Nck SH3 domain (DY pocket containing domain) in meditating NF-κB activation and endothelial inflammation. Pre-treatment of endothelial cells with the small molecule Nck1 SH3.1 inhibitor confirmed the critical role of this domain in flow-induced NF-κB activation and ICAM-1/ VCAM-1 expression.
Conclusions Taken together, our data reveal a hitherto unknown link between Nck1 signaling in endothelial cell activation and atherosclerosis development, highlighting the potential for targeting Nck1 to control atherogenic inflammation.
INTRODUCTION
Atherosclerosis, a chronic lipid-driven arterial inflammatory disease, develops at sites of local endothelial activation, a proinflammatory shift in endothelial cell phenotype. Local hemodynamic shear stress confers protection or susceptibility to endothelial activation, with atheroprotective laminar flow limiting endothelial activation and atheroprone disturbed flow stimulating endothelial activation, characterized by cytoskeletal remodeling, endothelial stiffening, and nuclear factor-κB (NF-κB)-driven proinflammatory gene expression1–3. Activated endothelial cells recruit monocytes from the circulation, and in the context of hypercholestermia, these monocytes accumulate lipid to drive early fatty streak formation4. Recruitment of smooth muscle cells from the underlying media contribute to plaque formation and drive the production of a collagen-rich protective fibrous cap that limits plaque vulnerability to rupture5. While recent results from the CANTOS trial highlight the important role of limiting inflammation in the treatment of atherosclerosis6, our understanding of the mechanisms regulating flow-induced endothelial activation remain limited.
The Nck family of adaptor proteins (Nck1 and Nck2) are ubiquitously expressed and share approximately 68% amino acids identity7. Nck adaptor proteins lack enzymatic activity but control the formation of signaling complexes through three tandem Src homology 3 (SH3) domains and one C-terminal SH2 domain8. SH2 domains bind with high affinity to tyrosine phosphorylated proteins, whereas the SH3 domains bind to proline-rich sequences (PXXP) in signaling partners, suggesting that Nck serves to couple tyrosine kinase signaling to the activation of downstream pathways9. The two highly similar Nck proteins (Nck1 and Nck2) are expressed by different genes9 that play redundant roles during development, as deletion of both Nck isoforms results in an embryonic lethal phenotype due to impaired vasculogenesis while deletion of only one isoform did not7. While Nck1 and Nck2 play redundant roles in regulating angiogenesis in mouse models of retinopathy10, Nck2 may play a dominant role in PDGF-induced actin polymerization in NIH3T3 cells11. In contrast, Nck1 plays a more dominant role in T cell receptor-induced ERK activation12, suggesting non-compensating roles during phenotypic regulation post-development. However, the signaling effects of Nck1/2 outside the context of cytoskeleton remodeling are incompletely understood.
We previously have shown that pretreatment of endothelial cells with a membrane-permeable peptide corresponding to a Nck-binding PXXP sequence significantly reduced inflammation and vascular permeability in atherosclerosis13, suggesting that Nck may regulate endothelial activation by flow. However, these studies cannot exclude the possibility of off-target interactions between this peptide and other SH3 containing adaptor proteins. Also, it cannot definitely prove that the anti-inflammatory effects observed in vivo are indeed due to direct inhibition of endothelial Nck14. The roles of Nck1/2 in atherogenic endothelial activation and the specific roles of Nck1 and Nck2 remain to be determined. Here we utilized both cell culture and animal models of disturbed blood flow and atherosclerosis to define the signaling roles of Nck1 and Nck2 in shear stress-induced endothelial activation.
METHODS
The authors declare that all the data supporting the current study are either available within the article or online data supplements. All reagents were provided from Gibco, USA, unless otherwise stated. All lentiviral vectors were designed and obtained using the VectorBuilder website, and site-directed mutagenesis performed by the COBRE Redox Molecular Signaling Core.
Cell culture
Nck1/2 were knocked down in human aortic endothelial cells (HAECs) using either SMARTPool siRNA (Dharmacon™) or shRNA (pLV-(shRNA)-mCherry:T2A:Puro-U6 (Nck1 target seq: GGGTTCTCTGTCAGAGAAA; Nck2 target seq: CTTAAAGCGTCAGGGAAGA)) with 3rd generation lenti components provided from Addgene; pMD2.G (#12259), pRSV-Rev (#12253), pMDLg/pRRE (#12251). To knock out Nck1/2 in HAECs, CRISPR-Cas9 was used. The dualguide RNA (sgRNA) sequences targeting Nck1/2 genes were as following: Nck1: GTCGTCAATAACCTAAATAC; Nck2: TGACGCGCGACCCCTTCACC; Scrambled sgRNA: GCACTACCAGAGCTAACTCA. Mouse aortic endothelial cells (MAECs) were isolated from the Nck1 knockout and Nck2 knockout mice as previously described15. Endothelial cells were either exposed to acute onset shear stress or chronic laminar and oscillatory shear stress (model of disturbed flow) as we previously described16.
Animal Studies
The LSU Health - Shreveport Institutional Animal Care and Use Committee has approved the experiments used in this study. Mice containing global Nck1 knockout (Nck1-/-) and conditional Nck2 knockout (Nck2fl/fl) alleles were a gift from the late Tony Pawson (Samuel Lunefeld Research Institute), whereas mice that contained tamoxifen-inducible endothelial-specific Cre recombinase (VE-Cadherin-CreERT2) were kindly provided from Dr Luisa Iruela-Arispe, (UCLA). Mice were crossed with ApoE-/- and bred in house. All mice were backcrossed onto a C57Bl/6J background for at least 10 generations. Male mice at 8-9-weeks of age were intraperitoneally injected with Tamoxifen (1mg/kg, Sigma, St Louis, MO) for five consecutive days to induce CreERT2 nuclear translocation and gene excision. After 2 week recovery, the four groups of animals, including the inducible and endothelial specific (iEC) control (iEC-Control; ApoE-/-, VE-cadherin CreERT2tg/?) Nck1 knockout (Nck1 KO; ApoE-/-, VE-cadherin CreERT2tg/?, Nck1-/-), IEC-Nck2 knockout (iEC-Nck2 KO; ApoE-/-, VE-cadherin CreERT2tg/?, Nck2fl/fl), and the iEC-Nck1/2 double knockout (iEC-Nck1/2 DKO; ApoE-/-, VE-cadherin CreERT2tg/?, Nck2fl/fl, Nck1-/-) were either subjected to partial carotid ligation (PCL) surgery as we previously reported17 or fed high fat diet (TD 88137, Harlan-Teklad, Madison, WI) for 12 weeks to induce spontaneous atherosclerosis.
