

ABSTRACT
The ANK3 gene, encoding the protein ankyrin-G (AnkG), is associated with a variety of neuropsychiatric and cognitive disorders including bipolar disorder, autism spectrum disorder, and schizophrenia. These diseases are characterized by abnormal dendritic and synaptic architecture. AnkG is a multifunctional scaffold protein with several isoforms: The 480 kDa and 270 kDa isoforms have roles at the axon initial segment and node of Ranvier, but the function of the 190 kDa isoform (AnkG-190) is less well understood. Moreover, these isoforms are regulated by palmitoylation, but palmitoylation of AnkG-190 has not been investigated in neurons. Here we show that AnkG is required for normal dendrite and spine architecture in vivo and that AnkG-190 stabilizes pyramidal neuron dendrites. We found that Cys70 palmitoylation stabilizes AnkG-190 in spine heads and at dendritic plasma membrane nanodomains, and is necessary for the maintenance of normal spine density, dendrite arborization, and for correct microtubule dynamics. Lithium, a commonly used mood stabilizer, reverses spine and dendrite deficits induced by AnkG knockdown in a manner that is dependent of palmitoylation. Finally, we found that lithium reduces AnkG-190 palmitoylation and increases its mobility in spines. Taken together, our data reveal a novel mechanism regulating dendritic architecture and mood stabilizer action on palmitoylation of an important psychiatric disorder risk factor.
INTRODUCTION
The human ANK3 gene, encoding the protein ankyrin-G (AnkG), has been associated with various neuropsychiatric diseases, including bipolar disorder (BD), schizophrenia, and autism spectrum disorder (1–3). Multiple independent genome wide association studies (GWAS) have strongly linked ANK3 to BD in a variety of different ethnicities (4–7) and have been corroborated by meta-analyses (8). In addition to a genetic link with ANK3, some studies have shown an increase of ANK3 expression in blood (9) and in lymphoblastoid cells of BD patients (10), revealing it as a potential biomarker. Furthermore, decreased expression of a specific AnkG isoform was observed within the cerebellum of a BD patient carrying a BD-associated ANK3 allele (11). Thus BD risk alleles may induce both increases and decreases of ANK3 expression, and may result in isoform-specific alterations in protein content.
Lithium is a mood stabilizer commonly used to treat BD patients as a first-line intervention (12). Responsiveness of BD patients to lithium treatment appears to be a heritable trait (13) linked to genetic markers in patients isolated from GWAS (14). The identification of 19 single nucleotide polymorphisms within the ANK3 regulatory regions that alter the response to lithium treatment suggest that AnkG may be involved in the lithium response (15). In accordance with this, alterations in murine AnkG levels through hemizygous Ank3 knockout (KO) (16), specific isoform KO (17) or within selected brain regions induced BD or behaviors related to schizophrenia (18). Interestingly, lithium was shown to rescue several of these behavioral deficits in AnkG KO mice (19–21).
The ANK3 gene encodes multiple isoforms of AnkG, of which the 190, 270, and 480 kDa isoforms are the most prominent in brain (22). The giant 270 and 480 kDa isoforms have been mostly studied for their function in the axon initial segment (23) or in nodes of Ravier (24), but the 480 kDa isoform has also been shown to prevent gamma-aminobutyric acid (GABA) receptor endocytosis and to have an important role in maintaining GABAergic synapses (25). The role of the 190 kDa isoform (AnkG-190) is less fully described in neurons, but it can be found in human postsynaptic density (PSD) fractionation, where it decreases in BD (26). Moreover, AnkG-190 was found to increase in rat PSD after 5 weeks of lithium treatment (27). AnkG-190 seems to play an important role in dendritic spine maintenance and long-term potentiation through subsynaptic nanodomains in dendritic spine heads and necks (28). However, the roles of AnkG-190 in dendrite architecture, and the regulation of its localization in dendrites and synapses, are not known.
S-palmitoylation is a reversible form of post-translational modification regulating protein attachment to lipid membranes (29) and localizing proteins to different neuronal subcompartments and organelles (30). Important synaptic proteins, including neurotransmitter receptors, scaffold proteins, transporters, adhesion molecules, SNAREs, and other trafficking proteins, are palmitoylated (31). However, much less is known about the role of palmitoylation in dendrite morphogenesis and in the mechanisms of action of therapeutic agents. Notably, the membrane-binding regions of all three main AnkG isoforms harbor a cysteine residue (C70) that can be palmitoylated, a process that is required for AnkG association with the membrane, appropriate cellular localization, and function in non-neuronal cells (32, 33). Structural analysis has also shown that the membrane anchoring of AnkG is facilitated by palmitoylation, defining a stable binding interface on the lipid membrane (34). Interestingly, non-palmitoylated AnkG also localized near the membrane but lacked a unique binding interface.
