

Abstract
The MreB actin-like cytoskeleton assembles into dynamic polymers that coordinate cell shape in many bacteria. In contrast to most other cytoskeletons, few MreB interacting proteins have been well characterized. Here we identify a small protein from Caulobacter crescentus, AimB, as an Assembly Inhibitor of MreB. AimB overexpression mimics inhibition of MreB polymerization, leading to increased cell width and MreB delocalization. Molecular dynamics simulations suggest that AimB binds MreB at its monomer-monomer protofilament interaction cleft. We validate this model through functional analysis of point mutants in both AimB and MreB, photo-crosslinking studies with site-specific unnatural amino acids, and species-specific activity of AimB. Together, our findings indicate that AimB promotes MreB dynamics by inhibiting monomer-monomer assembly interactions, representing a new mechanism for regulating actin-like polymers and the first identification of a non-toxin MreB assembly inhibitor.
Introduction
Maintenance of proper cell size is an important physiological process for all organisms. Changes in cell size are often strongly coupled to cell fitness in laboratory evolution experiments1, and mutations that affect cell size can be highly adaptive2. Cell size is also dynamically regulated, as in the example of rod-shaped bacteria whose dimensions are altered by environmental factors such as nutrient availability3. A recent study developed a biophysical model in which cell size is determined by the relative rates of surface area and volume synthesis; upon nutrient upshift, the increased rate of cytoplasmic synthesis reduced the surface area-to-volume ratio via an increase in cell width4. However, the molecular regulators of cell size remain largely unclear in most bacterial species.
Bacterial cell shape determination requires enzymes that directly synthesize and crosslink peptidoglycan chains in the periplasm and cytoskeletal factors that localize the activity of these enzymes. The actin homolog MreB serves this cytoskeletal function for cell elongation. Studies from Escherichia coli and Bacillus subtilis show that MreB forms filaments that localize and move5–7 along the membrane based on the local cell geometry8 and recruit cell wall enzymes to insert new cell wall and change the shape of those sites, resulting in a feedback loop that establishes rod shape9. In this model, MreB must dynamically assemble and disassemble to sample multiple cellular regions over time, and indeed in vivo analyses have indicated that MreB structures turn over rapidly10. Purified MreB filaments are quite stable in vitro11, suggesting that MreB dynamics may be stimulated by accessory factors that have yet to be discovered.
For other well characterized cytoskeletal systems such as eukaryotic actin and tubulin and bacterial FtsZ, there are multiple known regulators of filament dynamics (reviewed in12–14). For MreB, in contrast, RodZ is the only confirmed regulator and it functions to stimulate MreB assembly15 and regulate filament properties16, leaving MreB disassembly mysterious. There are several toxin-antitoxin systems whose toxins have been proposed to target MreB17–19, but the degree to which these toxins are expressed and function under standard growth conditions remains unclear. The only other factor proposed to interact with MreB is MbiA, a small C. crescentus protein that interacts with MreB through an unknown mechanism20. The effects of MbiA on MreB also remain unclear, as MreB localization was characterized using a non-functional N-terminal fluorescent fusion to MreB20.
Here, we address the lack of knowledge of MreB assembly inhibitors by directly screening for such factors with an overexpression library. We chose an overexpression approach since MreB and many of its known interactors are essential. Our overexpression screen identified a new factor that we named Assembly Inhibitor of MreB (AimB). As predicted for an important regulator of an essential gene, AimB appears to be essential. Overexpression of AimB resulted in wider cells that resemble the loss of MreB. To characterize the function of MreB, we developed a functional “sandwich” fusion of msfGFP to C. crescentus MreB and found that AimB inhibits its proper localization. Genetic and biochemical studies confirmed that AimB directly interacts with MreB. Finally, we used all-atom molecular dynamics simulations to develop a model for how AimB inhibits the assembly of MreB and confirmed predictions of this model biochemically.
Results
A C. crescentus protein overexpression screen identifies a novel cytoskeletal regulator
We previously constructed a C. crescentus Gateway entry vector library that includes 224 entry vectors containing ORFs encoding “conserved hypothetical” proteins21. To identify candidate MreB regulators among these previously uncharacterized proteins, we transferred these ORFs into a xylose-inducible overexpression destination vector using an in vivo Gateway cloning system, conjugated these constructs into C. crescentus, and imaged the strains at the single-cell level21. Among the various phenotypes observed, overexpression of cc_2490 resulted in a significant increase in cell width that was similar to that seen upon disruption of MreB assembly by the small-molecule inhibitor A2222 (Figure 1A).

A) CC_2490 expression was induced in wild-type C. crescentus for the indicated times. Phase-contrast images show disruption to cell width and cell shape in C. crescentus cells overexpressing CC_2490. Scale bar: 2 µm.
B) MreB-GFPsw cells with or without CC_2490 overexpression were imaged by phase and fluorescence microscopy at 9 h post-induction. MreB was delocalized in cells grown with CC_2490 overexpression. Scale bar: 2 µm.