Statistical Analysis
Data are analyzed as mean ± standard error of the mean (SEM) using GraphPad prism software (Version 7, GraphPad, San Diego, CA). Data was first tested for the Normality using Kolmogorvo-Smirnov test and then for multiple comparisons, 1-Way ANOVA followed by Tukey’s post-test or 2-Way ANOVA and Bonferroni’s post-test was performed for the normally distributed data. Statistical significance was achieved when p<0.05.
RESULTS
Nck1/2 deletion and their effects on shear stress-induced endothelial activation
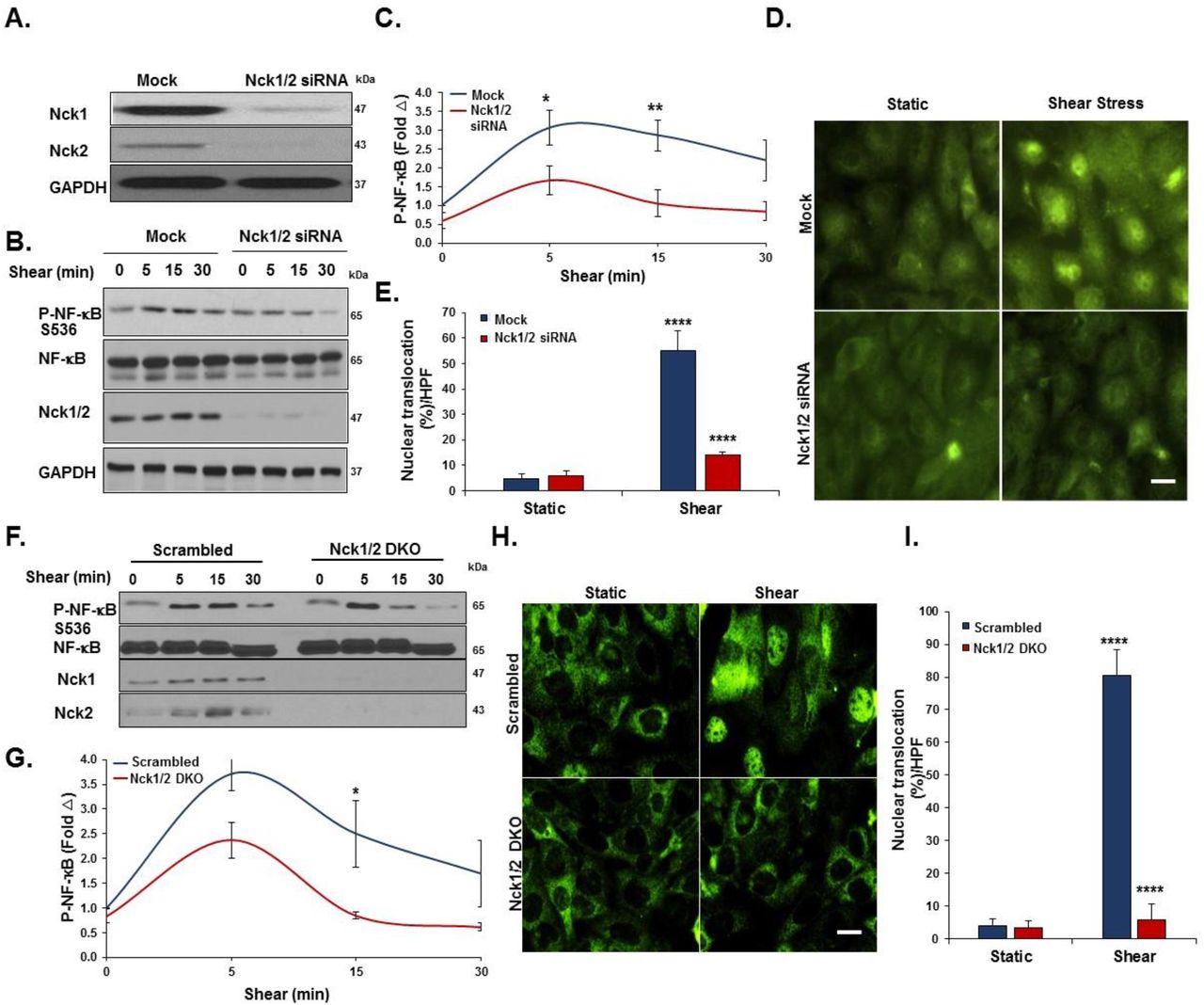
To investigate the direct roles of Nck1/2 in shear stress induced endothelial activation, we adopted a loss of function model where the cellular levels of Nck1 and Nck2 were reduced by transfection with specific siRNA oligonucleotides. The efficiency of transfection was confirmed using Western blotting, with a 70% knockdown of Nck1 and 84% knockdown of Nck2 (Figure 1A). Endothelial cells were subjected to acute shear stress for 5, 15, or 30 minutes or maintained as a static control, and NF-κB activation was assessed by measuring p65 S536 phosphorylation or nuclear translocation. Nck1/2 knockdown cells showed a significant reduction in both NF-κB phosphorylation (Figure 1B-C) and NF-κB nuclear translocation (55± 7.8% vs. 13±1.4%, p<0.0001) (Figure 1D-E). To confirm the combined effect of Nck1/2 knockdown, we used CRISPR/Cas9 editing to generate a stable HAEC line lacking in both Nck1 and Nck2. Nck1/2 double knock out cells (Nck1/2 DKO) show a similar decrease in shear-induced NF-κB phosphorylation (Figure 1F-G) and nuclear translocation (Figure1H-I) compared to scrambled controls (Figure 1I). Similar results were also observed in mouse aortic endothelial cells (MAECs) isolated from iEC-Nck1/2 DKO mice (Supplemental Figure I).

