

ABSTRACT
Variation in gene expression underlies inter-individual variability in immune response. However, the mutations responsible for gene expression changes remain largely unknown. In this work, we searched for transposable element insertions present at high population frequencies and located nearby immune-related genes in Drosophila melanogaster. We identified 12 insertions associated with allele-specific expression changes in immune-related genes. We showed that transgenically induced expression changes in most of these genes are associated with differences in survival to infection with the gram-negative bacteria Pseudomonas entomophila. We provide experimental evidence suggesting a causal role for five insertions in the allele-specific expression changes observed. Furthermore, for two insertions we found a significant association with increased tolerance to bacterial infection. Our results showed for the first time that polymorphic transposable element insertions from different families drive expression changes in genes that are relevant for inter-individual differences in immune response.
BACKGROUND
Innate immunity is the first barrier against infections, and many species rely solely in this response to cope with pathogens (1, 2). Mechanisms of pathogen recognition and activation of the innate immune response are conserved across animals (3, 4). In Drosophila melanogaster, several signaling pathways participate in the innate immune response (5–8). The Toll and the Imd are the main signaling pathways involved in recognizing and fighting pathogens (9, 10), while the JAK/STAT pathway is involved in cell proliferation (11, 12), and the JNK pathway is required for proper wound healing (13, 14). Cellular processes such as phagocytosis or melanotic encapsulation also play a critical role in the innate immune response, and studies in D. melanogaster are also highly relevant to understand them (5, 6).
One of the most likely infection routes happening in nature is oral infection, and the gut epithelium is the first barrier that bacteria encounter in the organism (15, 16). However, the gut immune response is still not completely understood, and it is likely more complex than the systemic immune response. First, both in insects and in vertebrates, the intestinal tract is a single tubule anatomically and functionally compartmentalized (17, 18). Second, the gut is constantly in contact with bacteria composing the microbiota. As such, the host has to differentiate between pathogenic bacteria and gut microbiota (15, 19–21). Thus, there must be a complex transcriptional regulatory toolkit in order to control the expression of immune responsive genes in the gut (22). Indeed, the analysis of gut immunocompetence variation in 140 D. melanogaster strains found that small but systematic differences in gene expression exists between resistant and susceptible strains to Pseudomonas entomophila, a natural pathogen of this species (23, 24). Variation in gene expression has been shown to underlay inter-individual variability in immune responses also in humans (25–27). However, the causal mutations responsible for these expression changes remain largely unknown (8, 28). Identifying the causal mutations is necessary to establish functional links between the expression phenotypes and the susceptibility/tolerance to infection.
Among the types of mutations that could be responsible for gene expression changes, transposable elements (TEs) are particularly likely to be important contributors. TEs can be a source of cis-regulatory elements that can influence gene regulation (29–32). For example, TEs have been shown to add transcription factor binding sites, and transcription start sites, leading to changes in expression of nearby genes (33–36). TEs can also influence gene expression by inducing changes in the chromatin structure (37–39). These changes in expression induced by TE insertions have been associated with several organismal phenotypes such as stress resistance and fertility (40–42). So far, only a few studies have linked individual TE insertions with pathogen-induced expression changes (40, 43). A recent study conducted in human lymphoblastoid cell lines established from European and African individuals showed that the regulatory effects of polymorphic TEs are associated with immune-related functions (44). These results suggest that TEs may contribute to regulatory variation between individuals, although no functional evidence was provided for the causal role of the insertions in the expression changes identified (44).
In this work, we aimed at assessing the role of polymorphic TE insertions in D. melanogaster gut immune response. We first identified polymorphic TE insertions present at high population frequencies and located nearby immune-related genes. We found that 12 of the 14 insertions analyzed were associated with allele-specific expression changes of their nearby immune-related genes. Transgenically induced expression changes in most of these genes were associated with differences in survival after infection, suggesting that expression changes in these genes are phenotypically relevant. Through a combination of experimental approaches including 3’RACE, qRT-PCR, ChIP-qPCR, and in vivo enhancer assays, we provided further evidence for the role of five of these insertions in the expression differences observed. Finally, we showed that two of these insertions are associated with increased survival to bacterial infection.
RESULTS
Nineteen TE natural insertions present at high population frequencies are located nearby genes with immune-related functions
To identify polymorphic TEs likely to affect gut immune response, we first looked for insertions present at high population frequencies, and located in genomic regions with a high recombination rate (see Methods). We analyzed 808 TEs annotated in the D. melanogaster reference genome and 23 non-reference TEs in four natural populations (Supplementary File 1A and 1B, see Methods) (45–47). We identified 128 insertions present at ≥ 10% frequency in at least one of the four populations analyzed: 109 reference TEs and 19 non-reference TEs (Supplementary File 1C, see Methods). We then surveyed the literature for the functional information available for the genes located nearby each one of these 128 TEs (Supplementary File 1D). We found that 19 of these 128 TEs were associated with 21 immune-related genes (Table 1). Note that for seven of these 19 TEs there is previous evidence suggesting that they have increased in frequency due to positive selection (Table 1) (47).
The 16 reference TE insertions are listed first, followed by the three non-reference insertions.
The functional evidence for the majority of the 21 genes nearby the 19 candidate immune-related TEs comes from transcriptional response to infection experiments (11 genes), infection survival experiments (five genes), or both (three genes) (Table 1). The other two genes, TM4SF and ken, are members of the JAK-STAT signaling pathway involved in immune response (11). Before investigating whether the identified TEs could be affecting the expression of nearby immune-related genes, we first tested whether transgenically induced changes in the expression of these genes affect survival to bacterial infection. We focused on nine genes: six genes for which survival experiments were not previously available, and three genes for which survival experiments were performed using a different pathogen (Table 2). When available, two different backgrounds were tested (Supplementary File 2A). For three of the 14 strains analyzed, we did not detect differences in expression of the target gene (Supplementary File 2A and 2B). Thus, we did not perform survival experiments for these three strains. For the other 11 strains, survival experiments were performed with the gram-negative bacteria P. entomophila (24). As a natural D. melanogaster pathogen, experiments with P. entomophila have the potential to identify specialized immune responses derived from antagonistic co-evolution (48). We found that mutant, RNAi knockdowns, and overexpression strains of seven of these genes showed differences in survival after oral infection with P. entomophila: NUCB1, CG2233, Bin1, and cbx showed higher survival, ken, CG8008, and TM4SF mutants showed lower survival (Table 2 and Supplementary File 2C). For CG10943 results were significant, but the mutation effect size was not significant. Finally, CG15829 RNAi flies did not show differences in survival (Table 2 and Supplementary File 2C).
Survival of mutant strains orally infected with P. entomophila compared with flies with a similar background (Supplementary File 2A). P-values obtained using log-rank test. OR (CI): odds ratio and confidence interval (95%)
Overall, we provide additional evidence linking changes in expression with survival differences for four of the six genes for which no phenotypic evidence was previously available, and for the three genes that were previously tested with a different pathogen (Table 2, Supplementary File 2B and 2C). Thus changes in the expression of the genes located nearby TEs present at high population frequencies affect survival to infection. These results suggest that if TEs also affect the change in expression of these genes, TEs could be associated with differences in survival to bacterial infections.