We expected that a factor that disrupts MreB assembly would have a strong effect on MreB localization. Since previous analyses of MreB localization in C. crescentus used N-terminal fluorescent fusions that we now know to be non-functional23, we first developed a functional reporter for C. crescentus MreB localization. To this end, we inserted monomeric-superfolder GFP (msfGFP), which is less prone to aggregation than most commonly used fluorescent proteins, into the same surface-exposed loop that tolerates functional fusion insertions in E. coli MreB24. We replaced mreB at its native chromosomal locus under its native promoter to generate a strain in which the only copy of MreB is this new “sandwich” fusion (MreB-GFPsw). The MreB-GFPsw fusion does not affect proliferation rate (Figure S1A), suggesting that it is functional with regards to regulating cell growth and division. As observed in the homologous msfGFP-fusion in E. coli, C. crescentus cells expressing MreB-GFPsw were slightly wider and shorter than wild-type cells (Figure S1B,C).
Consistent with its effects on cell shape, overexpression of cc_2490 strongly disrupted MreB localization. Whereas wild-type cells showed MreB-GFPsw foci distributed in patches or at midcell in dividing cells, cc_2490 overexpression caused MreB-GFPsw to disperse and become diffuse or to accumulate at the poles (Figure 1B). Based on these morphological and MreB-localization phenotypes, we renamed CC_2490 AimB for Assembly Inhibitor of MreB. AimB is a member of the Domain-of-Unknown-Function (DUF) superfamily DUF1476 and is widely conserved among Alphaproteobacteria but has no other known activity.
AimB and A22 have additive effects
Since A22 treatment is lethal to C. crescentus cells, we examined whether AimB overexpression is toxic. After only a few hours of overexpression, we observed a significant drop in growth rate as measured by optical density and colony forming units, confirming that AimB overexpression is lethal (Figure 2A,B). Western blots for MreB showed no change in MreB protein levels when AimB is overexpressed (Figure S2A), demonstrating that AimB toxicity was not due to a reduction in MreB protein concentration. To compare the toxicity of AimB overexpression with that of A22, we measured growth with AimB overexpression and A22 treatment individually or in combination. Both treatments were toxic, and the combination of AimB overexpression and A22 treatment further enhanced lethality (Figure 2C,D).

A) AimB overexpression inhibited growth as measured by optical density. Cells containing either the empty vector (pBXMCS-2) or an AimB overexpression vector were grown with 0.03% xylose (induced) or 0.3% glucose (uninduced).
B) AimB overexpression was toxic to cells as measured by colony forming units (CFUs). Samples were removed every 2 h from the cultures in (A) and plated to measure CFUs.
C,D) AimB overexpression and A22 treatment synergistically resulted in toxicity as measured by optical density (C) and CFUs (D). In (C), cells containing either pBXMCS-2 or an AimB overexpression vector were grown in the presence of 0.03% xylose and either 10 µg/mL A22 or methanol (MeOH). Samples were removed every 2 h from the cultures in (C) and plated to measure CFUs (D).
E) Depletion of aimB mRNA using CRISPRi did not affect population growth as measured by optical density. dCas9 expression was induced with 0.5 mM vanillate in cells harboring either a control plasmid (psgRNA-base) or sgRNA-aimB.
F) The rate of width increase for cells grown on PYE-agarose pads containing 2.5 µg/mL A22 and 0.5 mM vanillate was higher in cells depleted for AimB than in wild-type cells.
The similarities between A22 treatment and AimB overexpression suggested that AimB functions to destabilize MreB. Thus, we hypothesized that loss of AimB would stabilize MreB filaments. AimB appears to be essential for C. crescentus survival, as we were unable to generate a clean aimB deletion. Depletion of aimB using CRISPRi25 resulted in a 73.7 ± 2.0% (standard error of the mean; n=3) knockdown of aimB mRNA without having an effect on cell growth (Figure 2E). By contrast to the increased cell width upon AimB overexpression, depletion resulted in narrower cells when compared to controls (Figure 2F, initial time point). We hypothesize that the incomplete knockdown of aimB mRNA permitted cell proliferation despite having an effect on cell width. If loss of AimB stabilizes MreB filaments, we would expect these cells to have increased resistance to A22 treatment. Time-lapse imaging of control and aimB-depleted cells grown on A22-containing agarose pads demonstrated that the initial rate of cell-width increase was faster in the control cells (Figure 2F). Consistent with their opposing effects on MreB, aimB depletion doubled the minimum inhibitory concentration (MIC) of A22 compared to the control (control MIC = 2 µg/mL; aimB-depletion MIC = 4 µg/mL). Thus, AimB modulates MreB function in a manner consistent with that of a negative regulator of MreB assembly.
