

Abstract
The considerable microbial diversity of soils, their variety and key role in biogeochemical cycling has led to growing interest in their global distribution and the impact that environmental change might have at the regional level. In the largest study of Arctic soil bacterial communities to date, we used high-throughput sequencing to investigate the bacterial diversity from 200 widely dispersed Arctic soil samples. We identified a core microbiome, composed of 13 OTUs present at over 95% of sites, regardless of geographical location and environmental conditions. pH was identified as the key environmental driver structuring Arctic soil communities, while total organic carbon, moisture and conductivity had little effect. We were able to identify specialist, generalist and indicator taxa. Only one core biogeographical region was apparent (East Greenland, Svalbard and Iceland), although strong similarities did exist between Arctic sites separated by substantial geographical distances. We suggest that while pH might appear as the primary factor structuring soil bacterial community composition, dispersal may drive community structure in some parts of the region. Overall, Arctic soil bacterial communities, while driven by the same environmental factors as those elsewhere, were fundamentally different from those of temperate and tropical soils.
Introduction
Biogeography, the study of biodiversity across space and time, gives insights into ecological mechanisms such as speciation, extinction, dispersal and species interactions (Martiny et al., 2006; Fierer, 2008). Theoretically, distant and isolated habitats are expected to present high endemicity as a consequence of intrinsic dispersal limitations and environmental filtering (Mittelbach and Schemske, 2015; Kleinteich et al., 2017; Bahram et al., 2018). Thus, isolated, pristine ecosystems with limited human presence, such as the Arctic region, should harbour endemic communities. However, microbial communities may be less constrained by geographical barriers and thus, have long been considered ubiquitous (Finlay, 2002; O’Malley, 2007). Yet, recent studies have uncovered patterns of microbial biogeography on global scales (Fierer and Jackson, 2006; Lauber et al., 2009; Tedersoo et al., 2014; Henschel et al., 2015; Bahram et al., 2018; Delgado-Baquerizo et al., 2018). The study by Delgado-Baquerizo et al. (2018) illustrated the high number of OTU associations with soil pH and thus, the importance of pH in structuring bacterial communities globally. It followed a previous global study by Tedersoo et al. (2014) which identified pH as a major predictor of fungal richness and diversity worldwide. These studies, however, had a low number of Arctic samples despite the Arctic tundra covering over 5% of Earth land surface (Nemergut et al., 2005). Thus, the application of their predictions to the Arctic region is difficult to assess, especially considering that Arctic microbial communities generally cluster away from other terrestrial regions (Fierer et al., 2012; Tedersoo et al., 2014), suggesting a different character for these communities. Previous Arctic studies on various spatial scales have also identified pH as a primary factor structuring microbial communities (Chu et al., 2010; Siciliano et al., 2014). However, these studies generally have a low number of samples over restricted sampling areas. The study by Metcalfe et al. (2018) illustrated the sampling bias of Arctic studies, focused on Abisko, Sweden and Toolik lake, Alaska. This study identified large areas of Northern Canada and Siberia as being largely under-cited across all disciplines, including microbiology. The review by Malard and Pearce (2018) further illustrated this bias by identifying all studies investigating microbial diversity across the Arctic, and highlighting the need for increased research effort, sampling site number and standardized protocols.
Frozen soils in the Arctic region store over 1500 Pg of carbon (Koven et al., 2011; Mackelprang et al., 2011) and as Arctic warming is exacerbated and permafrost thaw accelerates, the depth of the active layer is increasing. As previously frozen carbon becomes available, it is expected that microbial activity will increase, which may lead to increased atmospheric release rates of climate active gases such as carbon dioxide (CO2), methane (CH4) and nitrous oxide (N2O) (Ma et al., 2007; Mackelprang et al., 2011). Carbon-climate feedback studies of permafrost affected regions use temperature, soil moisture and precipitation as the main drivers controlling decomposition rates (Koven et al., 2011; Schuur et al., 2015). While models are useful to gain a global understanding of the impact of climate change on permafrost thaw and greenhouse gas release, the accuracy of results obtained is highly variable when compared with data collected in the field or laboratory (Schuur et al., 2015) due to empirical and modelling uncertainties still needing to be addressed (Bradford et al., 2016).
Microorganisms drive biogeochemical cycling and participate in the uptake and release of CO2, CH4 and N2O, so microbial data should be incorporated in climate models. Current models use soil properties to model changes in fluxes, without considering microbial communities and the changes in community composition induced by climate change (Bardgett et al., 2008; Nazaries et al., 2013). Adding microbial information into models will improve their predictions; however, detailed microbial data is still required, with a focus on microbial community, diversity, function and long-term changes in these communities (Graham et al., 2012; Nazaries et al., 2013). While global surveys of microbial diversity have already been conducted (Tedersoo et al., 2014; Delgado-Baquerizo et al., 2018), the number of Arctic samples is restricted (Malard and Pearce, 2018) and therefore, microbial data is still lacking for permafrost-affected regions.