Because understanding of synaptic palmitoylation is likely to provide insights into normal neuronal function but also pathophysiological and therapeutic mechanisms (35), we investigated the physiological function of AnkG palmitoylation and its roles in the actions of lithium. BD and schizophrenia are highly correlated to a decrease of dendritic spines and dendrite length in cortex (36) and in induced pluripotent stem cell (iPSC)-derived neurons (37). We show that AnkG-190 stabilizes pyramidal neuronal basal dendrites. Palmitoylation stabilizes AnkG-190 in spines and at dendritic plasma membrane nanodomains. Lithium reverses spine and dendrite deficits induced by AnkG knockdown. Finally, lithium reduces AnkG-190 palmitoylation and increases its mobility in spines. Taken together, our data reveal a novel mechanism of regulation of dendritic architecture and lithium action on the cycling of AnkG-190 palmitoylation.
METHODS
See Supplementary Methods for details.
Conditional KO mice
Ank3-floxed mice (Ank3flox/flox) (The Jackson Laboratory; Bar Harbor, ME, USA; #029797) were crossed with Camk2a-Cre mice (The Jackson Laboratory; #005359) for forebrain-specific homozygous deletion of Ank3 (Ank3flox/flox, Camk2a-cre). The resulting mouse strain was called Ank3 cKO. To maintain the colonies, Ank3 cKO mice were mated with Ank3flox/flox mice. All experiments were performed following protocols approved by the Institutional Animal Care and Use Committee at Northwestern University.
Pharmacological treatments
2-Bromopalmitate (2-BrP), a palmitoylation inhibitor, and S-methyl methanethiosulfonate (MMTS), a thiol blocker, were from Sigma (St. Louis, MO, USA). All other chemicals were from ThermoFisher Scientific (Waltham, MA, USA) and were of the highest reagent grade. 2-BrP was used at 20 mM, palmostatin B (which inhibits depalmitoylation) at 50 μM, lithium chloride at 2 mM, and all were applied for stimulation over 24 h.
Microtubule plus-end-binding protein 3
Neurons were imaged at 37°C, 5% CO2, with a Nikon A1R confocal microscope using a 100x oil immersion objective with NA = 1.2 in an OKOLAB stage-type CO2 incubator. One-plan images were captured with a GaAsP detector every 2 s. Images were analyzed and kymographs were built in ImageJ, and microtubule plus-end-binding protein 3 (EB3) puncta were tracked with the plugin MTrack2.
Fluorescence recovery after photobleaching