A) Human aortic endothelial cells (HAECs) were transfected with siRNA specific for Nck1/2, and transfection efficiency was assessed using Western blot. B-C) HAECs were subjected to acute shear stress for the indicated times, and NF-κB activation was assessed by detection of p65 serine 536 phosphorylation by Western blot. D-E) p65 nuclear translocation was measured after 45 minutes of shear stress in Nck1/2 siRNA and mock control cells F-I) Nck1/2 was deleted from HAECs using CRISPR/Cas9 editing, and shear stress-induced NF-κB activation was assessed by (F-G) Western blotting for p65 phosphorylation and (H-I) staining for p65 nuclear translocation. Densitometric analysis was performed using Image j. Images were analyzed using NIS Elements software. Data are mean+/- SEM, n=4, analyzed by 2-Way ANOVA followed by Bonferroni’s post-test, *p<0.05, **p<0.01, ****p<0.0001.
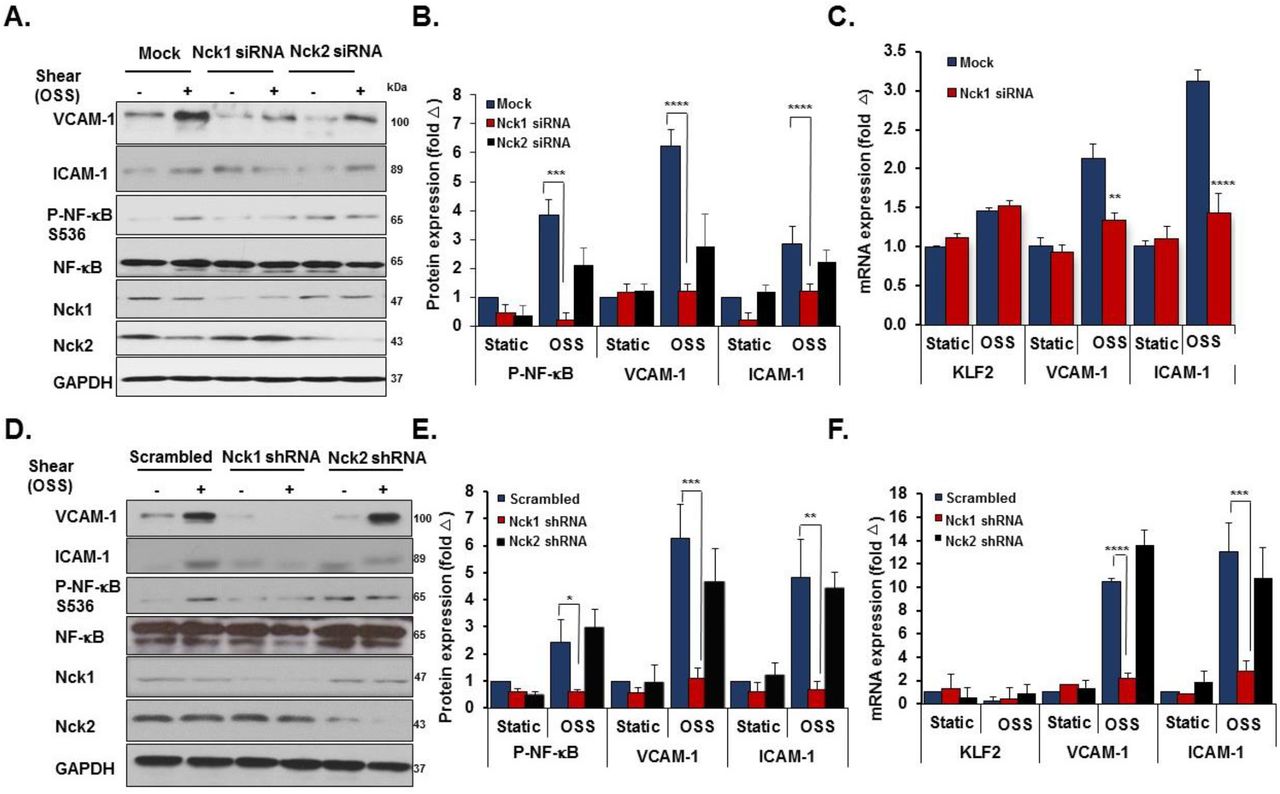
In response to chronic oscillatory shear stress (OSS), NF-κB activation drives proinflammatory gene expression, including ICAM-1 and VCAM-118. We found that siRNA-mediated Nck1/2 depletion reduced oscillatory shear stress-induced VCAM-1 and ICAM-1 protein expression (Figure 2A/B). Compared to the mock controls, Nck1/2 siRNA depleted cells showed significantly less NF-κB activation (Figure 2B) and mRNA expression of ICAM-1 and VCAM-1 (Figure 2C). Shear-induced expression of the atheroprotective gene KLF2 was not affected by Nck1/2 knockdown. Consistent with these data, Nck1/2 DKO endothelial cells show a similar reduction in oscillatory shear stress-induced NF-κB activation (Figure 2D/E) and VCAM-1/ICAM-1 expression (Figure 2D-F). Taken together, these data suggest that Nck1/2 expression is required for NF-κB activation and proinflammatory gene expression associated with atheroprone hemodynamics.