Here, we conducted a Pan-Arctic survey of bacterial communities in Arctic soils to provide a baseline database, characterize Arctic soil bacterial communities and identify biogeographical patterns of diversity across the region. The most straightforward demonstration of biogeography is demonstrating that microbial composition across a landscape is non-random (Martiny et al., 2006). To do so, we evaluated the influence of environmental conditions, known to impact global microbial communities, on bacterial composition and diversity in the Arctic region. We identified specialist, generalist and indicator taxa to evaluate the impact of dispersal and environmental factors on the structure of these communities. We also characterized cosmopolitan OTUs representing the Arctic core microbiome. Our sample collection is widespread across 43 core sites and orders of magnitude larger than previous Arctic studies.
Methods
Sample collection
Soil samples were collected at 43 sites across the Arctic region between April 2017 and September 2017 [Fig. 1], the GPS coordinates of each site was recorded with a portable GPS and photographs were taken. At each site, 3 to 5 soil samples were collected within a 100 m2 area under the most common vegetation, for a total of 200 unique Arctic samples. Approximately 150 g of soil per sample was collected in Whirl-Pak bags (Nasco, WI, USA), from the top 15 cm. Plant roots and rocks were removed, samples were homogenized thoroughly and frozen at −20 °C before transportation to the United Kingdom. Samples were conserved at − 20 °C until analysed.

Map of sampling sites. A total of 200 soil samples in 43 different sites were collected for this study.
Soil properties
Moisture content was measured gravimetrically on soils after drying at 150 °C for 24 h and total organic content (TOC) was measured gravimetrically by heating previously dried soils to 550 °C for 4 h. pH and conductivity were measured in the laboratory in a 1:5 freshly thawed soil to water ratio, using a Mettler-Toledo FE20 pH meter (Mettler-Toledo Instruments co., Shanghai, China) and a CMD500 conductivity meter (WPA, Cambridge, UK).
DNA extraction
Soil DNA was extracted in duplicate for each sample using the PowerSoil kit (Qiagen, Hilden, Germany), for a total of 400 DNA extracts. Each sample was PCR amplified using the universal primers 515F-806R, as per the Schloss lab standard operating Procedure (Kozich et al., 2013) and the Earth Microbiome Project (Thompson et al., 2017), under the following conditions: initial denaturation at 95°C for 2 min then 30 cycles of 20 s denaturation at 95°C; primer annealing at 55°C for 15 s; elongation at 72°C for 5 mn then a final elongation at 72°C for 10 min. Negative controls, DNA extraction kit controls and ZymoBIOMICS mock communities (Zymo Research, Irvine, CA, USA) were included alongside the soil DNA and sequenced. PCR amplicons were cleaned and normalized using SequalPrep Plate Normalization Kit (Invitrogen, Carlsbad, CA, USA) and combined into four pools. Each pool was quantified using fragment size determined by BioAnalyzer hsDNA assay (Agilent technologies, Santa Clara, CA, USA) and concentration by Qubit hsDNA kit (Invitrogen). The library was supplemented with 5% PhiX and loaded on an Illumina MiSeq V2 500 cycles cartridge.
Illumina Sequencing and Data Processing
Raw amplicon sequences were demultiplexed with the associated barcodes. Cutadapt (Martin, 2011) was used for adaptor and primer clipping. Forward and reverse reads that were long enough were merged (98 % ± 0.8 % / sample) using FLASH (fast length adjustment of short reads) (Magoč and Salzberg, 2011) for a total of 20 million reads (~50,000 ± 30000 reads/sample) initially. Vsearch (Rognes et al., 2016) was used for downstream analyses. Quality filtering was carried with an expected error > 1.5. Dereplication was performed to identify unique sequences. A two-step chimera detection method was used, first by aligning against ChimeraSlayer Gold database provided with SILVA (Pruesse et al., 2007), second by using the denovo detection module in Vsearch. An open-reference operational taxonomic unit (OTU) calling was performed on high-quality trimmed sequences at 97% similarity level using the USEARCH (Edgar, 2010) algorithm for clustering implemented in Vsearch to generate operational taxonomical units (OTUs). Unique chimera filtered sequences were aligned using the Python Nearest Alignment Space Termination (PyNAST) (Caporaso et al., 2009) tool with a relaxed neighbour-joining tree built using FastTree (Price et al., 2010). The taxonomy was determined using the Classification Resources for Environmental Sequence Tags (CREST) (Lanzén et al., 2012) classifier with a confidence threshold of 0.80 against SILVA release 128 as a reference database.