Neurons were imaged at 37°C, 5% CO2, with a Nikon C2 confocal microscope using a 63x oil immersion objective with NA = 1.4 in an OKOLAB stage-type CO2 incubator. Single plane images were captured with an EMCCD camera every 10 s for 200 s. 100% laser power pulses of 1 ms (2 iterations) were used to bleach GFP in the spine. After background substation and validation of maximum 5% remaining fluorescence after photobleaching, data were normalized with the prebleach value. Recovery data points were then fitted to a one-phase association exponential in GraphPad Prism. The mobile fraction was calculated as an average of the plateaued fluorescence level and expressed as a percentage of the pre-bleached level.
RESULTS
AnkG-190 maintains dendrite complexity in pyramidal neurons
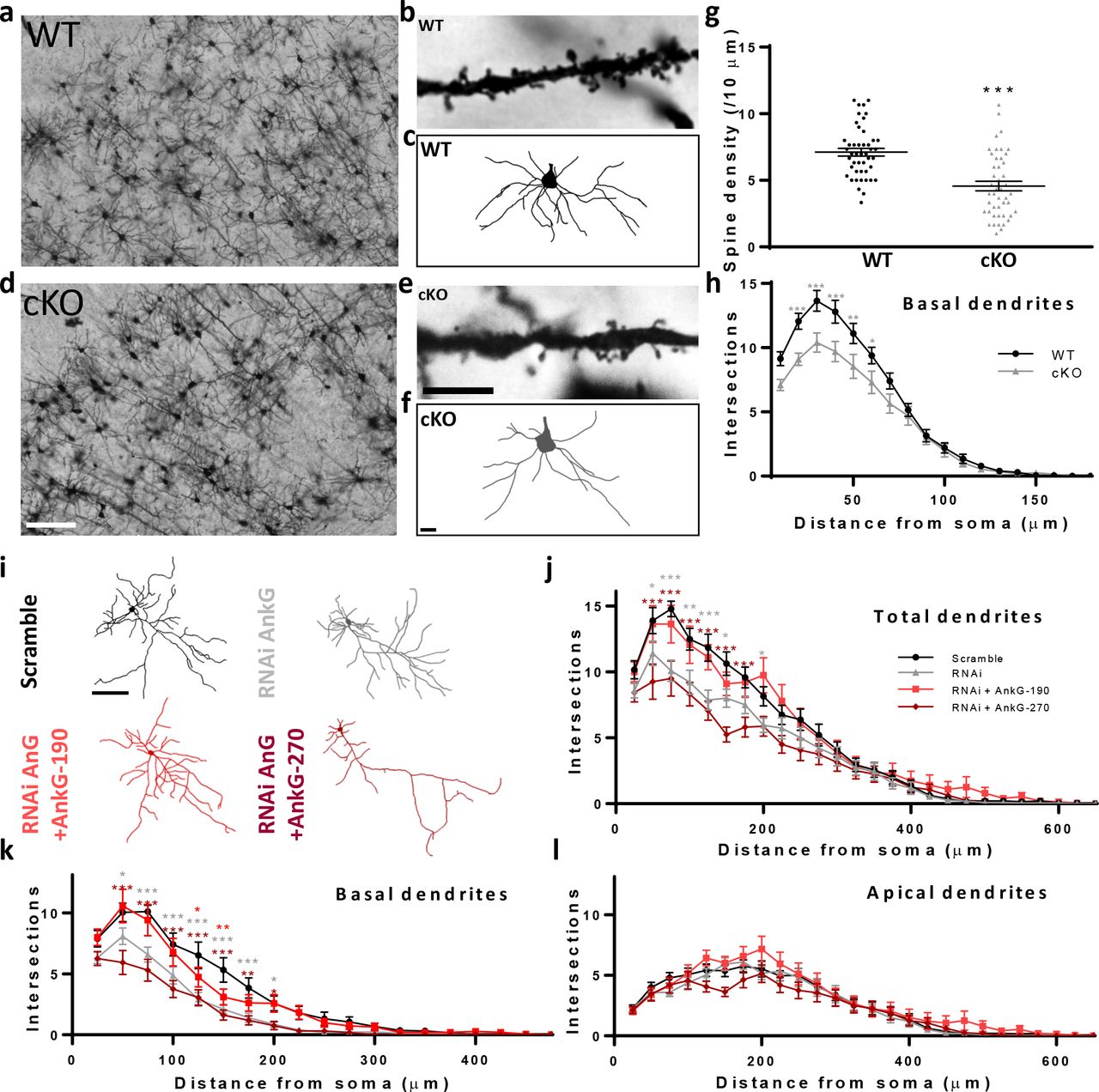
We have previously shown that AnkG-190 has an essential role in dendritic spine maintenance in vitro (28), but whether AnkG is similarly important in vivo has not been addressed. To investigate this question, we performed Golgi-Cox staining in Ank3 cKO (Ank3flox/flox, Camk2a-Cre) mouse brain slices. In this model, Cre recombinase is expressed under a Camk2a promoter to target the Ank3 gene in the postnatal forebrain (21). We analyzed dendrites of pyramidal neurons from layer 2/3 of the somatosensory cortex, a region of the cortex where AnkG is abundant (Fig. 1a, d). Quantitative analysis of spine number showed a decrease in spine density in Ank3 cKO brains, in support of our in vitro findings (Fig. 1c). We also observed that Ank3 cKO mice had striking alterations in dendrite morphology with decrease in basal dendrite complexity (Fig. 1c). To specifically determine whether the effects in dendrites were cell-autonomous, we knocked down AnkG expression in rat primary cortical neurons using a previously described Ank3 RNAi which induces a 50% decrease of AnkG staining in dendrites after 3 days of expression (28). By separating apical and basal dendrites (see supplementary methods), we again found reduced complexity in the basal dendritic arbor of AnkG knockdown pyramidal neurons (Fig. 1g), with no change in apical dendrites (Fig. 1h). Interestingly, coexpression of an RNAi-resistant AnkG-190 construct restored dendritic complexity, whereas coexpression of an RNAi-resistant 270 kDa isoform exacerbated the phenotype (Fig. 1g). Notably, AnkG knockdown also reduces dendrite complexity of interneurons, but this effect is only partially rescued by the overexpression of AnkG-270, and not at all by AnkG-190, suggesting isoform-specific requirements for different neuronal subtypes (Fig. S1a, b). Together, these data indicate that the AnkG-190 isoform may have a specific role in maintaining basal dendrite complexity in addition to maintaining spine architecture in cortical pyramidal neurons (28).