A/B) HAECs were transfected with Nck1/2 siRNA, and oscillatory shear stress (OSS)-induced proinflammatory gene expression (VCAM-1, ICAM-1) and proinflammatory signaling (P-NF-κB Ser536) was assessed by Western blotting. C) HAECs were treated as in (A), and mRNA expression was assessed by qRT-PCR. D/E) Nck1/2 was deleted from HAECs using CRISPR/Cas9 and OSS-induced proinflammatory gene expression and signaling was assessed by Western blotting. F) HAECs were treated as in (D), and mRNA expression was assessed by qRT-PCR. Data are mean ± SEM, n=4, analyzed by 2-Way ANOVA followed by Bonferroni’s post-test, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Deletion of Nck1, but Not Nck2, ameliorates shear stress-induced endothelial activation
Even though they are expressed by different genes, Nck1 and Nck2 proteins share a high sequence identity (68% overall)8 and their functions are generally regarded as overlapping19. However, emerging evidence has suggested the independent contribution of the two isoforms in a variety of responses, including T cell activation, cytokinesis, and podocyte cytoskeletal dynamics20–22. To investigate the selective roles of Nck1 and Nck2, we utilized Nck1 and Nck2 selective siRNAs that result in a 75% and 85% knockdown respectively without affecting the expression of the other isoform (Figure 3A). In response to shear stress, Nck1 depleted cells showed significantly less NF-κB phosphorylation (Figure 3B/C) and nuclear translocation (Figure 3D/E), whereas Nck2 depletion did not affect NF-κB activation by flow. To confirm these effects, we utilized lentiviral shRNA constructs to selectively deplete Nck1 (100% knockdown) and Nck2 (90% knockdown) (Figure 3F). Similar to siRNA data, only HAECs expressing Nck1 shRNA showed significant amelioration of NF-κB phosphorylation (Figure 3G/H) and nuclear translocation (Figure 3I/J), whereas cells expressing Nck2 shRNA did not significantly differ from cells expressing scrambled shRNA. MAECs isolated from Nck1 KO mice showed similar results with remarkable reduction in NF-κB activation following shear stress (Supplemental Figure II), whereas MAECs from iEC-Nck2 KO mice showed normal shear stress-induced NF-κB activation. To assess the specific role of Nck1 in atheroprone disturbed flow models, HAECs transfected with Nck1 and Nck2 siRNA or shRNA were exposed to oscillatory shear stress for 18 hours, and proinflammatory signaling (NF-κB) and proinflammatory gene expression (VCAM-1/ICAM-1) were assessed. Oscillatory flow-induced NF-κB activation (p65 Ser536 phosphorylation) and VCAM-1/ICAM-1 expression were blunted by Nck1 siRNA (Figure 4A-C) and Nck1 shRNA (Figure 4D-F), whereas Nck2 depletion (siRNA and shRNA) had no significant effects on any of these responses.
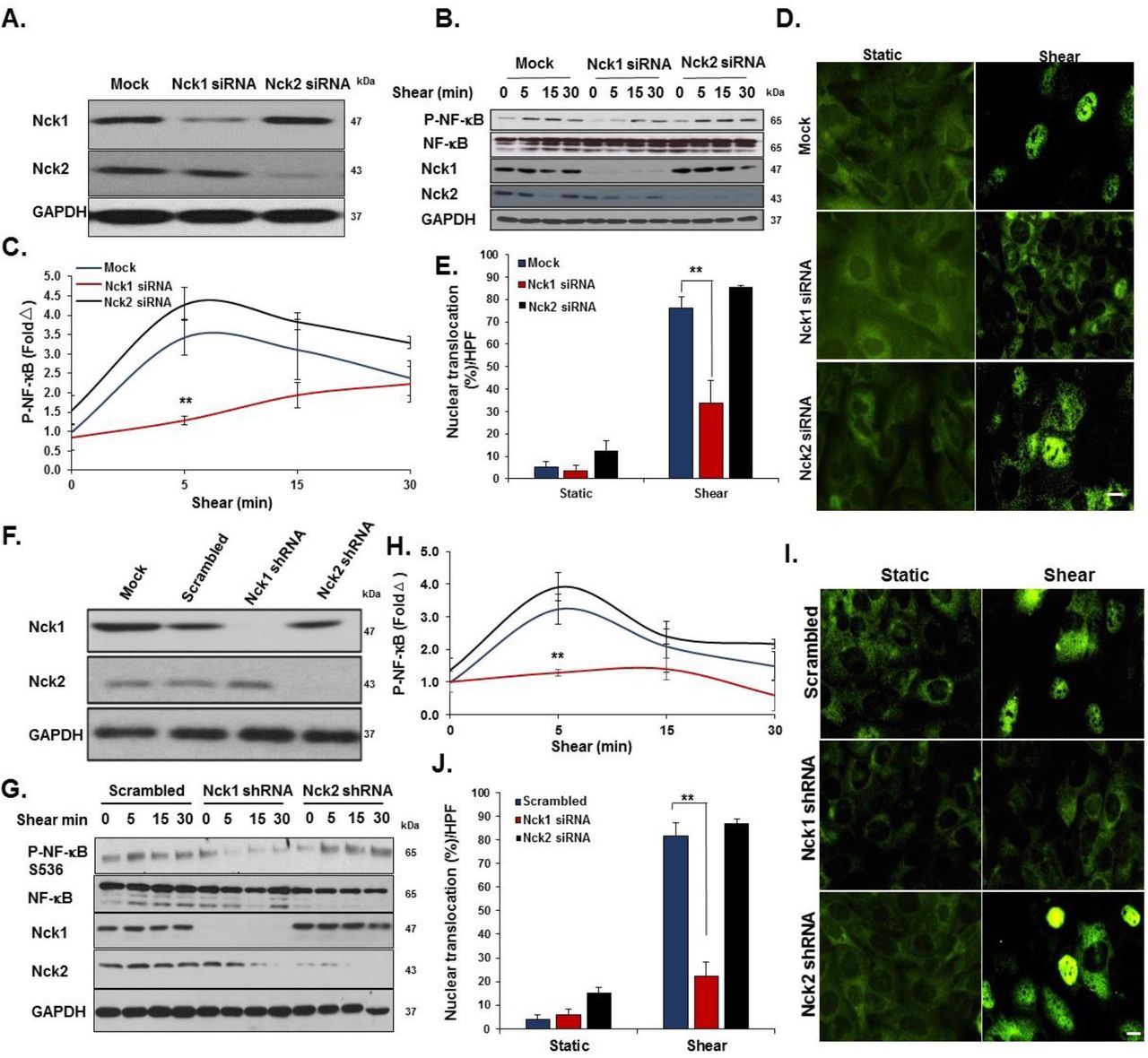
A) Transfection efficiency of selective Nck1 and Nck2 knockdown in HAECs using siRNA. B-E) HAECs lacking either Nck1 or Nck2 were subjected to acute shear stress for the indicated times, and NF-κB activation was assessed by measuring (B/C) NF-κB phosphorylation and (D/E) nuclear translocation. F) Lentiviral Nck1 and Nck2 knockdown by shRNA. G-H) HAECs expressing either Nck1 shRNA or Nck2 shRNA were subjected to acute shear stress for the indicated times, and NF-κB activation was assessed by measuring (G/H) NF-κB phosphorylation and (I/J) nuclear translocation. Data are mean ± SEM, n=4, analyzed by 2-Way ANOVA followed by Bonferroni’s post-test, **p<0.01.

A/B) HAECs were transfected with either Nck1 or Nck2 siRNA, and oscillatory shear stress (OSS, 18h) induced proinflammatory gene expression (ICAM-1/VCAM-1) and signaling (P-NF-κB Ser536) were assessed by Western blotting. C) HAECs were treated as in (A), and mRNA expression was assessed by qRT-PCR. D/E) HAECs were transfected with either Nck1 or Nck2 shRNA, and oscillatory shear stress (OSS, 18h) induced proinflammatory gene expression (ICAM-1/VCAM-1) and signaling (P-NF-κB Ser536) were assessed by Western blotting. F) HAECs were treated as in (D), and mRNA expression was assessed by qRT-PCR. Data are from n=4, mean ± SEM, analyzed by 2-Way ANOVA followed by Bonferroni’s post-test, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