Immune-related candidate TEs are associated with gene expression changes
In order to explore whether the 19 candidate adaptive TEs were associated with expression changes of their nearby immune-related genes, we measured allele-specific expression (ASE) in flies heterozygous for the presence of each candidate adaptive TE. Because both alleles in the heterozygous shared the same cellular environment, differential expression of the two alleles indicates functional cis-regulatory differences (76). We performed the analysis in flies with two different genetic backgrounds in order to detect possible background-dependent effects in allele-specific expression changes. We were able to analyze with this technique a total of 16 genes located nearby 14 TEs. In non-infected conditions, 10 out of the 16 genes showed statistically significant allele-specific expression differences in at least one of the two genetic backgrounds analyzed (Figure 1, Supplementary File 3). For five of these 10 genes, we found that the allele with the TE was more highly expressed compared to the allele without the TE, and for the other five genes, the allele with the TE was less expressed. In infected conditions, eight out of the 16 genes showed statistically significant allele-specific expression differences in at least one of the two genetic backgrounds analyzed (Figure 1, Supplementary File 3). For three genes, we found that the allele with the TE was more highly expressed, and in the other five genes the allele with the TE was less expressed. Considering both non-infected and infected conditions, five genes showed allele-specific expression differences in the two conditions: for CG10943 the allele with the TE was more highly expressed, and for CG8628, CG8008, CG15096 and cbx the allele with the TE was less expressed (Figure 1, Supplementary File 3).

Results from female guts in non-infected conditions (in green) and in infected conditions (in purple). Each dot represents the average ratio of gene expression levels between the allele with the TE and the allele without the TE for the three replicas analyzed. Each gene has two dots representing each one of the two genetic backgrounds analyzed. Statistically significant differences are depicted with dark color (t-test p-values < 0.05, corrected for FDR 5%). Error bars represent SEM.
We also checked whether the genetic background affected the allele specific expression differences. In 10 analyses, both backgrounds showed changes in expression in the same direction, more highly expressed or less expressed, and in two of them the differences were statistically significant in the two backgrounds analyzed (Figure 1, Supplementary File 3). On the other hand, seven analyses differed in the direction of the change of expression in the two backgrounds. However, results were always statistically significant in only one of the two backgrounds analyzed (Figure 1, Supplementary File 3).
Overall, we found that most of the candidate immune-related TEs, 12 out of 14, are associated with changes in expression of their nearby gene, in at least one of the two conditions analyzed (Figure 1). While some expression changes are significant only in infected or only in non-infected conditions, a significant proportion of genes (38%) showed consistent changes in expression in both conditions (Figure 1). Finally, we detected an effect of the genetic background on the allele specific expression differences as has been previously reported (77–79). However, statistically significant results were always consistent between genetic backgrounds (Figure 1).
Candidate TEs affected expression of nearby genes through different molecular mechanisms
We performed several experiments to identify the molecular mechanisms behind the expression changes observed and to further test whether the TEs are the most likely causal mutation behind these changes (Figure 1). We focused on studying the four TEs located in promoter regions and associated with ≥ 1.5-fold higher expression: FBti0019386, FBti0018868, tdn8, and FBti0061506. We also studied in detail two other insertions: FBti0019985 that showed genetic background dependent effects, and FBti0020057 associated with lower allele-specific expression. In addition, for FBti0019386 and FBti0018868, we also performed survival experiments to bacterial infection.
FBti0019386 provides a transcription start site to Bin1 that is only used in infected conditions in the female gut
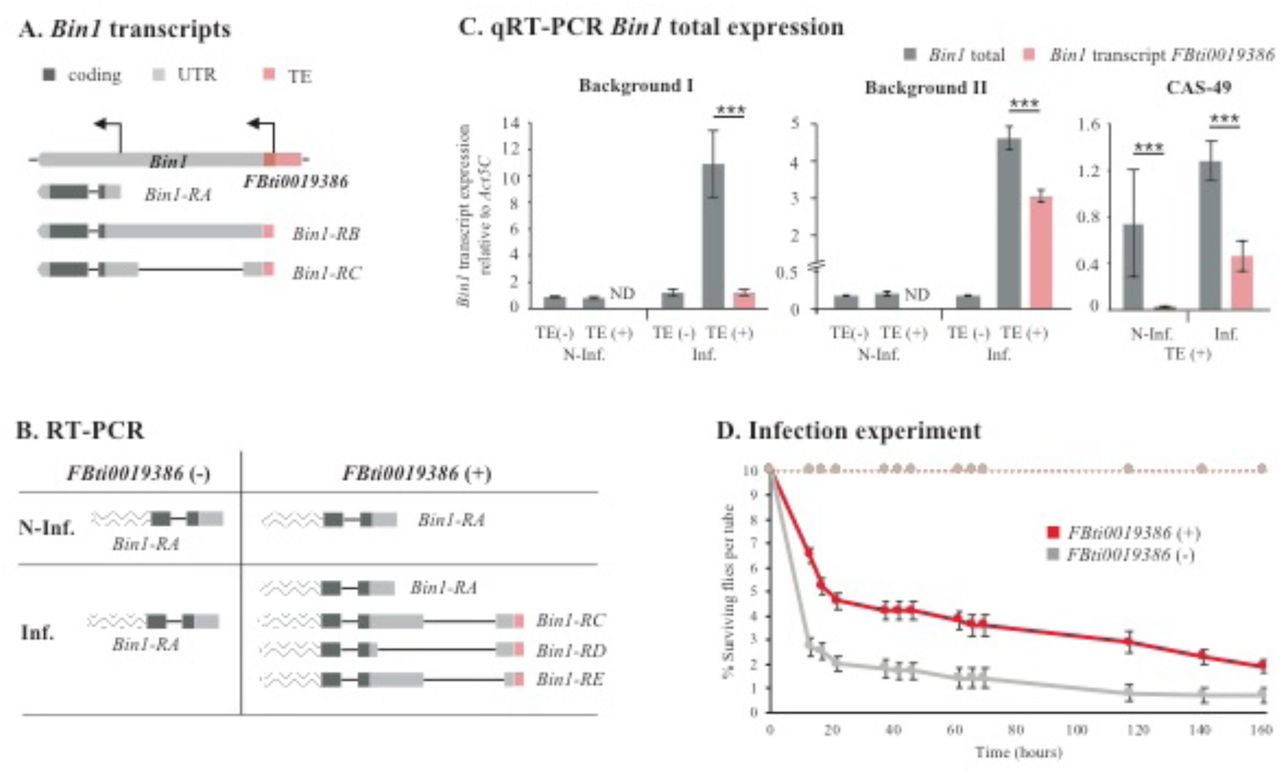
FBti0019386 is an invader4 element inserted in the 5’UTR region of Bin1, a gene required for the expression of immune- and stress-response genes (49), and associated with shorter developmental time (Table 2, Figure 2A) (80). There are two annotated Bin1 transcripts with the transcription start site (TSS) located in the FBti0019386 insertion (Figure 2A) (81). We found that homozygous flies with and without FBti0019386 expressed only the short Bin1-RA transcript in non-infected conditions (Figure 2B). However, in infected conditions, flies without FBti0019386 insertion only expressed Bin1-RA, while flies with FBti0019386 expressed Bin1-RA, and three transcripts starting in the TE: Bin1-RC, Bin1-RD and Bin1-RE. We confirmed these results by performing the experiments in a second genetic background (see Methods). Note that Bin1-RD and Bin1-RE transcripts were not described previously and differ in the 5’UTR length (Figure 2B).