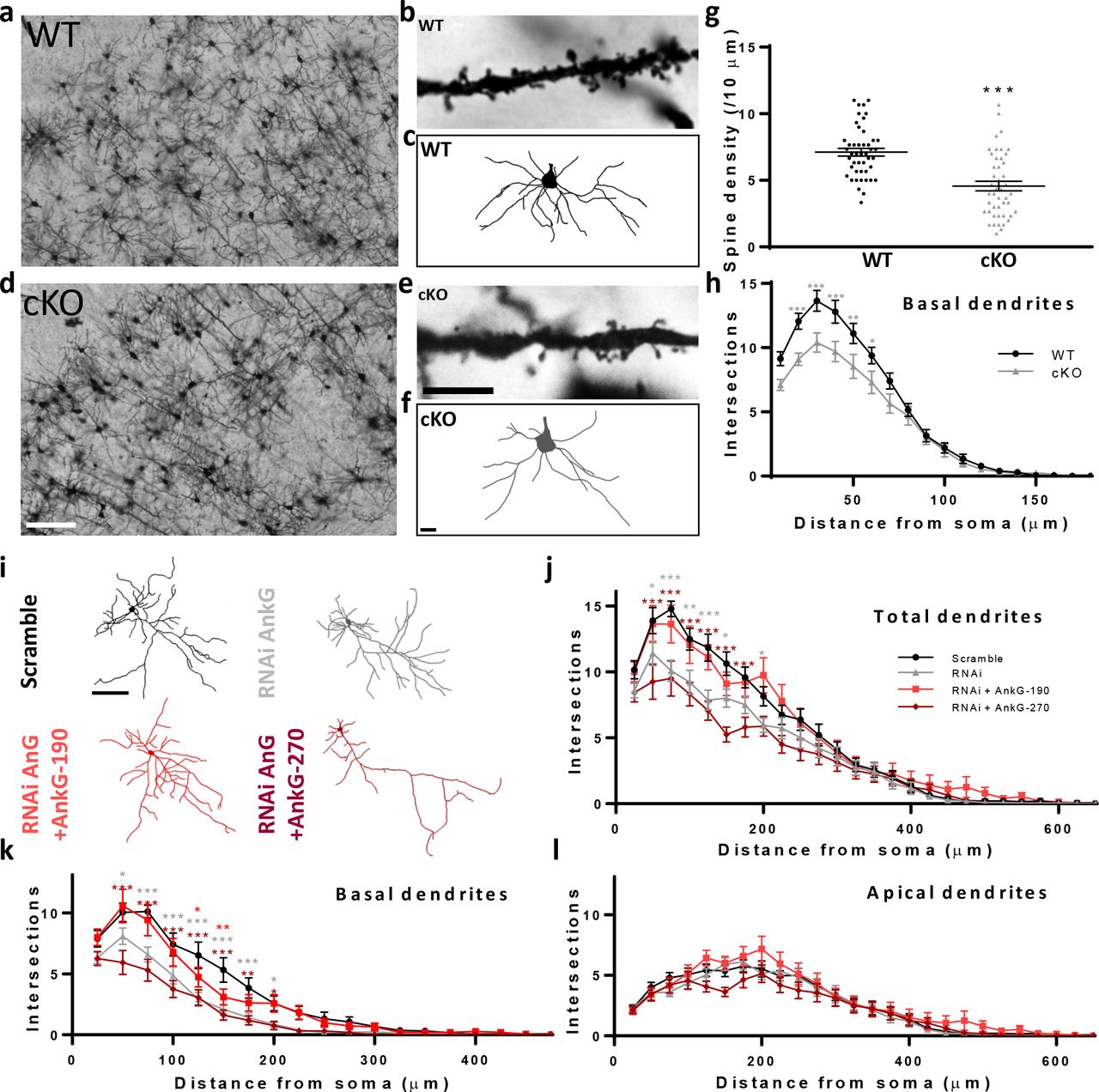
(a-f) Bright field images of Golgi-Cox staining in somatosensory cortical slices from (a) wild-type (WT) and (d) Ank3 cKO mice, scale=100 μm. Dendrite pictures (b and e, scale=5 μm) and representative traces of basal dendrites (c and f, scale=20 μm) from pyramidal neurons from cortical layer 2/3 (g) Scatter plot of spine density from WT and AnkG cKO mice (46 cells, 3 brains per group, t-test, ± SEM, ***p≤0.001). (h) Graph showing Sholl analysis for basal dendrites (20 neurons, 3 brains per group, 2-way ANOVA with Dunnett’s post-test, *p≤0.05, **p≤0.01, ***p≤0.001, ±SEM. (i) Representative traces of cultured rat neurons expressing scramble or AnkG RNAi with GFP (no AnkG), GFP-AnkG-190, GFP-AnkG-270. Scale=100 μm. (j-l) Graphs showing Sholl analysis for total (j), basal (k), and apical (l) dendrites (15-22 neurons, 2-way ANOVA with Dunnett’s post-test, *p≤0.05, **p≤0.01, ***p≤0.001, ±SEM).
Palmitoylation of C70 stabilizes AnkG-190 in dendritic spine head and dendritic nanodomains
The specific role of AnkG-190 in dendritic spine and dendrite maintenance led us to ask what post-translational modifications can regulate these specific functions of AnkG-190. Because palmitoylation is a general mechanism for regulation of synaptic proteins, and palmitoylation of AnkG in non-neuronal cells was shown to be essential for the association of AnkG with plasma membranes (32–34), we hypothesized that this modification may also modulate AnkG’s regulation in dendrites.