While these data identify Nck1 as a critical regulator of NF-κB activation and proinflammatory gene expression by atheroprone hemodynamics, Nck1 did not affect all shear stress responses. Neither Nck1 nor Nck2 depletion affected KLF2 expression under oscillatory shear stress (Figure 4C/F) or activation of other classic shear stress-induced signaling pathways, such as Akt, eNOS, and ERK1/2 phosphorylation (Figure 5A). Nck1/2 adaptor proteins classically regulate pathways involved in cytoskeletal remodeling, and steady laminar shear stress induces alignment of actin cytoskeleton in the direction of flow23. While siRNA-mediated knockdown of both Nck1 and Nck2 inhibited the endothelial alignment response to laminar shear stress (LSS) (Supplemental Figure III), depletion of only Nck2, and not Nck1, was sufficient to prevent flow-induced cytoskeletal alignment (Figure 5B/C). Collectively these data demonstrate a critical role for Nck1 in endothelial activation by atheroprone flow, whereas Nck2 expression is required for the cytoskeletal alignment response to atheroprotective laminar flow.

A) Endothelial cells subjected to acute shear stress for the indicated times, and activation of Akt, eNOS, and ERK1/2 assessed by Western blotting. Representative blots are shown. n=4. B) HAECs transduced with Nck1, Nck2 or scrambled shRNA were subjected to laminar shear stress (LSS; 10 dynes/cm2 for 18h) and alignment of the actin cytoskeleton assessed by phalloidin-Alexa488 staining. Nuclei were counterstained with DAPI. Arrow indicates the direction of flow. Scale bar=100μm. C) % Cytoskeletal alignment angle was measured using NIS Elements software and compared among experimental groups. Data are from n=4, mean ± SEM, analyzed by 2-Way ANOVA followed by Bonferroni’s post-test, **p<0.01.
Nck1 mediates disturbed flow-induced endothelial activation in vivo
Having shown that Nck1 regulates endothelial activation by atheroprone flow in vitro, we sought to investigate the differential effects of Nck1 and Nck2 in an in vivo model of disturbed flow. Following tamoxifen induction, inducible endothelial-specific control mice (iEC-Control; VE-cadherinCreERT2tg/?, ApoE-/-), Nck1 knockout (VE-cadherinCreERT2tg/?, Nck1-/-, ApoE-/-), endothelial-specific Nck2 knockouts (iEC-Nck2 KO; VE-cadherinCreERT2tg/?, Nck2fl/fl, ApoE-/-), and endothelial-specific Nck1/2 double knockout mice (iEC-Nck1/2 DKO; VE-cadherinCreERT2tg/?, Nck1-/-, Nck2fl/fl, ApoE-/-) were subjected to partial carotid ligation (PCL) to induce disturbed flow-associated endothelial activation specifically in the left carotid artery17,24. Changes in endothelial mRNA expression was assessed after 48 hours, whereas changes in inflammatory gene expression and macrophage recruitment was assessed after 7 days (Figure 6A). To assess endothelial activation, endothelial mRNA was isolated from the left and right carotid vessels by TRIzol flush after tissue harvesting25. The purity of intimal mRNA and medial/adventitial mRNA was confirmed by measuring platelet endothelial cell adhesion molecule-1 (PECAM-1) and a-smooth muscle actin (SMA) expression (Supplemental Figure IVa/b). Endothelial-specific deletion of Nck2 was confirmed in iEC-Nck2 KO and iEC-DKO mice, as Nck2 mRNA expression was depleted in the intimal but not the medial/adventitial fractions (Supplemental Figure IVc/d). KLF2 showed decreased expression in the ligated left carotid compared to the right carotid control, confirming a disturbed flow-associated gene expression profile (Figure 6B). However, this downregulation did not differ among experimental animals. Nck1 knockouts showed a pronounced reduction in disturbed flow-induced VCAM-1 and ICAM-1 mRNA expression (Figure 6B). However, Nck2 deletion (iEC-Nck2 KO) did not affect disturbed flow-induced VCAM-1 and ICAM-1 expression, and VCAM-1/ICAM-1 mRNA expression did not significantly decrease in iEC-Nck1/2 DKO mice compared to Nck1 KO mice (Figure 6B).