AimB and MreB interact genetically
To identify the cellular targets of AimB, we performed a screen to identify suppressors of toxicity associated with AimB overexpression. Suppressors of overexpressed AimB-FLAG were isolated and subsequently screened by Western blot to filter out mutants with reduced AimB expression. This screening eliminated suppressors that decreased AimB production as well as nonsense and frameshift mutations in the aimB gene. For each isolated suppressor, we sequenced aimB from the overexpression vector and the chromosomal mreB gene. Three point mutations were identified in the overexpressed aimB that resulted in the residue changes V66M, L74Q, and A97P. Interestingly, 13 unique single point mutations were also found in mreB, demonstrating a genetic interaction between AimB and MreB.
To gain insight into the potential interaction between MreB and AimB we mapped the altered residues in MreB and AimB suppressors onto structures of C. crescentus MreB and an AimB homolog from Jannaschia sp. (Figure S3A-C). Two mutations, MreBK236T and MreBT277A, were located at what is predicted to be the MreB longitudinal polymerization interface26. These changes may suppress the effects of AimB overexpression by stabilizing MreB filaments via increasing the interaction strength between MreB monomers or disrupting MreB-AimB interaction. The remaining 11 mreB mutations map near the ATP binding pocket and are reminiscent of mutations that suppress the effects of A22 treatment27.
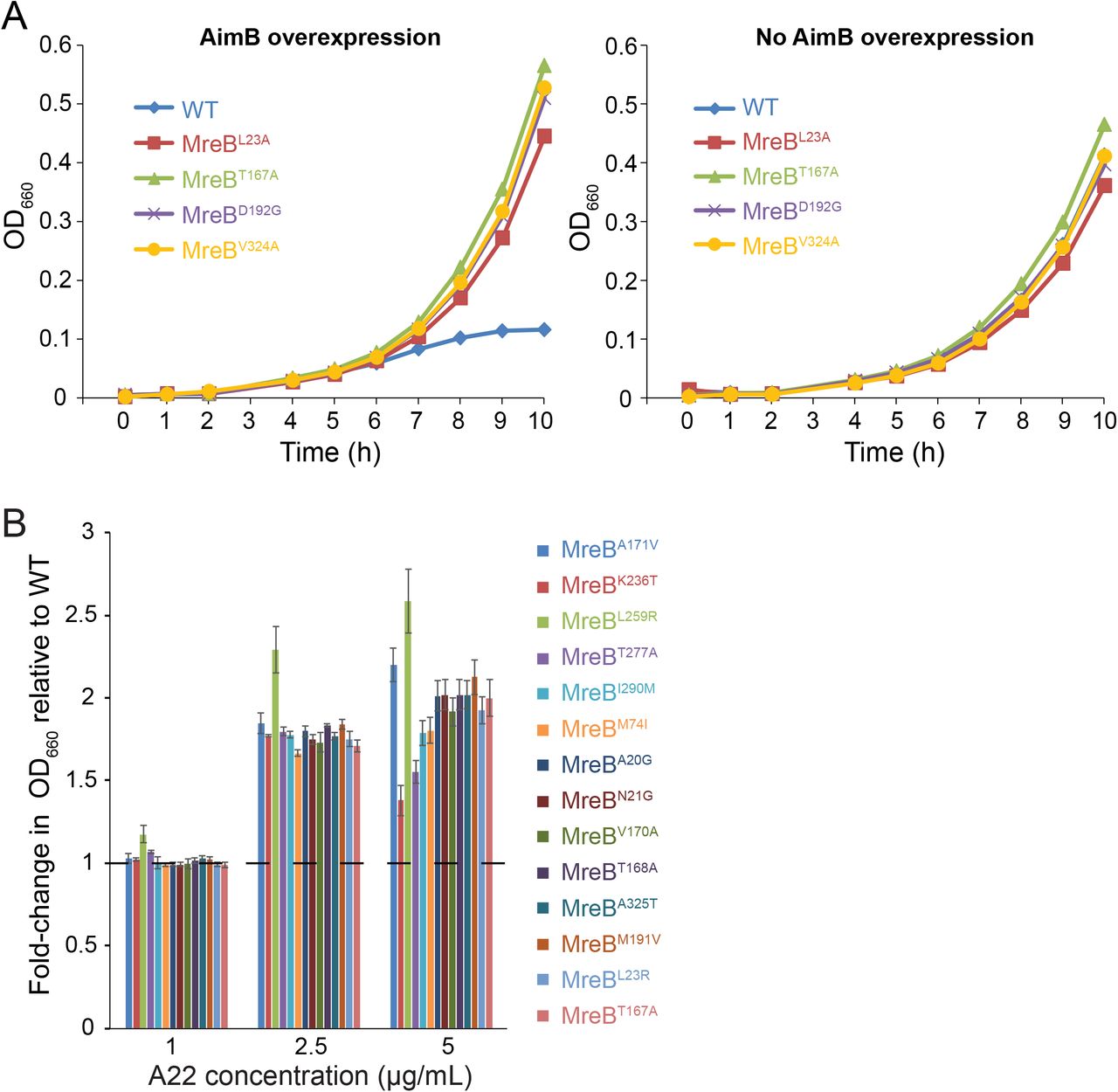
Since similar mreB mutations can confer resistance to A22 treatment and AimB overexpression, we tested previously characterized strains with A22-resistant point mutations in mreB27 for their ability to suppress AimB overexpression. C. crescentus producing chromosomally-encoded MreBT167A, MreBL23A, MreBD192G, or MreBV324A were resistant to AimB overexpression (Figure 3A). These mutations are predicted to inhibit ATP hydrolysis, thereby stabilizing MreB filaments. Conversely, bacteria expressing AimB-resistant mreB mutants also exhibited increased resistance to A22 (Figure 3B). Interestingly, the two MreB mutations at the longitudinal polymerization interface (K236T and T277A) had the highest sensitivity to 5 µg/mL A22 of all the mutants. This variability in A22 sensitivity demonstrates that mutations in the ATP binding pocket likely suppress the effects of AimB overexpression by a different mechanism than the mutations involved in MreB subunit-subunit interactions. Thus, while A22 treatment and AimB overexpression both appear to destabilize MreB polymers, they may act through distinct molecular mechanisms, which would explain their synergistic effects on cell growth.