We first validated the presence of palmitoylated AnkG-190 in rat cortex utilizing the acyl-biotinyl exchange assay (see supplementary methods) (38), which exchanges palmitoyl modifications with biotin (Fig. 2a). We therefore mutated the palmitoylated residue (32) to alanine (C70A) in a GFP fusion construct (GFP-AnkG-190-C70A) to assess how palmitoylation-deficient AnkG-190 would behave in neuronal dendrites and spines (Fig. 2b). Confocal images revealed that overexpressed GFP-AnkG-190 was enriched in the spine head, as previously observed (28), and this enrichment was lost when C70A was mutated, matching the levels of soluble GFP (Fig. 2c, d). We then reasoned that mislocalization of palmitoylation-deficient AnkG-190 may lead to alterations in spine morphology. t. We found that while AnkG-190 expression increased spine density, as shown in Smith et al (28), GFP-AnkG-C70A had no effect on spine density (Fig. 2e, f). GFP-AnkG-190 expression also increased spine width but not length, whereas GFP-AnkG-C70A further increased both spine width and length.

(a) AnkG-190 is palmitoylated in rat forebrain. Solubilized rat forebrain homogenates were subjected to ABE to purify palmitoylated proteins. ABE fractions and a portion of input homogenate (Inp.) were western blotted to detect AnkG. Exclusion of NH2OH was used as a control for assay specificity. (b) Schematic of 190 kDa Ankyrin-G isoform showing the mutated cysteine which prevents palmitoylation. (c) Confocal images of cultured cortical neurons expressing mCherry with GFP, GFP-AnkG-190, or GFP-AnkG-190-C70A, scale=5 μm. (d) Linescan analysis of spine:dendrite ratio of expressed GFP, GFP-AnkG-190, or GFP-AnkG-190-C70A (16-20 neurons per condition, 2-way ANOVA with Dunnett’s post-test, ***p≤0.001, ±SEM). (e) Confocal images of cultured cortical neurons expressing mCherry with GFP, GFP-AnkG-190, or GFP-AnkG-190-C70A, scale=5 μm. (f) Scatter plots showing quantification of spine density, spine width, and spine length for neurons in (e) (n=19-22 neurons, 1-way ANOVA with a Dunnett’s post-test, *p≤0.05, **p≤0.01, ***p≤0.001, ±SEM). (g) Spine SIM imaging 3D reconstruction of cultured rat neurons expressing mCherry (white), GFP-AnkG-190 or GFP-AnkG-190-C70A (both green), and immunostained for PSD-95 (pink). (h) SIM imaging of dendrites from cultured cortical neurons expressing GFP-AnkG-190 or GFP-AnkG-190-C70A, scale=0.5 μm, in gray or pseudocolor (middle and bottom) and their associated normalized linescan (top). (i) Scatter plot of the puncta index average (7-9 neurons per condition, Mann Whitney, *p≤0.05, ±SEM) for GFP-AnkG-190 or GFP-AnkG-190-C70A overexpression. (j) Representative time lapse images of GFP-AnkG-190 and GFP-AnkG-190-C70A during a FRAP experiment, scale=2 μm. (k) Quantification of GFP fluorescence in spines over time. Data are fitted with single exponentials (colored lines). Data are represented as mean ±SEM. (l) Scatter plot of mobile fraction (left) or half time recovery (right) of GFP-AnkG-190 and GFP-AnkG-190-C70A (n=15-22 neurons, ANOVA with Dunnett’s post-test, ***p≤0.001, ±SEM).
To access better resolution of dendritic spines, we used super-resolution structured illumination microscopy (SIM) imaging (28, 39). SIM imaging of dendrites showed that the C70A point mutation did not alter the presence of AnkG-190 at the membrane, but changed its localization pattern (Fig. 2g). We found that AnkG-190-C70A has a more diffuse distribution pattern compared to AnkG-190 (Fig. 2h and i) measured by a smaller variation in fluorescence intensity (Puncta index), suggesting palmitoylation is important to maintain nanodomains along the dendrite. This observation suggested that palmitoylation may alter AnkG-190 mobility. To test this hypothesis, we used fluorescence recovery after photobleaching (FRAP) to measure the mobility of GFP-AnkG-190 compared with GFP-AnkG-190-C70A in dendritic spines (Fig. 2j). FRAP analysis showed that ~60% of the GFP-AnkG190 in the spine was mobile, compared with ~80% of GFP-AnkG-C70A, suggesting that preventing AnkG-190 palmitoylation increases AnkG mobility in spines (Fig. 2k, l). We also analyzed the spine content of PSD-95, which complexes with AnkG in vivo (40). Here we showed a reduction in PSD-95 accumulation in spines when we overexpressed GFP-AnkG-C70A (Fig. S2a, b), which did not affect the size of the spine compared to wild-type AnkG, as shown in the correlation profile (Fig. S2c).