A) Schematic of the study in which four groups of mice were subjected to the ligation surgery as indicated animal genotypes and time of surgery. Endothelial mRNA analysis from iEC-Control, Nck1 KO, iEC-Nck2 KO, and iEC-Nck1/2 DKO mice. mRNA from the left carotid was normalized to the unligated right carotid and to the housekeeping gene ß-microglobulin. Data analyzed by 2-Way ANOA and Bonferroni’s post-test, *p<0.05, **p<0.01. C-E) ICAM-1 (red) and VCAM-1 (white) in the ligated left carotid compared to the unligated right carotid arteries among experimental groups. Endothelial cells stained with Von Willebrand factor (vWF) and the nuclei counterstained with DAPI. F-H) Macrophage staining (Mac-2, green) in the ligated and the unligated carotid arteries among experimental groups. Scale bars=100μm. Images analyzed using Nis Elements software, from n=7-10 mice/group. Data are mean ± SEM, analyzed by 1-Way ANOVA and Tukey’s post-test, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
To examine early atherogenic remodeling in the ligated carotid arteries, tissue was collected 7 days post-ligation and assessed for markers of inflammation by immunohistochemistry. Consistent with early changes in mRNA expression, VCAM-1 and ICAM-1 protein levels were significantly reduced in Nck1 KO mice after PCL compared to iEC-Control mice (Figure 6 C-E) whereas iEC-Nck2 KO animals were similar to controls. Similarly, Nck1 KO mice showed a significant reduction in intimal macrophage recruitment (Mac2-positive area) compared to iEC-Control mice (Figure 6F/G). Nck1 KO also significantly reduced adventitial macrophage content (Figure 6F/H), but endothelial Nck2 deletion (iEC-Control vs iEC-Nck2 KO; Nck1 KO vs iEC-Nck1/2 DKO) did not affect intimal or adventitial macrophage infiltration (Figure 6F-H). Taken together, our data suggest a direct role for Nck1 in regulating endothelial activation and macrophage recruitment under atheroprone hemodynamics.
Nck1 deletion reduces atherosclerotic plaque formation
Atheroprone flow establishes local susceptibility to endothelial activation and to diet-induced atherosclerotic plaque development26. Having observed differential effects for Nck1 and Nck2 in response to atheroprone flow-induced endothelial activation, we sought to determine if atherosclerotic plaque development was altered in Nck1 KO mice. iEC-Control, Nck1 KO, iEC-Nck2 KO, or iEC-DKO mice were fed high fat diet (HFD) for 12 weeks to induce atherosclerosis. No significant differences were observed in body weight over the 12 weeks of HFD feeding, though Nck1 KO mice tended to be smaller (Supplemental Figure Va), and no changes were noted for heart weight (Supplemental Figure Vb) or for plasma cholesterol, triglycerides or HDL levels among the experimental groups (Supplemental Figure Vc-e). However, Nck1 KO mice showed significant reductions in the plasma levels of several proinflammatory mediators, including interleukin-1α (IL-1α), IL-1ß, TNF-α, and MCP-1 (Supplemental Figure VI), highlighting the proinflammatory role of Nck1. Atherosclerotic lesion formation was assessed in four different vascular sites, including the aorta, the aortic sinus, the brachiocephalic artery, and the right and left carotid sinuses. En face analysis of atherosclerosis in the aorta was assessed by Oil red O staining and calculated as the percent lesion area compared to the total surface area of the aorta. While iEC-Nck2 KO mice did not differ from iEC-Controls, Nck1 KO mice show a significant reduction in plaque burden in the aorta (Figure 7A/B), brachiocephalic artery (Supplemental Figure VIIa/b), and the carotid sinus (Figure 7C/D). Atherosclerosis did not decrease further in the iEC-Nck1/2 DKO compared to the Nck1 KO, suggesting the endothelial Nck2 does not significantly contribute to atherogenic endothelial activation.

iEC-Control, Nck1 KO, iEC-Nck2 KO, and iEC-Nck1/2 DKO mice were fed high fat diet (HFD) for 12 weeks. A) Representative en face morphometric images of total aortic lesion area and (B) calculated whole aortic atherosclerosis (% of the total surface area). C) Representative stained H&E images of carotid atherosclerosis and (D) quantification of carotid atherosclerotic area among experimental groups. E-F) Analysis of plaque cellular content following staining for macrophages (Mac-2, green), smooth muscle cells (a-smooth muscle actin (SMA), red), and endothelium (vWF, white). G) Lipid core area quantification in carotid atherosclerosis. H) Aortic root atherosclerosis and (I-K) analysis of plaque cellular content following staining for macrophages (Mac-2, green), smooth muscle cells (a-smooth muscle actin (SMA), red), and endothelium (vWF, white) in plaques of the aortic roots. Analysis was performed using NIS-Elements software and data are represented as mean ± SEM, n=6-10/group, analyzed by 1-Way ANOVA, and Tukey’s post-test, **p<0.01, ***p<0.001, ****p<0.0001, ns=not significant. Scale bars=1mm or 50-200μm. ns=not significant.