(A) Transcripts annotated for Bin1. FBti0019386 overlaps with two of the annotated transcripts. (B) Transcripts detected by RT-PCR in flies with and without FBti0019386, both in non-infected (N-Inf) and infected (Inf) conditions. Transcript regions wave-patterned are inferred from FlyBase transcript annotation and were not sequenced in this work. Bin1-RD and Bin1-RE transcripts are, respectively, 318 bp and 172 bp shorter compared to Bin1-RC transcript. (C) Bin1 expression levels in the gut in the two backgrounds analyzed in the ASE, and a third homozygous background with FBti0019386 insertion (CAS-49). Error bars represent SEM. ND: not detected. (D) Survival curves in non-infected (discontinuous lines) and infected conditions (continuous lines) for flies with FBti0019386 insertion (red) and without this insertion (grey). Error bars represent SEM.
To test whether the transcripts that start in FBti0019386 insertion are associated with increased expression of Bin1, we quantified the expression of the transcripts starting in the TE and the total Bin1 transcript levels (Figure 2C). In non-infected conditions, flies with and without FBti0019386 did not differ in Bin1 expression levels (t-test, p-value > 0.05). In infected conditions, flies with FBti0019386 overexpressed Bin1 compared to flies without FBti0019386 in the two backgrounds (t-test, p-value < 0.001). The contribution of the transcripts starting in FBti0019386 to the total Bin1 expression is background dependent: 11.2% in background I, and 66.3% in background II (Figure 2C). To confirm this result, we analyzed a third genetic background homozygous for the presence of FBti0019386, and we found that the TE-transcripts contributed 36.2% to Bin1 total expression (Figure 2C).
Overall, we found that FBti0019386 adds a TSS for Bin1 that is only used in infected conditions in the gut (Figure 2B and 2C). We also found that increased expression of Bin1 in response to infection is only observed in flies with FBti0019386 insertion, and that the contribution of the transcripts starting in the insertion to the overall level of Bin1 expression is background dependent (Figure 2C). These results suggest that, besides adding a new TSS for Bin1, FBti0019386, which is a 347 bp solo-LTR insertion, could also be acting as an enhancer in infected conditions. Moreover, these results are in agreement with the ASE analysis that showed that FBti0019386 is associated with increased Bin1 expression only in infected conditions, further suggesting that the TE is the causal mutation (Figure 1). Finally, we found that flies with FBti0019386 had higher survival to bacterial infection compared with flies without this insertion (Figure 2D).
FBti0018868 adds a TSS both in infected and non-infected conditions
FBti0018868 is a 297 element annotated 1 bp upstream of one of the TM4SF transcripts, and 310 bp upstream of the other two transcripts (Figure 3A). TM4SF encodes a tetraspanin protein, which plays a role during immune response in Drosophila and humans (71). A previous work identified a new TSS for TM4SF inside FBti0018868 (81). We performed RT-PCR to check whether homozygous flies with FBti0018868 insertion expressed the transcript starting in the TE in non-infected and/or in infected conditions. We detected the presence of the transcript starting in the TE in both conditions (Figure 3A).

(A) FBti0018868 is located upstream of TM4SF gene and it has three annotated transcripts described. We detected a new TM4SF transcript overlapping with FBti0018868 both in flies in non-infected and infected conditions. Transcript regions wave-patterned are inferred from FlyBase transcript annotation and were not sequenced in this work. (B) TM4SF total expression levels in the gut in the two different backgrounds used in the ASE (I and II) with and without FBti0018868. Error bars represent SEM. ND: not detected. (C) Vector constructions with the empty vector as a negative control, and a vector carrying FBti0018868 in front of the reporter gene lacZ. Below, β-GAL immunostaining (in green), and DAPI staining (in grey) of guts from transgenic females with the empty vector and with the FBti0018868 construct. The scale bar represents 500 µm. (D) Survival curves in non-infected (discontinuous lines) and infected conditions (continuous lines) for flies with (red) and without FBti0018868 (grey). Error bars represent SEM.
To check whether flies with and without FBti0018868 differ in the expression level of the different TM4SF transcripts in infected and non-infected conditions, we performed qRT-PCR. We found that TM4SF expression can only be detected in the strains with FBti0018868 insertion, although at very low levels (Figure 3B). The primers designed to specifically detect the expression of the transcript starting in FBti0018868 insertion did not detect any expression (Figure 3B).
To test whether FBti0018868 could be acting as an enhancer, we generated transgenic flies in which FBti0018868 was cloned in front of the reporter gene lacZ (Figure 3C, see Methods). We did not detect lacZ expression by qRT-PCR in non-infected or in infected conditions. The β-GAL protein expression localization did not differ either from the negative control (Figure 3C).
Overall, we found that FBti0018868 that was associated with increased expression of TM4SF in infected conditions (Figure 1) adds a TSS for its nearby gene TM4SF, which is detected both in infected and non-infected conditions (Figure 3A). Only flies with FBti0018868 insertion showed TM4SF expression in both conditions, although the total level of expression was low, and we could not detect the transcripts starting in the insertion using qRT-PCR (Figure 3B). FBti0018868 is not driving the expression of a reporter gene (Figure 3C) suggesting that the insertion sequence by itself is not enough to increase the expression of a nearby gene, but probably needs additional regulatory sequences. A larger genomic region containing FBti0018868 insertion should be analyzed before discarding the effect of the insertion on TM4SF expression changes. Finally, we found that flies with FBti0018868 had higher survival to bacterial infection compared with flies without this insertion (Figure 3D).
tdn8 drives the expression of a reporter gene in non-infected and infected conditions
tdn8 is a Transpac element located 816 bp upstream of CG10943, a gene that is up-regulated in response to immune challenge with different pathogens including P. entomophila (Figure 4A and Table 2) (19, 23, 55). We tested whether tdn8 could be acting as an enhancer (Figure 4B). We found that transgenic strains in which the upstream region of CG10943 contained the tdn8 insertion showed higher expression than transgenic strains in which the same region without the insertion was cloned in front of the reporter gene (Figure 4C). Differences in expression were only statistically significant in infected conditions (p-value = 0.046 respectively) (Figure 4C). We found no differences between the two transgenic strains in the localization of the β-GAL protein expression in non-infected or infected conditions (Figure 4D).

(A) tdn8 is located upstream of CG10943 gene. (B) Vector construction without tdn8 and with tdn8 in the promoter region of the reporter gene lacZ. (C) Expression levels of the reporter gene lacZ in transgenic female guts without and with tdn8, both in non-infected and in infected conditions. (D) β-GAL immunostaining (in green), and DAPI staining (in grey) from female non-infected and infected guts. The scale bar represents 500 µm.