AnkG-190 palmitoylation modulates dendrite complexity and microtubule dynamics
We next assessed whether palmitoylation of AnkG-190 was required to rescue the deficit in dendritic complexity in AnkG knockdown neurons. While wild-type AnkG-190 could rescue the complexity deficit in AnkG knockdown neurons in basal dendrites, coexpression with GFP-AnkG-C70A further reduced dendritic complexity below knockdown levels (Fig. 3a-d), suggesting that the ability of AnkG-190 to be palmitoylated is important for its role in regulating dendritic arborization. Interestingly, AnkG-190-C70A affected only basal but not apical dendrites, similar to AnkG knockdown. AnkG interacts with microtubule plus-end-binding (EB) proteins EB1 and EB3, and AnkG has been directly implicated in regulating microtubule dynamics (41–43). Furthermore, microtubules are key regulators of dendritic morphogenesis in neurons (44). EB3 forms clusters at microtubule tips and can be visualized as a “comet” on polymerizing microtubules when tagged with GFP (Fig. 3e) to assess the speed of microtubule growth (43, 45), which can be represented visually with a kymograph (Fig. 3f). AnkG knockdown neurons exhibited a decrease of EB3 comet speed in dendrites compared with wild-type cells, suggesting reduced microtubule dynamics (Fig. 3g). Interestingly, AnkG knockdown induces an increase of EB3 comet speed in axon (Fig. S3). In comparison, overexpression of GFP-AnkG-190 increased EB3 comet speed compared with GFP alone (Fig. 3h). Overexpression of AnkG-190-C70A significantly reduced the speed of EB3 comets, indicating a reduction in microtubule dynamics when AnkG is unable to be palmitoylated. Collectively, these data suggest that palmitoylation of Ank-190 is required for the maintenance of normal basal dendrite arborization and for correct EB3-mediated microtubule dynamics.

(a) Representative traces of cultured rat neurons expressing AnkG RNAi with GFP-AnkG-190 or GFP-AnkG-190-C70A for three days, scale=100 μm. (b-d) Graphs showing Sholl analysis for total (b), basal (c), and apical (d) dendrites (15 to 22 neurons, 2-way ANOVA with Dunnett’s post-test, *p≤0.05, **p≤0.01, ***p≤0.001, ±SEM). (e) Representative time lapse images of dendrites from cultured rat neurons expressing EB3-dtTomato for three days. (f) Annotated kymograph. (g-h) Representative kymographs of dendrites from neurons expressing for three days: EB3-tdTomato plus either scramble or AnkG RNAi (g), EB3-tdTomato plus GFP, GFP-AnkG-190, or GFP-AnkG-190-C70A (h). (i) Scatter plots showing EB3-GFP speed of movement in the presence of scramble or AnkG RNAi (top, n=10-13 neurons, t-test, **p≤0.01, ±SEM), or in the presence of GFP, GFP-AnkG-190, or GFP-AnkG-190-C70A overexpression (bottom, n=10-12 neurons, 1-ANOVA with Dunnett’s post-test, *p≤0.05, **p≤0.01, ±SEM).
Lithium rescues dendrite and spine deficits in AnkG–deficient neurons in a palmitoylation-dependent manner
Lithium has been shown to increase dendrite length in BD patients (36) and rescue spine decreases observed in iPSCs from BD patients (37). We therefore asked whether lithium could reverse dendrite and spine deficits caused by reduced AnkG expression, and what the role of palmitoylation is in this process. We treated control and AnkG knockdown neurons with lithium for 24 h and assessed spine density and morphology. As previously published, our AnkG shRNA induces a 50% decrease of AnkG immunostaining in dendrites (28). Lithium exposure rescued the dendritic spine deficits we observed in apical dendrites, causing a normalization of spine linear density and length, and also leading to a significant increase in spine width (Fig. 4a-d). Lithium failed to rescue spine deficits in the presence of 2-BrP, indicating that palmitoylation in general is necessary for the effects of lithium on these structures. Expression of GFP-AnkG-190-C70A during the treatment also prevented the rescue of spine density by lithium. Similarly, lithium rescued the deficits in dendrite arborization of basal dendrites induced by AnkG knockdown and induced a small increase in apical dendrite complexity (Fig. 4e-h). As previously observed in dendritic spines, 2-BrP and overexpression of GFP-AnkG-190-C70A prevented the rescue by lithium. Furthermore, we also observed a small decrease in apical dendrite complexity with 2-BrP.