A) A22 resistance mutations complemented the growth defect due to AimB overexpression. Growth curves for wild-type and A22-resistant strains containing an AimB overexpression plasmid grown with 0.03% xylose (AimB overexpression) or 0.3% glucose (no AimB overexpression).
B) AimB overexpression-resistant strains exhibited higher resistance to A22 than wildtype. Overnight cultures were diluted and grown in media containing 1, 2.5, and 5 µg/mL A22 for 8 h, at which time OD660 readings were taken and standardized to the wild-type culture grown in that concentration of A22. Error bars are standard error of the mean (n=3).
The activity of AimB is specific to C. crescentus MreB
AimB is highly conserved among Alphaproteobacteria but rarely found outside of this clade. Since AimB is essential in C. crescentus yet absent in E. coli, we tested whether AimB alters cell-shape and/or MreB localization in E. coli. Even when AimB was expressed at similar or slightly higher levels compared to those that have a strong impact in C. crescentus (Figure S2B), there was no effect on E. coli cell shape or on qualitative MreB localization (Figure 4A). This selectivity of AimB for C. crescentus MreB is particularly interesting given that the MreB orthologues in these organisms are 78% similar and 64% identical.

A) E. coli expressing MreB-GFPsw were transformed with low-copy plasmids for AimB-FLAG expression and induced with 1 mM IPTG or 100 ng/mL aTc for 6 h. Cells were back-diluted 1:500 at 3 h to maintain log-phase growth. Cells were imaged by phase and fluorescence microscopy (overlay on left) and cell widths were analyzed (right). No effect on cell width or shape was observed. Scale bar = 5 µm.
B) Definitions of opening angle for an MreB monomer. The centers-of-mass of the four subdomains are shown as colored spheres.
C) Snapshot of an ATP-bound CcMreB (PDB ID: 4CZM) at the end of a 100 ns simulation.
D) Snapshot of an ATP-bound EcMreB at the end of a 100 ns simulation demonstrating a larger opening angle than CcMreB in (C). The initial EcMreB structure was a homology model of E. coli MreB built from the CcMreB crystal structure.
E) EcMreB exhibited larger opening angles than CcMreB at the last 30 ns of MD simulations.
F) Docking of a homology model of the Jannaschia sp. protein Jann_2546 (PDB ID: 2KZC), a homolog of AimB, to the equilibrated open structure from a CcMreB MD simulation.
G,H) The interfacial area between MreB and AimB showed that the docked heterodimer of CcMreB and AimB in (F) remained stable throughout 100 ns of MD simulation (Movie S1), while the interfacial area of AimB docking to EcMreB decreased over time (G). The distribution of interfacial areas over the course of the MD simulation demonstrates that AimB interacts more stably with CcMreB (H).
I,J) Substantial overexpression of AimB in E. coli disrupts cell width and MreB localization. E. coli MreB-GFPsw strains harboring low-or high-copy vectors for AimB-FLAG expression were induced as in (A). In (I), cell lysates (normalized to OD600) were analyzed by immunoblotting. In (J), cellular dimensions were quantified by phase and fluorescence microscopy. Scale bar = 5 µm.
Structural modeling suggests that AimB binds in the cleft of MreB
To develop a molecular hypothesis for how AimB specifically affects C. crescentus MreB, we used molecular dynamics (MD) simulations28 to investigate whether the differential effects of AimB in E. coli and C. crescentus are due to different conformations of the two proteins. We generated a homology model of E. coli MreB (EcMreB) based on the C. crescentus MreB (CcMreB) structure (PDB ID: 4CZM)26, and performed all-atom MD simulations (see Methods) on ATP-bound EcMreB and CcMreB monomers. We previously observed for Thermatoga maritima MreB monomers that the opening angle at the polymerization interface (Figure 4B) was polymerization dependent, with a larger value for monomers relative to the subunits of a dimer28. Here, we found that the opening angle of an EcMreB monomer was significantly higher than that of CcMreB (Figure 4C-E). Thus, we hypothesized that AimB’s selectivity could be explained by binding within the gap formed at the MreB-MreB longitudinal polymerization interface. Specifically, binding of AimB when CcMreB monomers open would prevent binding by another MreB monomer and inhibit polymer assembly. Meanwhile, the larger opening in EcMreB would destabilize the binding of AimB, rendering it less active.