Overall, we found that tdn8 is acting as an enhancer. These results are in agreement with our ASE results that showed that tdn8 is associated with higher expression of CG10943 in the two genetic backgrounds analyzed (Figure 1).
FBti0061506 does not drive the expression of a reporter gene
FBti0061506 is a 1360 element located in the 5’UTR intron of Dif-RD transcript, and 3.8 kb upstream of the other three Dif transcripts (Figure 5A). Dif is a main transcription factor of the Toll-pathway, and it was found to be up-regulated in gut tissue after P. entomophila infection (Table 2) (23).

(A) FBti0061506 is located in the first intron of one of Dif transcripts, and upstream of the other transcripts. (B) Vectors construction for the enhancer assays with and without FBti0061506 in front of the reporter gene LacZ. (C) β-GAL immunostaining (in green), and DAPI staining (in grey) from transgenic female non-infected and infected guts. The scale bar represents 500 µm.
To test whether FBti0061506 could act as an enhancer sequence, we generated two reporter gene constructs containing part of the Dif intron with and without the insertion (Figure 5B, see Methods). None of the two constructs affected the expression of the reporter gene or the localization of the β-GAL protein (Figure 5C).
Overall, our results do not provide evidence for an enhancer role of FBti0061506. However, our ASE results showed that FBti0061506 was associated with Dif higher expression in non-infected conditions (Figure 1). Although it is also possible that Dif allele-specific expression is due to a cis-mutation different from the FBti0061506, it could be that the effect of FBti0061506 is context dependent. Therefore, a larger genomic region with and without the insertion should be analyzed to discard an effect of FBti0061506 on Dif allele-specific expression differences (Figure 1).
FBti0019985 drives the expression of a reporter gene both in non-infected and infected conditions
Besides the four TEs located in promoter regions and associated with ≥ 1.5-fold higher allele-specific expression, we also studied in detail FBti0019985 insertion that showed genetic background dependent effects (Figure 1). FBti0019985 is a roo element inserted in two nested genes: CG18446 and cbx. FBti0019985 provides a transcript start site for CG18446 and has been associated with increased cold tolerance (34). FBti0019985 is also located in the first 5’UTR intron of cbx-RA (CG46338-RA) transcript, and 700 bp and 5.5 kb upstream of the other two cbx transcripts (Figure 6A). cbx mutant flies are more tolerant to bacterial infection (Table 2). We first checked whether the TE affects the expression of the different cbx transcripts by performing RT-PCR from non-infected guts of homozygous flies with and without the TE. We detected two of the three annotated transcripts, cbx-RB and cbx-RC, in flies with and without FBti0019985 (Figure 6A). Thus, we did not find evidence of FBti0019985 affecting cbx transcript choice or transcript structure in non-infected conditions.

(A) Transcripts annotated for the gene cbx. (B) Vector constructs for the enhancer assays. (C) Expression levels of the lacZ reporter gene in transgenic female guts, both under non-infected and infected conditions. Empty vector showed no detectable expression levels in any of the two conditions. (D) β-GAL immunostaining (in green), and DAPI staining (in grey) from transgenic female guts with FBti0019985. Scale bar represents 500 µm.
The allele containing FBti0019985 could be acting as an upstream enhancer for cbx-RB and cbx-RC transcripts. Thus, we performed enhancer reporter assays, and we detected that FBti0019985 drives the expression of the reporter gene only in infected conditions (Figure 6B and 6C). β-GAL immunostaining showed that the expression was localized in the anterior part of the gut, both in non-infected and in infected conditions (Figure 6D). The localization of the expression only in the anterior part of the gut could explain why we could not detect expression with the qRT-PCR in whole guts in non-infected conditions (Figure 6C).
Overall, we showed that FBti0019985 does not modify cbx transcript structure under non-infected conditions but acts as an enhancer in the anterior part of the gut. These results suggest that the effect of FBti0019985 could be background dependent as the insertion was associated with lower expression of cbx in the second genetic background analyzed (Figure 1).
FBti0020057 down-regulates the expression of a reporter gene
We also studied in detail FBti0020057, one of the six insertions associated with lower allele-specific expression (Figure 1). FBti0020057 is a BS element annotated in the intergenic region between CG15829 and CG8628 (Figure 7A). Both genes are predicted to be involved in Acyl-CoA homeostasis, associated with lipid metabolism (82). Resources redistributions between metabolism and immune response are a key process during infection, and genes involved in lipid metabolism are repressed after infection (5, 83–85). We checked whether FBti0020057 could be affecting the transcript choice of its upstream gene CG15829 (Figure 7A). We performed 3’RACE using cDNA of non-infected guts from homozygous flies with and without FBti0020057. We only detected the expression of the shorter transcript, CG15829-RA, in both strains. Therefore, the TE is not affecting CG15829 gene transcript structure or transcript choice in the studied conditions.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) FBti0020057 is located in the intergenic region of CG15829 and CG8628. (B) Vectors construction for the enhancer assays with and without FBti0020057 in front of the reporter gene LacZ. (C) Expression levels of the lacZ reporter gene under non-infected and infected conditions. Error bars represent SEM. (D) β-GAL immunostaining (in green), and DAPI staining (in grey) from transgenic female non-infected and infected guts. The scale bar represents 500 µm. (E) ChIP qRT-PCR analysis for H3K9me3 in the genomic region were FBti0020057 is inserted. “Absent”: H3K9me3 enrichment in the reference strain that does not contain FBti0020057 (grey), “Left”: H3K9me3 enrichment in the left flanking region of FBti0020057 in RAL-903 that contains this TE (red), and “Right”: H3K9me3 enrichment in the right TE flanking region of FBti0020057 in in RAL-903 (red).
We then tested whether FBti0020057 could be down-regulating its downstream gene. To do this, we cloned the whole intergenic region with and without FBti0020057 in front of the reporter gene lacZ (Figure 7B). Consistent with the ASE results for CG8628, we found that the transgenic strains with the TE have less expression of the reporter gene both in non-infected and in infected conditions (Figure 7C). We also found that the expression of the reporter gene was localized mostly in the posterior midgut (Figure 7D), a region known to be dedicated to absorption that expresses genes encoding lipid transporters (18). Because TEs can recruit repressive histone marks, such as H3K9me3, that can lead to silencing of nearby genes (38, 86, 87), we then checked whether FBti0020057 was enriched for H3K9me3. We did not find enrichment for H3K9me3 when comparing a strain with and without this insertion (Figure 7E). Thus, the TE could be disrupting a regulatory sequence, or it could be adding a binding site for a repressor protein.
Taken together, our results showed that FBti0020057 is associated with the down-regulation of a reporter gene, consistent with the observed allele-specific expression differences of CG8628 (Figure 1). On the other hand, we did not find differences in CG15829 transcript choice associated to the TE. Further experiments are needed to determine whether FBti0020057 is also responsible for the increased expression of CG15829 observed in the allele-specific experiments (Figure 1).