(a) Confocal images of mCherry from cultured rat neurons transfected for three days with scramble or AnkG RNAi and treated with lithium chloride +/− 2-bromopalmitate, scale=10 μm. (b-d) Scatter plots showing spine density, spine length, and spine head width (21-30 neurons, Kruskal-Wallis with Dunnett’s post-test, *p≤0.05, **p≤0.01, ***p≤0.001, ±SEM). (e) Representative traces of cultured rat neurons expressing scramble or AnkG RNAi, +/− lithium chloride and 2-bromopalmitate, scale=100 μm. (f-h) Sholl analysis graph for total, basal, and apical dendrites (27 to 74 neurons, 2-way ANOVA with Dunnett’s post-test, *p≤0.05, **p≤0.01, ***p≤0.001, ±SEM).
Together, these data show that lithium can reverse the functional effect of partial AnkG loss on dendrite and spines. We show that palmitoylation in general is required in the lithium rescue and blocking AnkG-190 palmitoylation will prevent the effects of lithium.
Lithium reduces AnkG-190 palmitoylation and increases its mobility
We wanted to further investigate the mechanisms underlying the involvement of palmitoylation in lithium-dependent rescue of different neuroarchitectural features. With the ABE assay in cortical neuron cultures followed by western blotting of AnkG-190. Interestingly, treatment with lithium resulted in a ~58% decrease in palmitoylated AnkG-190 (Fig. 5a, b). Consistent with this, immunocytochemistry of cortical neuron cultures after lithium treatment revealed a decrease in endogenous AnkG staining in mature spines (width ≥0.8 μm), which are more likely to contain AnkG (28), compared to vehicle treatment (Fig. 5c, d). These data demonstrate that lithium reduces the amount of AnkG localized in spines, which is consistent with the localization of palmitoylation-deficient AnkG. This result led us to hypothesize that lithium could prevent palmitoylation of AnkG-190 and therefore increase AnkG mobility in spines. To test this, we performed FRAP in neurons overexpressing AnkG-190 and treated with lithium for 24 h (Fig. 5e). Interestingly, lithium treatment increased the mobile fraction of AnkG-190 to ~80% compared with ~60% for controls. Furthermore, this effect was blocked by co-treatment with palmostatin B (Fig. 5f, g), suggesting that lithium may induce depalmitoylation of AnkG-190 rather than preventing its palmitoylation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(a) Lysates of cortical neurons treated with the indicated compounds were subjected to ABE to purify palmitoylated proteins. Levels of palmitoyl-AnkG-190 (top blot) and total AnkG expression in parent lysates (bottom blot) were detected with specific antibodies. Exclusion of NH2OH was used as a control for assay specificity. (b) Bar graph of 190 kDa AnkG isoform palmitoylation normalized with the input and relative to the untreated condition (5 independent experiments, t-test, *p≤0.05, ±SEM). (c) Representative maximal projection of confocal image of endogenous AnkG staining in cortical neurons transfected with GFP with or without lithium stimulation, scale=2 μm. (d) Scatter plot of AnkG mean intensity in mushroom spines on a single plane (26-30 neurons on 3 independent experiments, t-test, ***p≤0.001, ±SEM). (e) Representative time lapse images of GFP-AnkG-190 with lithium chloride +/− palmostatin B in FRAP experiments, scale=2 μm. (f) Quantification of GFP fluorescence in spines over time. Data are fitted with single exponentials (colored lines). Data are represented as mean ±SEM. (g) Scatter plots of mobile fraction (left) or half time recovery (right) of GFP-AnkG-190 in the presence of lithium chloride +/− palmostatin B (n=15-22 neurons, 1-ANOVA with Dunnett’s post-test, ***p≤0.001)
DISCUSSION
In this study, we demonstrate a role for AnkG in the arborization of pyramidal neuronal dendrites and dendritic spine density in vivo. Multiple Ank3 KO mouse models have severe behavioral deficits related to psychiatric disorders (16, 21). Our data support that those phenotypes may, at least in part, depend on alterations of dendrite and spine architecture. Interestingly, reduced dendritic length and spine density have already been described in BD patient brain tissue in post-mortem studies (36) and in iPSCs (37). Together these observations suggest that altered neuronal morphology has an central role in neuropsychiatric diseases (46).We identify the AnkG-190 isoform as being specifically required for the cell-autonomous maintenance of basal dendrite arborization in pyramidal neurons, as the 270 kDa form is unable to rescue arborization deficits caused by knockdown of all AnkG isoforms. This phenotype can be due the specific localization of isoforms since AnkG-190 is enriched in dendrites whereas AnkG-270 is enriched in axon (28). In addition, loss of AnkG also affects interneuron dendrites, but this dependence relies on other AnkG isoforms, indicating the cell-type specificity of AnkG-190.