Samples less than at least 2000 reads/sample were filtered from the OTU table in order to have sufficient reads to capture the accurate relationships among samples as described in Caporaso et al. (2010a). After filtering, a total of 386 samples were used for the statistical analyses, corresponding to 386 DNA extracts from 200 unique samples and ~19.5 million reads (50 609 ± 26 700 reads/sample) assigned against 49 057 OTUs.
Data Availability
The dataset is deposited at European Nucleotide Archive / SRA under the accession number PRJEB29109.
Statistical Analysis
All statistical analyses were performed with a combination of Qiime1 V 1.90 (Caporaso et al., 2010b) and R environment (Team, 2013) using phyloseq (McMurdie and Holmes, 2013), vegan (Dixon, 2003) and indicspecies (Cáceres and Legendre, 2009) packages. Alpha Diversity was calculated using matrices of richness (number of observed OTUs) and diversity (Shannon diversity) based on a rarefied OTU table to compensate for variation in sample depth. Multiple rarefaction was performed with the smallest sample size as maximum depth. The difference in alpha diversity indices was compared statistically using a non-parametric (Monte Carlo) test across different pH categories with Bonferroni correction. Beta Diversity using Bray-Curtis distance was calculated by normalizing the OTU table using cumulative-sum scaling (CSS) (Paulson et al., 2013). The dissimilarity matrix was plotted using principal coordinates analysis (PCoA). ANOSIM from vegan was used to analyze the similarities based on Bray-Curtis dissimilarity beta diversity matrix across pH categories with free permutations. Multivariate analysis by redundancy analysis (RDA) of bacterial communities and environmental variables was performed using Vegan (Dixon, 2003) to extract and summarize the variation in ordination explained by explanatory variables. Indicator species were determined by the Dufrene-Legendre indicator species analysis method (Cáceres and Legendre, 2009) to identify OTUs that were specifically associated with the different pH ranges. Spearman’s correlation coefficient was used to identify the possible correlations between the environmental variables.
Results
Overall bacterial community composition and drivers of diversity
We identified 48 147 bacterial taxa, of which 135 OTUs had abundances over 0.1% across all 386 samples (defined as abundant taxa). Abundant taxa represented 32% of all the reads, illustrating the dominance of a few taxa over the rest of the community. Of the abundant taxa only, Acidobacteria dominated the community at approximately 31% with Blastocatellia (12.8%) and Subgroup6 (7.2%) as abundant classes. Verrucomicrobia was the second most abundant phylum (23%), dominated by Spartobacteria (17.2%). Alphaproteobacteria (10.6%) and Betaproteobacteria (6.6%) were the most commonly identified Proteobacteria (20% overall). Actinobacteria (10.7%), Chloroflexi (7%) and Bacteroidetes (3.5%) were also among abundant phyla classified.
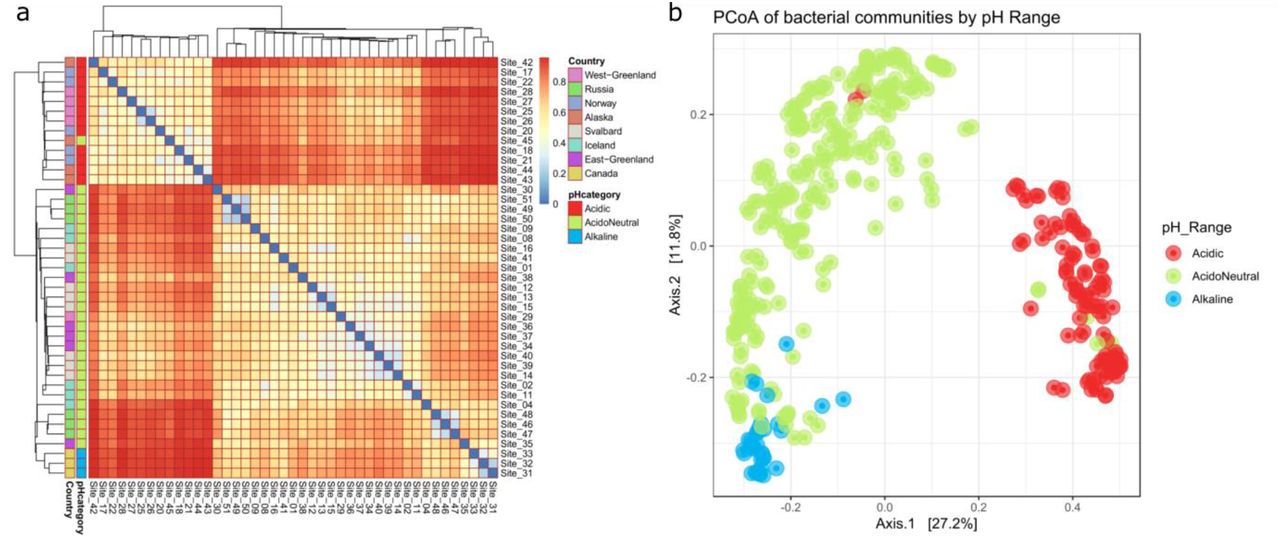
The Bray-Curtis dissimilarity heatmap and dendrogram [Fig. 2A] identified three main clusters illustrating community differences. The first cluster was composed of acidic samples in a gradient from Norwegian soils at pH = 4.07 (± 0.35) to samples from Alaska (pH = 4.64 ± 0.41) and West Greenland (pH = 4.90 ± 0.85). The second cluster included the lower acidoneutral range of samples from East Greenland (pH = 5.96 ± 0.69), Svalbard (pH = 5.65 ± 0.53) and Iceland (pH = 5.84 ± 0.46). Finally, the last cluster included the higher range of acidoneutral and alkaline soils from Russia (pH = 6.19 ± 0.22) and Canada (pH = 7.94 ± 0.67). The full spectrum of soil pH was covered from pH = 3.5 to pH = 9.0 and geographical locations often had samples from more than one pH category [Fig. S1].

Bray-Curtis dissimilarity matrix by sampling site. Each site was composed of 3 to 5 soil samples for which DNA extracted and sequenced in duplicates. In this analysis, all sequenced samples within a site were combined for ease of visualisation, and only the dominant pH category was displayed. Figure 2B: PCoA of microbial communities. Samples were individually considered to conserve accurate clustering.