DISCUSSION
In this work, we found 19 TE insertions present at high frequencies in D. melanogaster natural populations, and located nearby genes enriched for immune-related functions (Table 1). The majority of these insertions, 13 out of 19, have increased in frequency in out-of-Africa populations (Supplementary File 1C). D. melanogaster has recently colonized out-of-Africa environments (88, 89). Among the many stressors faced by D. melanogaster, our results suggest that response to pathogens has been an important biological process in the colonization of the new environments (Table 2). Immune response has previously been reported to be relevant for local adaptation not only in D. melanogaster but also in humans (67, 90–94). Our results are based on the analysis of four natural populations: one population from the ancestral range of the species, and three out of Africa populations: one North American and two European populations (95, 96). Although the three out-of-African populations analyzed come from locations with contrasting climates, analysis of natural populations from other geographical locations is needed to get a more general picture of the biological processes that are relevant for out-of-Africa adaptation. Indeed, a recent analysis of 91 samples from 60 worldwide natural populations suggested that response to stress, behavior, and development are shaped by polymorphic transposable element insertions (47).
We found that our candidate TEs were associated with allele-specific expression differences in 13 out of the 16 immune-related genes analyzed (Figure 1). Recent studies performed in several strains estimated that ∼8% to 28% of D. melanogaster genes showed allele-specific expression (97, 98). Thus, our results suggest that our candidate TEs are more often associated with genes that show allele specific expression than expected by chance (81%, p-value < 0.0001). We, and others, have shown that transgenically changes in expression of nine of these 13 genes led to changes in D. melanogaster survival rate after infection (Table 1, Table 2, and references therein). Changes in the expression of these genes are thus likely to be relevant for the fly ability to cope with infections. As described previously, we found both gene up-regulation and gene down-regulation in gut immune response (17, 24). Most of the genes showed allele-specific expression changes either in infected conditions, Bin1, TM4SF and NUCB1, or both in non-infected and infected conditions, CG10943, CG8628, CG8008, CG15906, and cbx (Figure 1). However, we also identified five genes that showed allele-specific expression changes only in non-infected conditions: CG2233, Dif, AGO2, CG15829, and Mef2 (Figure 1). Differences in the basal transcriptomic profile between tolerant and susceptible strains to P. entomophila infection have been described previously in D. melanogaster (23). Moreover, differences in gene expression pattern before parasitoid attack between control and selected lines for increased resistance to Asobara tabida, a D. melanogaster endoparasitoid, have also been reported (99). Taken together these results suggested that besides the genes that change their expression level in response to the immune challenge, gene expression variability in non-infected conditions also affects the susceptibility of the flies to immune-challenges.
We identified the molecular mechanism underpinning the changes in expression induced by four of the six insertions studied in more detail. We found TEs that add TSS (Figure 2), act as proximal enhancers (Figure 4 and Figure 6), and repressed the expression of nearby genes (Figure 7). These results add to an increasing body of literature showing the multiple ways in which TE insertions affect the expression of nearby genes (30, 40). On the other hand, the other two insertions analyzed did not affect the expression of a reporter gene (Figure 3 and Figure 5). However, it is known that enhancer reporter assays select for compact regulatory elements that can function in an autonomous manner (100). Thus, before discarding the causal role of these insertions in the observed allele-specific expression differences, a larger genomic region including these insertions should be tested for enhancer activity. Indeed, we further showed that FBti0018868 was associated with increased expression of TM4SF (Figure 3C) suggesting that this insertion is likely to be the causal mutation of the allele-specific expression differences previously observed (Figure 1). Moreover, for this insertion we found that it was associated with increased survival to infection (Figure 3E). Still, it could also be that differences in expression are due to polymorphism other than the TE insertions identified. Although we could not identify any other cis-variant in the proximity of the analyzed genes, except for AGO2, regulatory regions might not be conserved and there might be other variants also contributing to the differences in expression (Supplementary File 4, see Methods).
The expression changes associated with several of the TE insertions analyzed in this work were consistent with a role of the nearby genes in increased survival after infection (Figure 1 and Table 2). Indeed, for two of the insertions we showed that this is the case (Figure 2 and 3). We showed that FBti0019386 is associated with increased Bin1 expression in infected conditions, and increased survival after infection (Figure 1, 2C and 2D) (49). We also found that FBti0018868 is associated with TMS4F higher allele-specific expression and increased survival after infection (Table 3 and Figure 3E). Bou Sleiman et al (23) found that CG10943 and Dif were up-regulated and CG8628 was down-regulated in strains resistant to P. entomophila infection. We indeed found that the candidate adaptive insertion tdn8 and FBti0061506 were associated with increased expression of CG10943 and Dif respectively, and FBti0020057 insertion was associated with CG8628 lower expression. Finally, we also found that FBti0019985 located upstream of cbx could act as an enhancer (Figure 6). A cbx knockout was more sensitive to gram-positive bacterial infection (53), and we found that the same mutant stock was associated with increased survival to P. entomophila infection (Table 2). Thus, in this case the change of expression associated with the candidate adaptive TE is also likely to lead to increased survival after infection (Figure 1). Thus, based on results already available in the literature and in our own results, we found that several of the TE insertions analyzed induced changes in nearby genes that are likely to lead to increased P. entomophila infection survival.
Overall, we have shown that TEs contribute to immune-related gene expression variation, which could be crucial for a rapid process of adaptation to new environments. For two of the insertions analyzed, we further showed that they are associated with increased survival to bacterial infection. TEs are likely to be key players in immune response in other organisms as well, as has been shown for a particular fixed TE family and a fixed TE insertion in mammals (40, 43). Besides, several polymorphic insertions have been found to be associated with expression changes in immune-related genes in human populations, and our results suggest that polymorphic insertions could also be relevant for immune response variability (44).
METHODS
Fly strains
DGRP strains
141 DGRP strains (96) were used to estimate the frequencies of TEs annotated in the D. melanogaster reference genome using the data in Rech et al (47) (Supplementary File 5). Besides, we used 37 DGRP strains to analyze by PCR a subset of TEs not annotated in the reference genome (46). Finally, DGRP strains were also used to perform allele specific expression analyses (ASE), transcription start site identification (TSS), and enhancer assays (Supplementary File 5). Note that it has previously been shown that differences in the presence/absence of the endosymbiont Wolbachia, differences in commensal bacteria and/or feeding behavior has no major effect in the susceptibility of DGRP strains to P. entomophila infection (23).
African strains
Frequency estimates for reference TE insertions for a subset of 66 African strains collected in Siavonga (Zambia, (95)) with no evidence of cosmopolitan admixtures were obtained from Rech et al (47) (Supplementary File 5).
European strains
Frequency estimates for reference TE insertions for 73 European strains, 57 from Stockholm (Sweden) and 16 from Bari (Italy), were obtained from Rech et al (47) (Supplementary File 5). Additionally, one strain from Bari (CAS-49) was used for ASE and TSS experiments and one strain from Munich (MUN-8) was used for ASE experiments (Supplementary File 5).