We show here that AnkG-190 is palmitoylated in brain tissue, and that this modification is important for AnkG-190 subcellular localization to dendrites and spines. Unpalmitoylated AnkG has a higher mobility within spines, indicating that palmitoylation restricts its mobility to specific sites, while depalmitoylation increases the AnkG dynamics. Interestingly, loss of palmitoylation causes a selective impairment to the function of AnkG in dendritic spines; removing its ability to increase spine density, but preserving its ability to increase spine size. In contrast, mutation of AnkG’s β-spectrin-binding site reduces both spine density and size (28). These data suggest that only a subset of AnkG is palmitoylated at C70A and other subpopulations of AnkG-190 exist in spines with distinct roles. We show that AnkG palmitoylation has a dendritic subcompartment-specific role, as it affects only basal but not apical dendrites. This may have implications for pathogenesis of ANK3-related disorders and bipolar disorder, as basal dendrites receive different inputs, integrate distinct signals, and are selectively regulated in physiological, as compared to apical ones (36, 47). Mechanistically, the role of AnkG-190 in dendritic arborization is likely related to its interaction with the microtubule plus-end-binding protein EB3. Dendrite arborization depends on microtubule growth, and AnkG has been shown to interact with EB3 in axon initial segments (41, 42). We found that AnkG knockdown and C70A mutation reduces the movement speed of EB3 puncta, consistent with reduced dendrite arborization. Knockdown of AnkG has been shown to enhance microtubule dynamics in the axon (43), which is supported by our own data, but suggests a differential role for AnkG isoforms in microtubules regulation in axons and dendrites. AnkG palmitoylation may thus increase EB3 mobility in dendrites and promote basal dendrite arborization.
Lithium is commonly used for the treatment of mood disorders and has been shown to rescue mood-related behavioral deficits in different Ank3 KO mouse models (19–21). Studies have shown that lithium can rescue dendritic alterations in BD, suggesting a possible cellular mechanism for lithium action (36, 37). However, the cellular and molecular basis for this rescue is not well understood. In our AnkG knockdown model, we show that lithium rescues dendritic spine and dendrite arborization deficits, supporting the view that lithium acts to restore dendritic complexity in neurons, and that this may be part of its therapeutic effect. Moreover, we demonstrate that the biological process of palmitoylation in general is required for the beneficial effects of lithium on dendritic structure. However, we find that AnkG-190 palmitoylation is only required for specific dendritic phenotypes rescued by lithium, such as the increase in spine density and basal dendrite complexity, but not for restoring spine size. Importantly, we found that lithium treatment leads to a net reduction of AnkG palmitoylation likely causing a more mobile fraction of dendritic AnkG. Based our findings we propose that dynamic palmitoylation/depalmitoylation is necessary for the normal physiological function of AnkG and the lithium-dependent rescue of neuroarchitecture. This is consistent with a report of dynamic cycling of palmitoylation of Cdc42 and other synaptic proteins, where drug-induced activity caused rapid changes in palmitoylation, indicating that cycling may be important for a role in spines (31). In addition to AnkG palmitoylation, other factors not explored here may be required for the lithium-dependent rescue of neuroarchitecture in AnkG-deficient neurons. For instance, palmitoylation of synaptic proteins such as GRIP1 or PSD-95, have been shown to regulate dendritic spines (38, 48) which may occur through a lithium-dependent re-localization (49).
In summary, we present evidence for a novel regulatory mechanism of dendritic architecture controlled by the AnkG-190 isoform which is dependent on its palmitoylation. We provide support for the cellular basis of the therapeutic actions of lithium in the Ank3 KO mouse model, and demonstrate that this process relies on post-transcriptional palmitoylation. Our work describes a new mechanism of mood stabilizer action and its effects on an important psychiatric disorder risk factor ANK3. These findings open new directions for understanding basic control of neuroarchitecture, but may also provide new opportunities for treatment.
CONFLICT OF INTEREST
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
NHP performed and analyzed cellular and biochemical experiments, confocal imaging, SIM experiments, live imaging, Golgi staining imaging, and led the project. FID and SSS performed and analyzed the ABE assays and biochemistry. SY performed conditional knockout mouse breeding, and Golgi staining studies. RG performed biochemistry and generated neuronal cultures. KEH and LED generated and maintained neuronal cultures. JCG and TLP advised on the project. GMT supervised the ABE assays and advised on the project. KRS performed cellular experiments. PP supervised the project. All authors contributed to the writing and/or critical revision of the manuscript.
ACKNOWLEDGMENTS
This work was supported by R01MH107182 to P.P. Imaging work was performed at the Northwestern University Center for Advanced Microscopy, generously supported by NCI CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. We are grateful to members of the Penzes lab for helpful discussions, especially Dr. Marc P. Forrest for his help on the manuscript. Dr. Antonio Sanz-Clemente is gratefully acknowledged for providing access to his lab equipment. All experiments involving animals were performed according to the Institutional Animal Care and Use Committee of NU.
REFERENCES