The clustering of samples by pH range was clearly observed on the principal coordinate analysis (PCoA) of bacterial communities [Fig. 2B]. The overlapping acidoneutral and alkaline samples in the PCoA illustrate the third cluster of samples, which is composed of the higher range of acidoneutral and alkaline samples. The multivariate analysis [table 1] further indicated that pH explained the largest variability (R2=0.789, p<0.001) while site location accounted for only 5% (R2=0.063, p=0.001) of the variance. Pearson’s correlation coefficients identified positive and negative correlations between environmental variables [table S1]. The redundancy analysis [Fig. S2] also suggested that pH, amongst all explanatory variables, can most significantly explain the variation of bacterial communities’ ordination and composition. This trend was consistent in pH binned samples (ANOSIM: |R| = 0.748, P< 0.001; table S2); further confirming pH accounted for the observed variations in community composition.
MetaMDS results of the influence of environmental variables on bacterial communities. NMDS1 and NMDS2 illustrate the nature of the correlation between environmental variables and bacterial communities. R2 indicates the percentage of variance explained by each variable and Pr indicates the significance of the results.
In the literature, alkaline soils are consistently identified as pH>7 (Clark and Baligar, 2000; Rousk et al., 2010) while the differentiation between acidoneutral and acidic soils is less distinct, and as such, we based the cut-off on the study by Gubry-Rangin et al. (2011), which showed ecological clustering of archaeal ammonia oxidizers within these pH categories.
Abundant taxa diversity by pH range
The characterisation of bacterial communities across pH ranges identified clear differences. The observed richness and Shannon diversity index were significantly lower in acidic samples than in acidoneutral and alkaline soils [Fig. S3], which were not significantly different from each other [table S3]. At the phylum level [Fig. 3A], the main differences observed were the increase in Acidobacteria from 22% in acidic to 33% in acidoneutral and 42% in alkaline soils, along with a decrease in Actinobacteria, from 17% in acidic soils, to 8% in acidoneutral and less than 5% in alkaline samples. We also observed a drop in Verrucomicrobia from 25% in acidic and acidoneutral soils, to 16% in alkaline samples, along with changes in Chloroflexi, Bacteroidetes, Gemmatimonadetes and Planctomycetes. The abundance of Proteobacteria remained relatively stable across the gradient, with a slight drop from 21% to 18% in acidoneutral soils. At the class level [Fig. 3B], the differences in bacterial communities appeared more clearly, with large differences in presence, absence and relative abundance of certain classes [Fig. 3B]. For instance, acidoneutral and alkaline soils harboured high proportions of Blastocatellaceae (17% and 27% respectively) while they represented 1.39% in acidic soils. Acidic soils had a higher diversity of Acidobacteria, which were dominated by Acidobacteria group 1 (8%) and Acidobacteria group 2 (5.7%). We could also observe the decrease in Acidobacteria group 6 from alkaline (10%) to acidic (2.3%) soils. A decrease in Actinobacteria (12% to 3%) and Thermoleophilia (4% to 1%) could be observed from acidic to alkaline soils. Similarly, Alphaproteobacteria and Gammaproteobacteria were identified in higher abundance in acidic soils, while Betaproteobacteria were more abundant in alkaline soils. Acidic soils also harboured some classes that could not be identified or only in very low abundances in other pH ranges. Notably, the candidate Methylacidiphilum which composed over 7% of the acidic bacterial communities but was present in <0.5% in acidoneutral and <0.005% in alkaline soils. Overall, acidoneutral and alkaline soil bacterial communities showed similarities when considering OTUs with over 0.1% abundance.