In support of our hypothesis, we were able to dock an AimB homology model of the Jannaschia sp. protein Jann_2546 (PDB ID: 2KZC) to the equilibrated open structure of our CcMreB MD simulations (Fig. 4F), and this docked heterodimer remained stable throughout 100 ns of MD simulation (Figure 4G and Movie S1). By contrast, after a docking of the C. crescentus AimB homology model to an EcMreB with a similar opening angle to that of the equilibrated CcMreB-ATP structure, the AimB gradually dissociated during the simulation (Figure 4G and Movie S2), coincident with further MreB opening. Quantification of the MreB-AimB interfacial area over 100 ns of simulation showed that AimB consistently had greater contact with CcMreB as compared to EcMreB (Figure 4H).
Our MD simulations suggested that AimB can form a stable interaction within the opening cleft at the longitudinal polymerization interface of CcMreB, while AimB has decreased affinity for EcMreB. Therefore, we hypothesized that the decreased affinity of AimB for EcMreB could be overcome by increasing its expression. Consistent with this prediction, when we expressed aimB from a high-copy E. coli expression vector, we observed an increase in E. coli cell width (Figure 4I,J), similar to the effects of sublethal A22 treatment29. Importantly, the residues of CcMreB that interact with AimB (within 5 Å) are highly conserved in EcMreB (79% identical and 96% similar; Figure S3D); thus, the relative affinities for CcMreB and EcMreB appear to be due to their opening angles rather than differences in binding-site amino acids.
AimB and MreB interact directly
The isolation of AimB-resistant strains with MreB mutations suggested that MreB and AimB interact directly, and our MD docking simulations further predicted specific regions of the two proteins that may interact. To test these predictions, we used a photo-crosslinking assay. Specifically, we created an expression plasmid with C. crescentus MreB driven by the lac promoter and AimB driven by an arabinose-inducible promoter. Based on the CcMreB crystal structure, we selected 26 surface-accessible residues (Figure 5A) to probe for AimB interactions. Each of the 26 residues was individually mutated to the amber stop codon TAG to enable the incorporation of the unnatural amino acid p-benzoylphenylalanine (pBPA). Each amber mutant plasmid was transformed into an E. coli ΔmreB strain carrying the plasmid pEVOL-pBpF, which encodes the tRNA synthase/tRNA pair for pBPA incorporation30. We chose to use a ΔmreB strain so that the only potential MreB-AimB interaction would be that of the C. crescentus proteins. Following cross-linking, an interaction was only observed when pBPA was incorporated at residue 185 of MreB (Figure 5B). Probing this interaction with an anti-FLAG antibody to detect AimB-FLAG confirmed the interaction (Figure 5C). The size of the shifted band indicated a 1:1 interaction stoichiometry between MreB and AimB. Strikingly, this position is at the base of the cleft where AimB and MreB are predicted to interact based on MD simulations; analysis of our simulations showed that the intermolecular distance between MreBR185 and AimBG64 remained small in CcMreB whereas the distance was larger and more variable in EcMreB (Figure 5D). These crosslinking data provide compelling evidence that AimB directly interacts with MreB in vivo in a manner that validates the conclusions of our MD simulations.

A) The location of the 26 residues of MreB that were mutated to the amber codon for in vitro crosslinking assays are highlighted in blue on the CcMreB crystal structure. Arginine 185 is highlighted in red.
B) In vitro crosslinking experiments were performed by incorporating the UV-crosslinkable non-natural amino acid p-benzoylphenylalanine at various positions in MreB (Materials and Methods). Crosslinked samples were analyzed by immunoblotting for MreB. A crosslinked band was observed for position R185 (blue rectangle).
C) UV-crosslinking of R185 was performed as in (A) using a FLAG-tagged AimB construct. Immunoblotting for MreB or the FLAG-tagged AimB showed similar crosslinked bands.
D) R185 is at the base of the cleft where AimB and MreB are predicted to interact. The intermolecular distance between MreBR185 and AimBG64, the nearest AimB residue, was quantified over the course of our CcMreB-AimB and EcMreB-AimB MD simulations. AimB interacts with CcMreB more stably compared to EcMreB, as shown by a smaller distance between the two residues.