To assess atherosclerotic plaque characteristics in this model, plaques were stained for macrophage (Mac2 positive area) and smooth muscle (α-smooth muscle actin (SMA) positive). Compared to iEC-Control mice, Nck1 KO mice show a significant reduction in macrophage area in both the carotid sinus (Figure 7E/F) and brachiocephalic arteries (Supplemental Figure VIIc/d). Similarly, Nck1 deletion reduces plaque smooth muscle (SMA positive) area (Supplemental Figure VIIe/f) and lipid core area (Figure 7G) at these sites, consistent with the very early stages of plaque formation observed in Nck1 KO and iEC-Nck1/2 DKO mice. Atherosclerotic lesions in the brachiocephalic artery and carotid sinuses tend to be less well developed than the plaques in the aortic root, potentially due to delayed onset of plaque formation at these sites27. Unlike other sites, we did not observe any differences in plaque size in the aortic root (Figure 7H). However, the plaques that formed showed reduced macrophage area (Figure 7I/J) and enhanced smooth muscle area (Figure 7I/K), suggestive of enhanced plaque stability.
Nck1 regulates inflammation via its SH2 and SH3.1 domains
To understand why Nck1 but not the highly homologous Nck2 regulates endothelial activation, we conducted domain swap experiments mixing the Nck1 SH2 domain with Nck2 SH3 domains and the Nck2 SH2 domain with Nck1 SH3 domains (Figure 8A). We confirmed similar expression levels of transfected constructs encoding Nck1, Nck2, the Nck1 SH2/Nck2 SH3 chimera, and the Nck2 SH2/Nck1 SH3 chimera in Nck1/2 DKO cells by Western blotting (Figure 8B). Nck1, but not Nck2, re-expression restored oscillatory shear stress-induced NF-κB activation (Figure 8C), VCAM-1 expression (Figure 8D) and ICAM-1 expression (Supplemental Figure VIIIa) in the Nck1/2 DKO cells. However, only the Nck1 SH2/Nck2 SH3 chimera showed a similar restoration, suggesting that the Nck1 SH2 domain is essential to form the signaling complex required for oscillatory flow-induced endothelial activation. However, the redundancy of the Nck1 and Nck2 SH3 domains does not exclude them as important for the activation of this response. To gain further insight into the Nck1 domains regulating oscillatory flow-induced NF-κB activation, we introduced single point mutations (Figure 8E/F) to inactivate the Nck1 SH2 domain (R308M, Nck1 SH2*), the first SH3 domain (W38K, Nck1 SH3.1*), the second SH3 domain (W143K, Nck1 SH3.2*), or the third SH3 domain (W229K, Nck1 SH3.3*). Following oscillatory shear stress exposure, NF-κB activation and VCAM-1 expression were only blunted in cells expressing the Nck1 SH2* and the Nck1 SH3.1* constructs (Figure 8G-I), suggesting critical roles for Nck1 SH2-based phosphotyrosine binding and Nck1 SH3.1-based binding partners. The Nck SH3.1 domain binds to an atypical proline rich region containing the sequence PxxDY, and the small molecule inhibitor AX-024 binds to the Nck1 SH3.1 DY pocket to prevent its interaction with the T cell receptor28. Treatment with AX-024 reduces NF-κB activation by acute shear stress (Figure 8J/K) and oscillatory shear stress (Figure 8L), suggesting that Nck1 SH3.1 inhibition may reduce endothelial activation at atheroprone sites (Supplemental Figure VIIIb).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A) Schematic showing the domain structure of Nck1 and Nck2 and the two chimeras of Nck1 SH2/ Nck2 SH3 (1-3) and Nck2 SH2/ Nck1 SH3 (1-3). B) Western blot analysis showing comparable transduction efficiency of Nck1/2 chimeras following introducing the constructs in Nck1/2 DKO cells. n=3. C-D) Nck1/2 DKO HAECs were transduced with constructs in (A), and oscillatory shear stress-induced proinflammatory signaling (P-NF-κB Ser536) and gene expression (VCAM-1) were assessed. E) Schematic of Nck1 domain point mutations, and (F) Western blot analysis showing the efficiency of re-expression of different Nck1 mutants in Nck1/2 DKO HAECs. G-I) Nck1/2 DKO HAECs were transiently transfected with Nck1 or Nck1 variants described in (E), and OSS-induced proinflammatory signaling and gene expression assessed as indicated above. J-K) HAECs were pre-treated with AX-024 (10 nM) or DMSO control 1h before the cells were subjected to acute shear stress for the indicated times. Western blot analysis of NF-κB activation as assessed by Ser536 phosphorylation by Western blotting, n=3. L) NF-κB activation was assessed in HAECs pre-treated with DMSO or AX-024 (10 nM) 1h before oscillatory shear stress for 18h. Data are n=4, represented as mean ± SEM, analyzed by 2-Way ANOVA, and Bonferroni’s post-test, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
DISCUSSION
Atherogenic endothelial activation promotes vascular permeability and enhances adhesiveness for circulating leukocytes26. In atherosclerosis, shear stress critically regulates endothelial activation, with atheroprotective laminar flow reducing activation and atheroprone disturbed flow promoting activation29. In this manuscript, we provide the first description of the Nck family of signaling adaptors (Nck1 and Nck2) as novel regulators of atherogenic endothelial activation in vitro and in vivo. Our data identify Nck1 as a critical signaling mediator of shear stress-induced NF-κB activation and proinflammatory gene expression, and show important roles for the phosphotyrosine-binding Nck1 SH2 domain and the proline-rich region-binding first SH3 domain. In contrast, Nck2 expression is dispensable for flow-induced endothelial activation but critically required for laminar flow-induced endothelial alignment. In vivo, only Nck1 deletion reduces endothelial proinflammatory gene expression, monocyte recruitment, and early plaque formation in both the partial carotid ligation model of disturbed flow and in diet-induced spontaneous atherosclerosis. Taken together, these data identify a novel isoform-specific role for Nck1 in mediating endothelial activation under atheroprone hemodynamics.