Relative abundance of bacterial phyla with abundances over 0.1%, by pH range. Figure 3B: Relative abundance of bacterial classes with abundances over 0.1%, by pH range.
Generalist vs specialist taxa
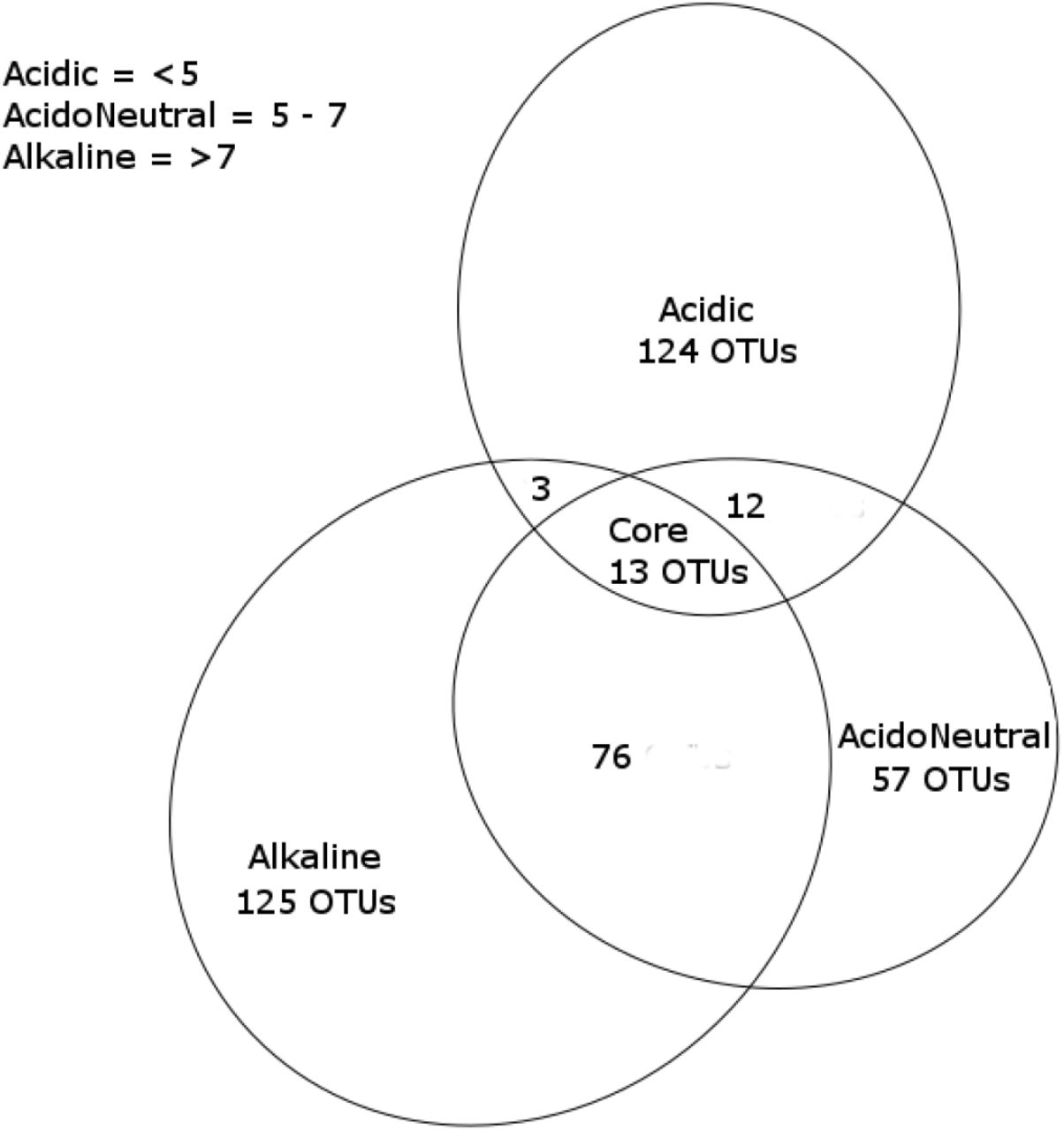
The differentiation of specialist from generalist taxa associated with pH range was conducted by considering all phylotypes identified in this study and present in a minimum 95% of all samples from each pH category [Fig. 4]. 125 acidic specialist OTUs were identified and unique to acidic soil samples. Of these 125 unique OTUs, most belonged to the Acidobacteria (27%), Verrucomicrobia (20%), Actinobacteria (14%), Planctomycetes (14%) and Proteobacteria (14%). At the class level, Acidobacteria group 1 dominated at 14%, while the rest of the identified classes had balanced relative abundances oscillating between 5 and 8%. Shared OTUs, or generalists were present in low numbers in acidic soils. Only 12 OTUs were shared with acidoneutral soils only, which were dominated by Alphaproteobacteria (65%), mainly Rhizobiales, while 3 OTUs were shared with alkaline only, belonging to the Spartobacteria (Verrucomicrobia), Phycisphaerae (Planctomycetes) and Chitinophagia (Bacteroidetes). In comparison, acidoneutral soils had 76 shared taxa with alkaline soils, against 12 shared OTUs with acidic. Taxa shared with alkaline soils mainly identified as Blastocatellia (Acidobacteria), Spartobacteria and Acidobacteria subgroup 6. Taxa exclusively found in acidoneutral soils belonged mainly to Actinobacteria (20%), Verrucomicrobia (20%) and Acidobacteria (19%). At the class level, unique taxa living in acidoneutral soils were dominated by Spartobacteria, Alphaproteobacteria and Holophagae. Alkaline soils presented a combination of both, a high number of shared (79 OTUs in total) and 125 exclusive taxa. Alkaline unique taxa were mostly composed of Acidobacteria (36%) and Proteobacteria (22%). From these unique taxa, the Acidobacteria subgroup 6 (20%), Blastocatellia (12%) and Alphaproteobacteria (12%) dominated the community.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Venn diagram of unique (specialists) and shared (generalists) OTUs by pH range. Only OTUs present in at least 95% of samples for each category are taken into account.
Indicator species