Discussion
As the number of sequenced bacterial genomes rapidly increases, a striking feature of virtually all genomes is the lack of comprehensive annotation, leading to an overwhelming number of “hypothetical genes” whose cellular functions are completely unknown. For even the best studied model organism, E. coli K-12, the fraction of hypothetical genes is >25% (UniProt “uncharacterized” or “putative” genes), roughly similar to other model organisms such as Pseudomonas aeruginosa PAO1 (39%), Vibrio cholera O1 El Tor (42%), B. subtilis 168 (43%), and C. crescentus (20%)31. Moreover, these fractions are likely an underestimate because automated genome annotation pipelines have difficulty distinguishing bona fide small proteins from random unexpressed open reading frames. Advanced transcriptomics and proteomics techniques, such as ribosome profiling32, have improved our ability to robustly confirm the expression of small proteins (<50 residues), some of which are critical regulators of protein kinases, membrane bound enzymes, transport, cell division, or spore formation (reviewed in33). Using the C. crescentus genome as an example, of the 762 genes annotated as hypothetical proteins, there are 34 ORFs shorter than 50 codons and 172 ORFs with 50-100 residues. Here we establish overexpression phenotypic screening as a rapid and robust platform to functionally characterize hypothetical proteins involved in the regulation of the bacterial cytoskeletal element MreB.
The turnover of eukaryotic actin filaments is accomplished by a variety of regulatory proteins that either sequester actin monomers or sever intact filaments34,35. While structural studies of actin-regulator interactions have yielded mechanistic insights into the modulation of actin polymerization, our understanding of MreB polymerization dynamics in general and polymer turnover in particular is quite limited. In C. crescentus, the protein MbiA binds directly to MreB, and its overexpression leads to a loss of proper cell shape and an increase in cell death20. The E. coli toxins YeeV and CptA inhibit MreB polymerization in vitro18,19, however their roles in normal physiology are unclear. Importantly, the mechanism of action for all three proposed MreB inhibitors is unknown. Here, we identified AimB as a novel inhibitor of C. crescentus MreB and provide the first mechanistic model for MreB assembly inhibition. Specifically, in vivo cross-linking experiments (Figure 5A-D) coupled with MD simulations (Figure 4B-H) suggest a novel mechanism for the interaction between AimB and MreB in the cleft of open MreB subunits that blocks MreB dimerization.
Overexpression of AimB results in an increase in cell width (Figure 1A) and mislocalization of MreB (Figure 1B) in a manner similar to the MreB inhibitor A22. A screen for AimB-overexpression suppressor mutants found mutations in MreB (Figure 3B), demonstrating a genetic interaction between MreB and AimB. To probe for a direct interaction between these proteins, we used a photo-crosslinking approach to discover that MreB residue 185 interacts with AimB (Figure 5B,C). These data are consistent with our MD simulations that propose a model in which AimB binds to the longitudinal polymerization interface of MreB. In this model, AimB would function as a pointed-end competitive inhibitor of MreB-MreB dimerization. This model represents a novel mechanism for destabilizing actin-like filaments; thymosin-β4 sequesters G-actin monomers by stretching across the actin molecule and interacting with both the pointed and barbed ends36, while twinfilin inhibits actin polymerization by binding G-actin barbed ends with high affinity37.
In addition to explaining how AimB inhibits MreB assembly, our model can also explain the specificity of AimB for C. crescentus MreB as well as the synergy between AimB overexpression and A22 treatment. Our simulations and crosslinking are consistent with AimB binding to the cleft that forms in MreB subunits at the longitudinal (intra-polymeric) polymer interface when the opening angle is large. Binding of AimB at this site would sterically prevent additional MreB monomers from adding to the polymer, thereby inhibiting MreB filament assembly. Furthermore, this binding site is conformationally distinct in C. crescentus and E. coli (Figure 4B-E), thereby explaining the species specificity. Finally, the structure of C. crescentus MreB filaments solved in the presence of A22 indicates that A22 disrupts the lateral (inter-polymeric) MreB filament interface26, which would explain how AimB and A22 use distinct mechanisms to inhibit MreB filament formation and therefore synergistically inhibit growth rate.
MreB coordinates peptidoglycan insertion to regulate cellular elongation in a variety of species, including Gram-negative E. coli7 and Gram-positive B. subtilis5,6. Although mreB is found across a wide range of bacterial lineages, the aimB gene is restricted to Alphaproteobacteria. Based on our overexpression studies and MD simulations, we suggest that AimB binds the longitudinal polymerization interface of C. crescentus MreB with higher affinity than E. coli MreB. This species specificity is demonstrated by the ability of AimB to disrupt the localization of E. coli MreB only when highly overexpressed (Figure 4J). Species-specific regulation of a bacterial cytoskeletal protein is not unexpected given that the highly conserved tubulin-ortholog FtsZ is regulated by a variety of divergent mechanisms. For example, placement of FtsZ and the divisome at midcell can be mediated by multiple, distinct mechanisms. In E. coli and most Gram-negative bacteria, oscillations of the MinC/D complex are facilitated by MinE38,39, whereas in B. subtilis and most Gram-positive bacteria MinC/D restricts FtsZ to the midline via interactions with DivIVA40. Similarly, nucleoid occlusion in E. coli and B. subtilis is directed by two different proteins, Noc and SlmA, respectively41,42. Interestingly, C. crescentus does not use either the MinC/D or nucleoid occlusion mechanisms for FtsZ localization; instead, a gradient of MipZ antagonizes FtsZ polymerization closer to the poles, leading to midcell Z-ring formation43. Thus, while the core MreB and FtsZ cytoskeletal proteins are widely conserved in bacteria, emerging evidence suggests that the regulation of these core cytoskeletons is largely performed by species-specific factors.