Outbred strains
We generated present and absent outbred strains for FBti0019386, and FBti0018868 insertions. First, we selected all the strains that were present or absent for these TEs based on data generated by Tlex2 in the DGRP, Zambia, Sweden, and Italy populations (47). Then, in these selected strains, we checked by PCR the presence/ absence of the other nine TEs identified is this work as they are likely to be involved in the immune response as well (FBti0019985, FBti0061506, FBti0019602, FBti0020119, FBti0018883, FBti0018877, FBti0020137, tdn4, and tdn8). For generating both present and absent outbred populations of each TE we chose between seven and nine strains present and absent of a specific TE, respectively (Supplementary File 5). Moreover, present and absent outbred populations have similar frequencies of all the other 10 TEs likely to be involved in immune responses in order to not mask the effect of the studied TE. In every outbred population, we placed 10 males and 10 virgin females of each selected strain in a cage with fresh food. We maintained the population by random mating with a large population size for over four generations before starting the experiments.
Mutant, RNAi knockdown, and overexpression strains
We used three RNAi mutant strains from the VDRC stock center (Supplementary File 2A). To generate the mutants, we crossed the strains carrying the RNAi controlled by an UAS promoter with flies carrying a GAL4 driver (a transcription activation system) to silence genes ubiquitously. We performed the experiments with F1 flies that were obtained from each cross. Based on the phenotypic markers, we separated the RNAi mutant flies from the rest of the F1 that do not carry the GAL4 driver. The flies without the GAL4 driver were used as the baseline of the experiment. To overcome the lethality of silencing CG15829 during development, we used an Act5c-GAL4 strain regulated by the temperature sensitive repressor GAL80. For this mutant, we transferred flies from 25°C to 29°C 24h before performing the experiment.
We also used nine mutant strains generated with different transposable element insertions and two overexpression strains. In this case, we used strains with similar genetic backgrounds as the baseline for the experiments (Supplementary File 2A).
Transposable element datasets
TEs annotated in the reference genome
There are 5,416 TEs annotated in the v6 of the D. melanogaster reference genome (101). In this work, we focused on polymorphic TEs present at high population frequencies and located in high recombination regions of the genome. Most TE insertions are expected to be deleterious. Due to its big effective population size, we expect most TE insertions to be present at low frequencies in D. melanogaster. Thus, TEs present at high population frequencies are likely to be adaptive. We did not consider the 2,234 INE-1 insertions that are fixed in D. melanogaster populations (102–104). We also discarded 1,561 TEs that are flanked by simple repeats, nested TEs, or TEs that are part of segmental duplications because frequencies cannot be accurately estimated for these TEs using T-lex2 (45). Finally, we discarded 813 TEs present in genomic regions with a recombination rate = 0 according to Fiston-Lavier et al (105) or Comeron et al (106). TEs present at low recombination regions are more likely to be linked to an adaptive mutation rather than being the causal mutation (107–110). Moreover, the efficiency of selection is low in these regions and, thus, slightly deleterious TEs could have reached high frequencies (111, 112). Hence, we ended up with a dataset of 808 annotated TEs for which we estimated their population frequencies using T-lex2 (45) (Supplementary File 1A).
231 of the 808 annotated TEs were fixed in the four populations studied. Although some of these fixed TEs might be adaptive, we did not consider them as we cannot perform comparative functional experiments between flies with and without the insertions. We considered high frequent TEs those present at a population frequency ≥ 10%: 109 TEs. Note that varying this threshold does not substantially alter the number of TEs present at high frequencies (e.g. 95 TEs if we consider ≥ 15%).
Non-reference TE insertions
We also analyzed a subset of TEs identified by Rahman et al (46) in DGRP strains that are not annotated in the reference genome (Supplementary File 1B). We analyzed 23 TEs that are present in regions with recombination rate > 0 (105, 106), and were inferred to be present in at least 15 DGRP strains out of the 177 strains analyzed by Rahman et al (46). Then, we obtained from Bloomington Drosophila Stock Center (BDSC) all the strains carrying each of the 23 insertions, and we confirmed by PCR the presence of the insertions in several strains (see below). For each TE, we sequenced at least one of the PCR products to confirm the presence and the family identity of the TE. For those insertions that we could verify, we estimated the frequency of each TE based on TIDAL results in the 177 DGRP strains and considered as high frequent those present at a population frequency ≥ 10%.
Presence/Absence of TEs in the analyzed strains
We performed PCRs to confirm the in silico results obtained with T-lex2 (45) and TIDAL (46). We designed specific primers for each analyzed TE using the online software Primer-BLAST (113) (Supplementary File 6). Briefly, we designed a primer pair flanking the TE (FL and R primers), which produces a PCR product with different band sizes when the TE is present and when the TE is absent. For those TEs that are present in the reference genome, we also designed a primer inside the TE sequence (L primer) that, combined with the R primer, only amplifies when the TE is present (114). To perform the PCRs, genomic DNA was extracted from 10 females from each analyzed strain.
Functional annotation of genes nearby candidate adaptive TEs
We looked for functional information of the genes associated to the TEs present at high population frequencies using FlyBase (101). We considered all the genes that were located less than 1kb from the TEs. If the TEs did not have any gene located in the 1kb flanking regions, we considered only the closest gene. We considered GO annotations based on experimental evidence, and we also obtained functional information based on the publications cited in FlyBase. Several lines of evidence were considered: genome-wide association studies in which SNPs in the analyzed genes were linked to a phenotypic trait, differential expression analyses, and phenotypic evidence based on the analyses of mutant strains (Supplementary File 1D).
qRT-PCR expression analysis of mutant, RNAi, and overexpressing strains
For RNA extraction, three replicates of 20-30 females, males, or guts from each mutant and wild-type strain were flash-frozen in liquid nitrogen and stored at −80°C until sample processing (Supplementary File 2A). RNA was extracted using the GenElute™ Mammalian Total RNA Miniprep Kit (Merck) following manufactureŕs instructions. RNA was then treated with DNase I (Thermo). cDNA was synthesized from a total of 250-1,000 ng of RNA using the NZY Fisrt-Strand cDNA synthesis kit (NZYTech). Primers used for qPCR experiments are listed in Supplementary File 2D. In all the cases, gene expression was normalized with the housekeeping gene Act5c. We performed the qRT-PCR analysis with SYBR Green (BioRad) on an iQ5 Thermal cycler. Results were analyzed using the dCT method following the recommendations of the MIQE guideline (115).
Infection experiments
We infected 5- to 7-day-old female flies with the gram-negative bacteria Pseudomonas entomophila (24). Flies were separated into food vials under CO2 anesthesia two days before the bacteria exposure, and were kept at 25°C. The experiments were performed as described in Neyen et al (116). Briefly, flies were starved for two hours and then they were flipped to a food vial containing a filter paper soaked with 1.25% of sucrose and bacterial pellet. The bacterial preparation was adjusted to a final OD600 = 100, corresponding to 6.5 x1010 colony forming units per ml (117). Flies were kept at 29°C and 70% humidity, which are the optimal infection conditions for P. entomophila. In parallel, we exposed non-infected flies to sterile LB with 1.25% sucrose.