The indicator species analysis of dominant taxa (abundance > 0.1%) identified 17 taxon-habitat patterns of associations [table S4]. In acidic soils, 10 indicator species were identified, mainly Acidobacteria group 1 and 2 and the candidate Methylacidiphilum. The 6 OTUs associated with Acidoneutral soils were mainly Acidobacteria group 4 (Blastocatellia) and Spartobacteria (Verrucomicrobia). Finally, only 1 OTU was identified as indicator species for alkaline soils and belonged to the Holophagae. We also conducted the indicator species analysis of abundant taxa to combine pH ranges and identified 84 OTUs. 21 were identified as indicator species of acidic and acidoneutral soils combined, and mainly belonged to the Verrucomicrobia. Only 2 OTUs were associated with acidic and alkaline soils, Acidothermus (Actinobacteria) and Candidatus Xiphinematobacter (Verrucomicrobia), further illustrating the low overlap of taxa between these ecosystems. Finally, 61 OTUs were associated with acidoneutral and alkaline soils, mainly belonging to the Acidobacteria and Proteobacteria phyla.
The Arctic soil core microbiome
While some taxa displayed unique patterns of distribution, others were cosmopolitan and identified in over 95% of all sequenced samples. The core Arctic microbiome represented 0.026 % of bacterial communities and accounted for 2.77 % of all reads. It was composed of 13 OTUs, mainly Alphaproteobacteria and Acidobacteria, notably belonging to the Rhizobiales and Acidobacteria SD6 orders (Fig. S4).
Discussion
The importance of pH
The characterisation of soil bacterial communities on the Pan-Arctic scale and the investigation of the impact of environmental factors identified pH as explaining the most variation across bacterial communities. While TOC and moisture also explained some variation [table 1], Pearson’s correlations of the physicochemical properties [table S2] indicated that both, moisture and TOC, were negatively correlated with pH and positively correlated with each other and thus, could not be used as sole predictors of microbial communities. Furthermore, soil moisture is largely dependent on seasonal variations as soil moisture increases during snow melt, active layer thaw and precipitation events (Godin et al., 2016).
The identification of pH as the main factor influencing Arctic soil bacterial community composition is in line with previous Arctic studies, over both, small and large scales (Männistö et al., 2006; Ganzert et al., 2014; Siciliano et al., 2014; Schostag et al., 2015), including the study by Chu et al. (2010) which also investigated Pan-Arctic diversity by analysing 47 samples. While pH has been identified globally as a major factor influencing microbial diversity and community structure (Fierer and Jackson, 2006; Lauber et al., 2009; Tedersoo et al., 2014; Delgado-Baquerizo et al., 2018), the underlying processes and mechanisms by which it does remain unclear.