Methods
Bacterial strains, plasmids, and growth conditions
The strains, plasmids, and primers used in this study are described in Tables S1, S2, and S3, respectively. Details regarding strain construction are available in the Supplementary Text. C. crescentus wild-type strain CB15N and its derivatives were grown at 30 °C in peptone-yeast-extract (PYE) medium (Poindexter, 1964). E. coli strains were grown at 37 °C in LB medium. When necessary, antibiotics were added at the following concentrations: kanamycin (Kan) 30 µg/mL in broth and 50 µg/mL in agar (abbreviated 30:50) for E. coli and 5:25 for Caulobacter; tetracycline (Tet) 1:2 for Caulobacter; chloramphenicol (Cm) 20:30 for E. coli; carbenicillin (Carb) 50:100 for E. coli. Gene expression was induced in Caulobacter (0.03-0.3% w/v xylose; 0.5 mM vanillate) or E. coli (100 ng/mL anhydro-tetracycline (aTc)); 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG)) as noted. Pharmacological inhibition of MreB was performed by adding 1-10 µg/mL A22 (methanol was used as the vehicle control).
CRISPRi-mediated gene depletion
C. crescentus CRISPRi was performed using the plasmids (Table S2) and methods developed by the Jacobs-Wagner lab25. Briefly, primers EK1003/1004 (Table S3), encoding the sgRNA mapping to the 5’-end of aimB, were phosphorylated and annealed. The annealed oligos were ligated into the BbsI site of plasmid psgRNA-Base. The resulting plasmid (pEK334) was transformed into a strain carrying a vanillate-inducible catalytically dead cas9 gene (CJW6270) to generate strain EK335 (ΔvanA::pV-dcas9hum-RBSmut1 with plasmid psgRNA-aimB). Gene-depletion was initiated with 0.5 mM vanillate and monitored by qRT-PCR. Cells carrying psgRNA-base were used as controls.
High-throughput cloning and microscopy
Xylose-inducible plasmids for overexpression of conserved hypothetical proteins were generated using an in vivo Gateway strategy, as described previously21,44. The resulting multicopy plasmids were conjugated into C. crescentus. Strains were induced with 0.3% xylose and imaged in high-throughput format using custom 48-pedestal agarose slides21,44. Cell morphology was compared to wild-type controls to identify overexpression plasmids resulting in aberrant cell shape.
Fluorescence microscopy and image analysis
Cells were spotted onto pads made of 1% agarose with the corresponding growth medium. Fluorescence microscopy was performed on a Nikon Ti-E inverted microscope equipped with a Lumen 220PRO illumination system (Prior), Zyla sCMOS 5.5-megapixel camera (Andor), CFI Plan Apochromat 100X oil immersion objective (NA 1.45, WD 0.13 mm), and NIS Elements software for image acquisition. Images were segmented using Morphometrics45. Cell width and length were calculated using custom Matlab scripts. For time-lapse imaging, coverslips were sealed with VALAP (1:1:1 vaseline:lanolin:paraffin) to prevent drying of the agarose pad.
Cell growth measurements
For experiments up to 12 h, cells were grown in standard culture tubes and aliquots were removed at the specified intervals for measurements of OD660 or colony forming units (CFUs). For experiments longer than 12 h, cells were aliquoted into a 96-well plate and the OD660 was measured on a ClarioSTAR plate reader (BMG Labtech) with shaking and temperature control.
Immunoblotting
Cell samples were normalized by optical density (1 mL of OD=0.5) and lysed in 1X SDS sample buffer. Samples were separated on a 4-20% gradient polyacrylamide gel, transferred to a PVDF membrane, and blotted with antibodies against MreB (1:1000)22, GFP (1:1000, Abcam ab6556), or FLAG (1:500, Santa Cruz sc-166355). Horseradish peroxidase-conjugated secondary antibodies (1:5000) and enhanced chemiluminescence reagents (GE Healthcare) were used to detect the bands on a Bio-Rad ChemiDoc MP system.
Quantitative RT-PCR (qRT-PCR)
RNA was extracted from bacterial cultures using the Qiagen RNeasy kit. Following DNase digestion, RNA (5 ng/µL) was reverse-transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). 1 µL of cDNA was used as template in a 10 µL qRT-PCR reaction performed with Power SYBR reagent (Applied Biosystems). qRT-PCR was performed on an ABI QuantStudio 6 using the ∆∆Ct method. rpoD expression was used as the loading control.