Survival experiments
We performed infection survival experiments with mutant strains, RNAi strains, and overexpression strains. We compared the mortality of these strains to the mortality of strains with similar genetic backgrounds (Supplementary File 2A). We also performed infection survival experiments comparing outbred flies with FBti0019386 and FBti0018868 with outbred flies without these insertions, respectively. Female flies were placed in groups of 10 per vial, and we performed the experiments with 5-12 vials (Supplementary File 2C), except for cn1 considered as a wild-type background for which we used 3 vials. As a control for each experiment, we exposed 3-4 vials containing 10 flies each to sterile LB with 1.25% sucrose.
Fly mortality was monitored at several time points until all the flies were dead. Survival curves were analyzed with log-rank test using SPSS v21 software. If the test was significant, we calculated the odds-ratio and its 95% confidence interval when 50% of the susceptible flies were dead, except for CG8008 and cbx that was estimated when 30% and 96% of the susceptible flies were dead.
RNA extraction and cDNA synthesis from non-infected and infected guts
We dissected 20-30 guts from both non-infected and orally infected 5- to 7-day-old females. Flies were infected with the gram-negative bacteria P. entomophila as mentioned above, and they were dissected after 12 hours of bacterial exposure. Samples were frozen in liquid nitrogen and stored at −80°C until sample processing. RNA from gut tissue was extracted using Trizol reagent and PureLink RNA Mini kit (Ambion). We treated RNA on-column with DNase I (Thermo) during the RNA extraction, and we did an additional treatment after the RNA purification. We synthesized cDNA from a total of 500 ng – 1,000 ng of RNA using the Anchored-oligo (dT) primer and Transcription First Strand cDNA Synthesis kit (Roche).
Allele-specific expression analysis (ASE)
For each TE analyzed, we first identified two strains homozygous for the presence of the TE and two strains homozygous for the absence of the TE according to T-lex2 or TIDAL (45, 46). We then looked for a synonymous SNP linked to the presence of the TE and located in the coding region of the nearby gene. Note that we only selected a SNP when it is present in the coding region of all the alternative transcripts described for that gene. To select the SNP, we downloaded the coding region of the nearby gene from the sequenced DGRP strains available in http://popdrowser.uab.cat/ (118). Once we identified a diagnostic SNP, we re-sequenced the region identified in the used strains to confirm the presence of the SNP, and we performed a PCR to confirm the presence or the absence of the TE. We selected a synonymous SNP that is not linked to the TE in all the strains analyzed (Supplementary File 7).
We also analyzed the coding region of the gene in order to discard the presence of nonsynonymous SNPs that could be linked to the TE (Supplementary File 4A). Additionally, we analyzed the flanking regions of each TE in order to discard other variants that could be linked to the TE, or that could be potentially modifying the gene regulatory regions (Supplementary File 4B). To do this, we used VISTA to define the conserved regions in the 1 kb TE flanking sequences between D. melanogaster and D. yakuba, which diverged approximately 11.6 Mya (119). We then checked whether there is any SNP linked to the presence of the TE in the DGRP strains. Only for the AGO2 gene, we found two SNPs in the coding region that were linked to the TE insertion (Supplementary File 4A). AGO2 is a gene showing a fast rate of adaptive amino acid substitutions (120, 121), and it is associated to a recent selective sweep (122). However, it is still not clear which is the genetic variant that is under positive selection (122). Thus for 13 out of the 14 TEs analyzed, we could not detect any other cis-regulatory change that could be responsible for the observed allele-specific expression differences suggesting that the TE is the most likely mutation.
We were not able to analyze five of the candidate TEs: for three TEs, FBti0019381, FBti0061105 and FBti0062242, we could not identify homozygous strains with and without the TE. For FBti0019564, we could not identify a diagnostic SNP. Finally, for tdn17, we could not design primers to validate the diagnostic SNP due to the presence of repetitive sequences in the nearby gene.
We then crossed a strain with the TE with a strain without the TE differing by the diagnostic SNP to obtain heterozygous flies in which allele-specific expression was measured (Supplementary File 7). Note that for each TE two crosses were performed so that ASE was measured in two different genetic backgrounds.
ASE was measured in non-infected and infected conditions. We obtained cDNA samples from three biological replicates. We also extracted genomic DNA (gDNA) from 15-20 heterozygous females for each cross, which is needed to correct for any bias in PCR-amplification between alleles (123). cDNA and gDNA samples were sent to an external company for primer design and pyrosequencing. We analyzed the pyrosequencing results as described in Wittkopp et al (123). Briefly, we calculated the ratios of the allele with the TE and the allele without the TE of the cDNA samples, and we normalized the values with the gDNA ratio. In order to perform the statistical analysis, we transformed the ratios with log2, and we applied a two-tailed t-test in order to check whether there were allele expression differences between the alleles. We corrected the p-values for multiple testing using Benjamini-Hochberg’s false discovery rate (5% FDR) (124).
Transcript start site detection
To detect whether FBti0019386 is adding a Transcription Start Site (TSS) to their nearby gene, as suggested by Batut et al (81), we performed RT-PCR in gut tissue of non-infected and infected flies. We used the forward primer 5’-ATCTGAAGCTCGTTGGTGGG-3’ and the reverse primer 5’ ATGAGACTCCTGTTTCGCCG-3’ to detect Bin1 transcript starting in the TE, and the same forward primer with the reverse primer 5’ AAGAGCAAAGAGAAGCCGGAA-3’ to detect Bin1 short transcript.
3’RACE
We performed 3’RACE to detect whether the FBti0020057 was affecting the transcript structure or the transcript choice of CG15829. We extracted total RNA from gut tissue of non-infected flies, and synthetized the cDNA using SuperScriptTM II Reverse Transcriptase (Invitrogen). We amplified the 3’ ends with the Universal Amplification Primer (UAP) (5’-CUACUACUACUACUAGGCCACGCGTCGACTAGTAC-3’) and nested PCRs specific for CG15829: outer primer 5’-CTGCCTAGCAAGGAGGAGTT-3’ and the inner primer 5’-GAGAAGAAGGCCCGCTACAA-3’.
Enhancer reporter assays
We generated transgenic flies carrying the TE sequence in front of the reporter gene LacZ by using the placZ.attB vector (accession number: KC896840) (125). In order to construct a clone with the correct orientation in the promoter region of lacZ, two cloning steps were necessary. We first had to introduce specific restriction sites into the flanking regions for each TE sequence. For that, we introduced the restriction sites with the primers used to amplify the region containing the TE sequence (Supplementary File 8). We used a high fidelity Taq DNA polymerase for DNA amplification (Expand High Fidelity PCR system from Sigma). After that, we cloned the PCR product into the vector pCR®4-TOPO® (Invitrogen). Finally, we digested both vectors and ligated the TE sequence into the placZ.attB, and we sequenced the cloned insert to ensure that no polymerase errors were introduced in the PCR step. We purified the vector with the GeneEluteTM Plasmid Miniprep kit (Sigma), and prepared the injection mix at 300 ng/μl vector concentration diluted with injection buffer (5 mM KCl, 0.1 mM sodium phosphate, pH 6.8). The injection mix was sent to an external company to inject embryos from a strain that contain a stable integration site (Bloomington stock #24749). After microinjection, surviving flies were crossed in pairs and the offspring was screened for red eye color, which was diagnostic for stable mutants. We established three transgenic strains for each analyzed TE, which were considered as biological replicates in the expression experiments. As a negative control, we also established transgenic strains with the placZ.attB empty vector, in order to control for possible lacZ expression driven by the vector sequence.