Studies have demonstrated that the soil pH is correlated with other elements of the geochemistry and has a strong impact on nutrient and water availability as well as solubility and adsorption. (Gray et al., 2014) For instance, acidic pH increases aluminium, hydrogen and manganese solubility, retarding plant root growth due to high toxicity (Clark and Baligar, 2000; Singh et al., 2017). Acidic soils also have nutrient deficiencies such as calcium, magnesium and potassium but also decreased phosphorus and molybdenum solubilities (Clark and Baligar, 2000; Gray et al., 2014). Alkaline soils are generally the result of low precipitation and high evapotranspiration, leading to low water availability and in common with acidic soils, nutrient deficiencies are found with, for instance, decreased phosphorus, iron, copper or zinc (Clark and Baligar, 2000). In similar ways, acidic and alkaline soils are generally considered harsh environments requiring a wide range of adaptations from microorganisms while acidoneutral soils are considered the optimum environment for microbial life (Fierer and Jackson, 2006; Rousk et al., 2010); such differences in soil composition are likely responsible for the observed differences in microbial community composition by pH range.
We investigated soil bacterial communities (abundant and rare taxa) but focused descriptions and bar charts on the abundant bacterial taxa, which includes taxa with abundance > 0.1 % for ease of visualisation. Rare taxa (< 0.1%) are characterized by Lynch and Neufeld (2015) as the rare biosphere and represent a large and diverse pool of taxa across the Arctic region. Rare microbes are a vast functional gene pool, which may be used by other microbes as a resource to respond to disturbance events or harsh environmental conditions and may play essential roles in ecosystem functioning, disproportionate to their abundance (Lynch and Neufeld, 2015; Jousset et al., 2017).
Dominant Arctic soil bacterial taxa appeared generally different from global diversity, as seen in Delgado-Baquerizo et al. (2018). Specifically, Verrucomicrobia were not clearly identified in the low pH cluster, and present in low abundance in the high pH cluster of global soils. In Arctic soils, they appeared dominant [Fig. 3A], with up to 25% in acidic soils. Proteobacteria consistently represented approximately 20% of communities, against almost 40% in global soils. Similarly, Acidobacteria comprised between 22% to 42% of Arctic soil communities, while only up to 15% in global soils. While there is likely an overlap of taxa between global and polar soils, Arctic bacterial communities seem to be dominated by different taxa than other biomes, likely reflecting the impact of polar environmental conditions on microbial communities. However, it should be noted that the differences of methods likely accounts for some of these differences, highlighting the need to include more Arctic samples in global studies to capture the full extent of worldwide soil microbial diversity.
Distribution of generalist and specialist taxa
Microbial communities are assembled by deterministic (selection) and stochastic (dispersal) processes. It has been hypothesized that communities primarily structured by deterministic processes will host more specialist taxa, highly adapted to the ecosystem, while communities influenced by dispersal will harbour primarily generalist taxa, more resilient to change (Pandit et al., 2009; Graham and Stegen, 2017; Sriswasdi et al., 2017). While specialist taxa are restricted to certain habitats, they can be locally abundant; shared taxa, or generalists, are distributed across many habitats (Barberán et al., 2012). In most cases specialist are more abundant because generalists rapidly become specialists to adapt to their ecosystems, despite generalists having evolutionary advantages (Sriswasdi et al., 2017). By identifying specialist and generalist taxa, we can speculate about the dominant processes structuring microbial communities, providing hypotheses for future studies.
In this study, bacterial communities changed from specialist-dominated in acidic soils, to generalist-dominated in acidoneutral, to a mixed community in alkaline samples. The higher abundance of specialists in acidic soils (considered the harshest systems) illustrates the need for environmental adaptations to survive in these ecosystems and suggests that deterministic processes likely structure microbial communities. Geographically, the first cluster identified by the Bray-Curtis dissimilarity matrix [Fig. 2] grouped all acidic samples from northern Norway, western Greenland and Alaska, illustrating the similarities of their bacterial communities despite the large distances separating these locations, further suggesting the strong influence of selection over dispersal. The dominance of generalists in acidoneutral soils illustrates the lower environmental pressure to have specific survival adaptations and infers the dominance of stochastic processes in community structuring. Eastern Greenland, Svalbard and Iceland were grouped together by the Bray-Curtis dissimilarity matrix. Geographically, these locations are influenced by the East Greenland current, East Icelandic current, Jan Mayen current, and West Spitzbergen currents, all connected through the Greenland sea and possible routes of dispersal, in addition to possible Aeolian dispersal. Finally, alkaline soils hosted a mixed-community of specialists and generalists, suggesting the combination of both, selection and dispersal, in community structuring. The abundance of generalist taxa in alkaline soils, generally considered a harsh system in many ways similar to acidic soils, highlights the adaptability of generalists to a wide range of environmental conditions. Interestingly, the Bray-Curtis dissimilarity matrix clustered the Canadian (alkaline) and Russian (acidoneutral) samples together. The Russian samples were on the higher end of the acidoneutral pH scale and the grouping of these samples illustrates the blurred boundary between acidoneutral and alkaline pH categories. While environmental selection may be driven by many variables, dispersal could occur via the Yukon current, the Beaufort gyre, aeolian dispersal and possibly winter sea ice.