Molecular dynamics simulations
All simulations (Table S4) were performed using the molecular dynamics package NAMD v. 2.1046 with the CHARMM27 force field, including CMAP corrections47. Water molecules were described with the TIP3P model48. Long-range electrostatic forces were evaluated by means of the particle-mesh Ewald summation approach with a grid spacing of <1 Å. An integration time step of 2 fs was used49. Bonded terms and short-range, nonbonded terms were evaluated every time step, and long-range electrostatics were evaluated every other time step. Constant temperature (T = 310 K) was maintained using Langevin dynamics50, with a damping coefficient of 1.0 ps−1. A constant pressure of 1 atm was enforced using the Langevin piston algorithm51 with a decay period of 200 fs and a time constant of 50 fs. Setup, analysis, and rendering of the simulation systems were performed with the software VMD v. 1.9.252.
Simulated systems
MD simulations performed in this study are described in Table S4. Simulations were initialized from the C. crescentus MreB crystal structure (PDB ID: 4CZM)26. The bound nucleotide was replaced by ATP, and Mg2+-chelating ions were added for stability. An AimB homology model was built based on Jannaschia sp. protein Jann_2546 (PDB ID: 2KZC) using Phyre253. Water and neutralizing ions were added around each monomer or dimer, resulting in final simulation sizes of up to 89,000 atoms. All simulations were run for 100 ns. For mean values and distributions of measurements, only the last 30 ns were used. To ensure simulations had reached equilibrium, measurement distributions were fit to a Gaussian.
Analysis of opening angles
The centers-of-mass of the four subdomains of each protein were obtained using VMD. For each time step, we calculated one opening angle from the dot product between the vector defined by the centers-of-mass of subdomains IIA and IIB and the vector defined by the centers-of-mass of subdomains IIA and IA. Similarly, we calculated a second opening angle from the dot products between the vectors defined by the centers-of-mass of subdomains IA and IB and of subdomains IA and IIA. The opening angles we report are the average of these two opening angles. Subdomain definitions are as in28.
In vitro crosslinking
A low-copy plasmid for induction of MreB and AimB was constructed using the pZS2-123 vector backbone54. The aTc-regulated CFP open reading frame was removed by inverse-PCR with primers EK644 and EK645 (Table S3). The arabinose-inducible RFP was replaced with AimB by Gibson assembly (primers EK646-649; Table S3). Wild-type MreB and a series of amber codon mutants (Table S1) were synthesized by Genscript (Piscataway, NJ) and used to replace the IPTG-inducible YFP to create a plasmid encoding Plac-MreB and Para-AiMB (pMreBXL1-26). A C-terminal FLAG tag was introduced into AimB in a subset of amber codon mutants by inverse PCR using primers EK679-680. In vitro crosslinking of MreB and AimB was performed essentially as previously described30. pMreBXL and pEVOL-pBpF30 were co-transformed into strain NO36 (MC4100 ∆mreB) and grown overnight in LB containing kanamycin and chloramphenicol. Cells were diluted 1:100 into fresh LB with antibiotics along with inducers (1 mM IPTG and 0.1% w/v L(+)-arabinose) and 1 mM p-benzoylphenylalanine (Bachem). After 4 h, 1 mL of each culture was pelleted, resuspended in 50 µL cold PBS, and transferred to a white 96-well plate. The samples were irradiated under a UV bulb (Norman Lamps CFL15/UV/MED) on ice for 15 min and 50 µL 2X SDS sample buffer was added to stop the reaction. Samples were boiled for 5 min and analyzed by immunoblotting.
Author contributions
J.N.W., H.S., J.H., K.C.H., Z.G., and E.A.K. designed the research, J.N.W., H.S., J.H., and E.A.K. performed research, and J.N.W., H.S., J.H., K.C.H., Z.G., and E.A.K. analyzed data. E.A.K. wrote the manuscript and J.N.W., H.S., K.C.H., Z.G., and E.A.K. edited the manuscript.
Acknowledgements
We thank Anna Konovalova (University of Texas, Health Science Center) for helpful discussions and assistance with the photo-crosslinking assay and Christine Jacobs-Wager (Yale University) for providing CRISPRi reagents. Funding was provided by NSF CAREER Award MCB-1149328, the Stanford Center for Systems Biology under Grant P50-GM107615, and the Allen Discovery Center at Stanford on Systems Modeling of Infection (to K.C.H.); NIH Ruth L. Kirschstein National Research Service Award 1F32GM100677 (to J.H.); an Agilent Fellowship and a Stanford Interdisciplinary Graduate Fellowship (to H.S.); NIH Grant R01GM107384 (to Z.G.); and NSF CAREER Award MCB-1553004 (to E.A.K.). K.C.H. is a Chan Zuckerberg Investigator. All simulations were performed with computer time provided by the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number OCI-1053575, with allocation number TG-MCB110056 (to K.C.H.).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}