The Nck adaptor proteins have been extensively studied in diverse signaling events, most often affecting pathways leading to cellular morphogenesis8. However, the role of Nck1/2 signaling in the context of atherogenic endothelial activation remains limited. We previously demonstrated that inhibiting Nck1/2 signaling by siRNA blunts oxidative stress induced NF-κB activation in models of ischemia/reperfusion injury13, but isoform-specific roles were not addressed. A peptide derived from the Nck1/2-binding sequence in p21 activated kinase reduces NF-κB activation13 and permeability in both cell culture models and in atherosclerosis30, but this peptide could be targeting other SH3 domain containing proteins or affecting non-endothelial cell types to mediate this effect. Our current data provide the first direct evidence that Nck1/2 meditates inflammation due to atheroprone flow and demonstrates an unexpected isoform selective role in mediating flow-induced NF-κB activation and cytoskeletal alignment. Deletion of Nck1 reduces early endothelial proinflammatory gene expression in disturbed flow models, reduces plasma levels of circulating proinflammatory cytokines, limits monocyte recruitment at sites of induced and endogenous disturbed flow, and significantly blunts atherosclerotic plaque formation. Intriguingly, the Nck1 gene resides in regions of chromosome 3q22.3 linked to the susceptibility of stroke31 and premature myocardial infarction (MI)32.
Nck1 and Nck2 are functionally redundant during organismal development, vasculogenesis, and post-natal angiogenesis7,10, but some non-redundant functions have also been described33. For example, T cell receptor-induced ERK and NFAT activation critically require Nck1, but not Nck2, due to interactions between the Nck1 SH3.1 domain and the T cell receptor12. In contrast, Nck2 but not Nck1 facilitates EGF receptor-induced actin polymerization34. We now provide the first evidence for non-redundant functions of Nck1 and Nck2 in the vasculature, with Nck1 mediating flow-induced endothelial activation and Nck2 mediating flow-induced endothelial alignment. While the modular structure of Nck1/2 allow for numerous individual and probably simultaneous protein-protein interactions9, our data specifically identify the Nck1 SH2 domain and the Nck1 SH3.1 (first SH3 domain) as critical mediators of endothelial activation by atheroprone flow. Previous studies have identified at least 60 Nck1 and Nck2 associated proteins mostly involved in cytoskeletal organization20,35. Although the consensus SH2 binding sequence for Nck1 and Nck2 are highly similar36, the Nck1 and Nck2 SH2 domains are able to bind distinct phosphotyrosine sequences on growth factor receptors36 and at sites of cell adhesion36. Consistent with this selectivity, phosphorylated Nephrin selectively recruits Nck1 and not Nck2 through Nck1’s SH2 domain in vivo37. To our knowledge the binding and signaling properties of individual Nck1 SH3 domains have yet to be systematically explored. The critical SH3.1 domain in Nck1 binds an atypical PxxDY motif that undergoes negative regulation by tyrosine phosphorylation, providing a potential negative feedback response that would be expected to limit atherogenic endothelial activation. However, future studies will be required to identify the Nck1 SH3.1 binding partners important for endothelial activation by atheroprone flow.
Due to its role in mediating inflammation and angiogenesis, therapeutic targeting of the Nck1/2 adaptor proteins has been an area of intense investigations. A blocking peptide to the Nck1/2 SH3.2 (second SH3) domain reduces angiogenesis in vivo38, blunts vascular permeability in models of I/R injury13 and atherosclerosis30, and limits inflammation in atherosclerosis and LPS-induced lung injury39,40. However, targeting both Nck1 and Nck2 with this inhibitor is unlikely to be beneficial to atherosclerotic disease, as the inhibitor would limit angiogenesis in ischemic regions affected by the plaque. Since inhibition of either isoform alone is not sufficient to reduce angiogensis10, targeting Nck1 may represent a realistic therapeutic to limit endothelial activation without affecting angiogenic tissue remodeling. A recently developed small molecule Nck1 SH3.1 inhibitor (A-X024) has shown prophylactic effects in the nanomolar range in experimental models of T cell-mediated autoimmune and inflammatory diseases, including psoriasis, asthma, and multiple sclerosis28. We demonstrate that A-X024 significantly reduces endothelial activation in models of disturbed flow, suggesting that this inhibitor may be beneficial to limit plaque formation without preventing ischemic angiogenesis.
In conclusion, our results reveal Nck1, but not Nck2, as a key regulator for endothelial activation and atherosclerotic plaque development. Furthermore, we identified the Nck1 SH2 domain and the first Nck1 SH3 domain as critical mediators of atheroprone flow-induced NF-κB activation and proinflamatory gene expression. Taken together, our findings extend our current understanding of endothelial cell activation in response to atheroprone hemodynamics and identify inhibition of Nck1 by AX-024 as a potential future therapeutic in atherosclerotic cardiovascular disease.
Sources of Funding
This work was supported NIH grants [HL098435, HL133497, HL141155] to AWO and by a Malcolm Feist Cardiovascular Research Endowment Post-doctoral Fellowship (to M A). Research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institute of Health under grant number P20GM121307.
Disclosures
None.
Acknowledgements
The authors acknowledge the COBRE Center for Redox Biology and Cardiovascular Disease and the Redox Molecular Signaling Core for site-directed mutagenesis of Nck1 constructs.
References