Identification of indicator species
The indicator species analysis determined OTU-pH range associations identifying different classes of Acidobacteria and Verrucomicrobia, primarily, characteristic of each pH range. The identification of abundant indicator species with strong habitat associations opens the possibility of predicting the presence and relative abundance of these taxa across the Arctic region. This is especially important for taxa such as Ca. Methylacidiphilum, a known methanotroph unlike others as it belongs to the Verrucomicrobia phylum instead of the Proteobacteria (Dunfield et al., 2007; Khadem et al., 2010). The high abundance of these phylotypes in acidic soils suggest that they may might play major roles in CH4 uptake, thus limiting methane release to the atmosphere. While natural CH4 emissions come primarily from wetlands, the identification of these phylotypes in acidic soils suggests wetland may not be the only major source of northern methane emissions. Northern latitudes are estimated to support over 53% of all wetlands (Aselmann and Crutzen, 1989) but estimations of CH4 emissions from northern wetlands can vary drastically, from 18 to 120 Tg CH4 yr−1 (Petrescu et al., 2010) and the rates and impact of methanotrophy on CH4 emissions are difficult to assess (Jørgensen et al., 2015). Understanding the distribution and abundance of such taxa, combined with field gas measurements may allow large-scale estimates of methanotrophic rates in the region, from soils and wetlands (Wartiainen et al., 2006; Jørgensen et al., 2015). In addition to the uptake of CH4, Ca. Methylacidiphilum is also able to fix N2, illustrating the essential role of this class not only in the carbon cycle but also in the nitrogen cycle (Khadem et al., 2010).
Arctic soil core microbiome
The scale of this study led to the identification of the Arctic core microbiome, composed of 13 OTUs only. The most abundant of these taxa belonged to the Bradyrhizobiaceae family. This is one of the most common families worldwide, as identified by Delgado-Baquerizo et al. (2018), for which most species are plant-associated bacteria. The identification of a core Arctic soil microbiome is novel and this low number of cosmopolitan OTUs illustrates the low microbial ubiquity in the region.
Concluding remarks
To our knowledge, this is the first Pan-Arctic soil study with this scale and diversity of samples. Here, we focused on bacterial communities but there is a lack of understanding for archaeal, fungal, viral and eukaryotic communities in the region as well. We also focused our investigations on variables known to influence global bacterial communities, however, it is likely that nutrient concentration, metal content and other factors participate in the structuring of microbial assemblages in the region, on different scales and magnitudes. By conducting a large Pan-Arctic survey of bacterial communities of Arctic soils and associated environmental variables, we identified pH as the main factor structuring these communities, emphasising the need to investigate the underlying mechanisms and processes by which pH directly or indirectly influences microbial communities. We also highlighted differences between Arctic soil communities and global communities, further suggesting the impact of Polar environmental conditions on prokaryotes. The investigation of specialist and generalist taxa highlighted the possible role of geographical dispersal across the region, which may be more important in acidoneutral soils dominated by generalist taxa. Finally, we identified the Arctic core microbiome, composed of only 13 OTUs across the entire region. While this study brings a deeper understanding of Arctic bacterial community assemblages, this is also a baseline for future functional studies in the region, which will be critical to forecast the ecological consequences of environmental change.
Conflict of interest
The authors report no conflict of interests.
Acknowledgements
This work was supported by a grant from the European Commission’s Marie Sklowdowska Curie Actions program under project number 675546. The authors also thank Dr Paul Mann, Dr Charles Greer, Dr Svetlana Evgrafova, Dr Bill Holben, Sam Pannoni, Edwin Sia and UNIS for their participation and support in fieldwork. MiSeq sequencing of the 16S rRNA gene was performed by the NU-OMICS sequencing service (Northumbria University).
Footnotes
LAM and DAP conceived and designed the study and sampling strategy. LAM carried the fieldwork and laboratory work. MZA conducted bioinformatics processing and statistical analysis. LAM drafted the manuscript and MZA, DAP and CSJ revised and approved the final version.
References