For two TEs, FBti0018868 and FBti0019985, we designed primers flanking the TE and cloned the PCR product in front of the reporter gene lacZ (Supplementary File 8). For the other three TEs, we constructed two different clones to generate two transgenic strains: one strain with the TE and the other strain without the insertion. For the TE FBti0061506, which spans only 48 bp, one strain carries the TE and part of the flanking intronic region, and the other strain contains the same genomic region without the TE. For the TE tdn8, one strain carries the upstream region of CG10943, including the 5’UTR, with tdn8, and the other strain carries the same genomic region without tdn8. Finally, for the TE FBti0020057, we cloned the whole intergenic region, including the UTRs of the flanking genes (Supplementary File 8).
We analyzed the flanking regions of FBti0020057 to check whether the insertion is disrupting a regulatory region. For that, we analyzed the 150 bp flanking sequence with and without the insertion looking for predicted transcription factor binding sites with JASPAR (126), using a relative profile score threshold of 90% (Supplementary File 9).
qRT-PCR expression analysis
For the transgenic strains generated in the enhancer assays, we checked lacZ expression in female guts in non-infected and infected conditions. We used the forward primer 5’-CCTGCTGATGAAGCAGAACAACT-3’, and reverse primer 5’-GCTACGGCCTGTATGTGGTG-3’ to check lacZ expression.
We measured the gut total expression of TM4SF and Bin1 genes in homozygous strains with and without FBti0018868 and FBti0019386, respectively. We used the following primers to detect Bin1 total expression: forward 5’-TGTCGTCCCGTAGAGCAGAA-3’ and reverse 5’-CAAGCAGATTGACCGCGAGA-3’, and TM4SF total expression: forward 5’-GCAGCGAGGATAACGGGAAA-3’ and reverse 5’-AGTAGACCGAGTGACCCCAG-3’.
We also designed primers to detect specifically those transcripts starting in the TE for each gene. For transcript starting in FBti0019386, we used forward 5‘-TGCAGCAGATGGCTCATATT-3’ and reverse 5’-AGTGCTCAAGACCCTAATGGAA-3’, and for transcripts starting in FBti0018868, we used forward 5’-CTTGGCGTTGTCCTTAGTCA-3’ and reverse 5’-ACTGATTTATATCGTATGGGGTGCT-3’. We analyzed the two pairs of genetic backgrounds used for the ASE experiments, and one extra genetic background for Bin1. In all the cases, gene expression was normalized with the housekeeping gene Act5c. We performed all RNA extractions, cDNA synthesis and qRT-PCR analysis as mentioned above.
Immunofluorescence staining
We performed immunofluorescence gut staining to localize β-GAL expression in the transgenic flies from the enhancer assays, both in non-infected and infected conditions. Flies were dissected and gut tissue was fixed with 4% Formaldehyde. The tissue was then stained by using the primary antibody mouse anti-βGalactosidase (Hybridoma bank 40-1a), and the secondary antibody anti-mouse Alexa Fluor ® 555 (Sigma). Images were analyzed and captured using a Leica SP5 confocal microscope.
Chromatin immunoprecipitation-qPCR
We performed ChIP-qPCR experiments to detect whether FBti0020057 that was associated with allele-specific lower expression was adding H3K9me3 repressive marks (38, 87). For that, we compared the histone mark levels in homozygous flies with the TE with the levels in homozygous flies without the TE. We used y1;cn1bw1sp1strain (127), the strain used to obtain the D. melanogaster reference genome sequence (128–130), as the homozygous strain with FBti0020057 insertion, and RAL-908, as the homozygous strain without those insertions (96). We first confirmed by PCR the presence or absence of FBti0020057 in these strains. To detect H3K9me3 levels associated to the TE, we designed primer pairs in the TE flanking regions (“left” and “right”): one primer inside the TE sequence and one primer outside the TE sequence (Supplementary File 10). To detect H3K9me3 levels in the strains without the TE, we used the left forward primer and the right reverse primer. Primer efficiencies ranged from 90-110%. We used a total of 45-55 guts per strain and performed three biological replicates for each strain. In order to obtain the chromatin, we followed Magna-Chip™ A/G kit (from Merck) protocol. After dissection, we homogenated the samples in the buffer A1 with a dounce 30 times, and we crosslinked the guts using formaldehyde at a final concentration of 1.8% for 10 minutes at room temperature. We stopped the crosslink by adding glycine at a final concentration of 125 mM, we incubated samples three minutes at room temperature, and kept them on ice. Then, we washed the samples three times with buffer A1, and we incubated the sample for three hours at 4°C with 0.2 ml of lysis buffer. After lysis, we sonicated the samples using Biorruptor® pico sonication device from Diagenode: 14 cycles of 30 seconds ON, 30 seconds OFF. We kept 20 µl of input chromatin for the analysis (see below), and we immunoprecipitated 80 µl of the sample with antibody against H3K9me3 (#ab8898 from Abcam). As a control for the immunoprecipitation, we checked the H3K9me3 levels in the genes 18S and Rpl32 that are expected to be, respectively, enriched and depleted for this histone mark (Supplementary File 10). We quantified the immunoprecipitation by qRT-PCR analysis with SYBR Green (BioRad) on an iQ5 Thermal cycler. We quantified H3K9me3 immunoprecipitation normalizing with the input chromatin for each sample. Results were analyzed using the dCT method and following the recommendations of the MIQE guideline (115).
DECLARATIONS
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its supplementary information files].
Competing interests
The authors declare that they have no competing interests.
Funding
This work was funded by the European Commission (H2020-ERC-2014-CoG-647900 and FP7-PEOPLE-2011-CIG-293860), by the MEC/FEDER (BFU2014-57779-P). A.U. was a FPI fellow (BES-2012-052999) and JG was a Ramon y Cajal fellow (RYC-2010-07306). The funding bodies had no role in the design of the study and collection, analysis, and interpretation of data or in writing the manuscript.
Authors’ contributions
AU participated in the conception and design of the experiments, performed the experiments, analyzed the results, and drafted the manuscript. MM performed experiments and analyzed the results. JG participated in the conception and design of the experiment, analyzed the results, and wrote the manuscript.
Acknowledgements
We would like to thank all the members of González Lab for help with the experiments, and for their comments on the manuscript.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.
- 32.↵
- 33.↵
- 34.↵
- 35.
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.
- 51.
- 52.
- 53.↵
- 54.
- 55.↵
- 56.
- 57.
- 58.
- 59.
- 60.
- 61.
- 62.
- 63.
- 64.
- 65.
- 66.
- 67.↵
- 68.
- 69.
- 70.
- 71.↵
- 72.
- 73.
- 74.
- 75.
- 76.↵
- 77.↵
- 78.
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.
- 92.
- 93.
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.
- 109.
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.
- 130.↵




