

ABSTRACT
The chromosome 3q29 deletion confers >40-fold risk for schizophrenia and related neurodevelopmental disorders. Here, we used quantitative methods to assay Drosophila melanogaster and Xenopus laevis models with knockdown of individual 3q29 homologs in different tissues. We identified developmental and neuronal phenotypes for multiple 3q29 homologs, and specifically observed cellular defects due to altered cell cycle and apoptosis mechanisms. We screened 316 pairwise knockdowns of 3q29 genes in the developing eye, and identified 44 interactions between pairs of 3q29 genes and 34 interactions with other neurodevelopmental genes. In particular, NCBP2 synergistically enhanced the phenotypes of other 3q29 homologs in both Drosophila and X. laevis, leading to significant increases in apoptosis that disrupt cellular organization and brain morphology during development. The NCBP2-driven defects were rescued with overexpression of the apoptosis inhibitor Diap1/XIAP in both models. Our study implicates NCBP2-mediated genetic interactions within apoptosis pathways as a potential mechanism for pathogenicity of the 3q29 deletion.
SIGNIFICANCE Rare copy-number variants, or large deletions and duplications in the genome, are associated with a wide range of neurodevelopmental disorders. For example, the 3q29 deletion confers a >40-fold risk for schizophrenia. To understand the biological mechanisms underlying the pathogenicity of this deletion, we systematically tested 14 individual homologs and 316 pairwise interactions of 3q29 genes for neuronal, cellular, and developmental phenotypes in Drosophila melanogaster and Xenopus laevis models. We found that a key modifier gene, NCBP2, synergistically enhances the neurodevelopmental phenotypes of other 3q29 genes through disruption of apoptosis pathways. This study establishes a novel paradigm for the role of modifier genes within the region towards CNV pathogenicity and provides strong evidence for a mechanistic association between apoptosis and schizophrenia.
INTRODUCTION
Rare copy number variants (CNVs), including deletions and duplications in the human genome, significantly contribute to complex neurodevelopmental disorders such as schizophrenia, intellectual disability/developmental delay, autism, and epilepsy (1, 2). Despite extensive phenotypic heterogeneity associated with recently described CNVs (3), certain rare CNVs have been linked to specific neuropsychiatric diagnoses. For example, the 22q11.2 deletion (DiGeorge/velocardiofacial syndrome), the most frequently occurring pathogenic CNV, is found in about 1-2% of individuals with schizophrenia (4, 5), and animal models of several genes within the region show neuronal and behavioral phenotypes on their own (6, 7). Furthermore, the 1.6 Mbp recurrent deletion on chromosome 3q29, encompassing 21 genes, was initially identified in individuals with a range of neurodevelopmental features, including intellectual disability, microcephaly, autism, craniofacial features, and speech delay, but further studies implicated this deletion as a major risk factor for schizophrenia (8, 9). In fact, a recent meta-analysis estimated that the deletion conferred a greater than 40-fold increase in risk for schizophrenia (10, 11).
Although PAK2, DLG1, and FBXO45 have been proposed as potential candidates within the 3q29 region based on their involvement in neuronal development (12), genetic studies alone have not identified a definitive causative gene (13, 14) or explained the effects of haploinsufficiency of multiple genes within the deletion (15, 16). Furthermore, the functional role of individual 3q29 genes and their interactions towards cellular pathways and mechanisms responsible for the observed neurodevelopmental defects are not completely understood. Therefore, an approach that integrates the function of individual genes within the CNV and their combinatorial effects on neuronal and cellular phenotypes is necessary to understand the pathogenicity of the deletion. Such an approach requires model systems that are amenable for rapid evaluation of developmental and cellular phenotypes, and at the same time allow for testing interactions between pairs of dosage-imbalanced genes without affecting the viability of the organism. Drosophila melanogaster and Xenopus laevis provide such powerful genetic models for studying neurodevelopmental disorders, with the ability to manipulate gene expression in a tissue-specific manner in Drosophila (17) and examine developmental defects in X. laevis (18). For example, Drosophila knockdown models of the candidate schizophrenia gene DTNBP1 showed dysregulation of synaptic homeostasis and altered glutamatergic and dopaminergic neuron function (19, 20), and fly models for UBE3A, the gene associated with Angelman syndrome, showed sleep, memory and locomotor defects (21). Furthermore, X. laevis models have been widely used to identify morphological and neuronal defects associated with developmental disorders (18), such as dendritic connectivity defects with overexpression of MECP2, the causative gene for Rett syndrome (22).
Here, we used a mechanistic approach to understand the role of individual homologs of 3q29 genes and their interactions towards pathogenicity of the deletion. We systematically characterized developmental, cellular, and nervous system phenotypes for 14 conserved homologs of human 3q29 genes and 316 pairwise interactions using Drosophila, and validated these phenotypes using X. laevis. We found that multiple genes in the 3q29 region, including NCBP2, DLG1, FBXO45, PIGZ, and BDH1, contribute to neuronal and developmental defects in both model systems by disrupting apoptosis and cell cycle pathways. The pathogenic effects of each of these genes were synergistically enhanced with concomitant reduced expression of the key modulator gene NCBP2, resulting in increased apoptosis and dysregulation of cell cycle genes leading to more severe cellular and neuronal defects. Our results establish an oligogenic and complex interaction-based model for pathogenicity of the 3q29 deletion, and provide evidence for apoptosis as an underlying mechanism for its associated developmental phenotypes.
RESULTS
Reduced expression of individual 3q29 homologs causes global developmental defects
We used reciprocal BLAST and orthology prediction tools (see Methods) to identify fly homologs for 15 of the 21 genes within the 3q29 deletion region (Figure 1, Table S1). We note that all fly homologs in this manuscript are represented as Humanfly gene names (for example, NCBP2Cbp20). We used RNA interference (RNAi) and the UAS-GAL4 system to knockdown expression levels of 3q29 homologs ubiquitously and in neuronal, wing and eye tissues (23) (Figure 1). Quantitative PCR (qPCR) confirmed knockdown of gene expression to an average of 50% for the tested homologs, mimicking the heterozygous condition of the deletion; fly lines for TCTEX1D2CG5359 did not show knockdown with qPCR and were therefore excluded from further analysis (Table S2). To identify genes essential for organism survival and neurodevelopment, we first assessed the effect of ubiquitous knockdown of 3q29 homologs using the da-GAL4 driver (Figure 2A). Seven of the 14 homologs, including DLG1dlg1, MFI2Tsf2, and NCBP2Cbp20, showed lethality or severe developmental defects with ubiquitous knockdown, suggesting that multiple 3q29 homologs are essential for viability during early development. Similarly, BDH1CG8888, MFI2Tsf2, NCBP2Cbp20 and PAK2Pak showed severe wing defects and DLG1dlg1 showed larval lethality after knockdown using the wing-specific beadexMS1096-GAL4 driver (Figure S1A).
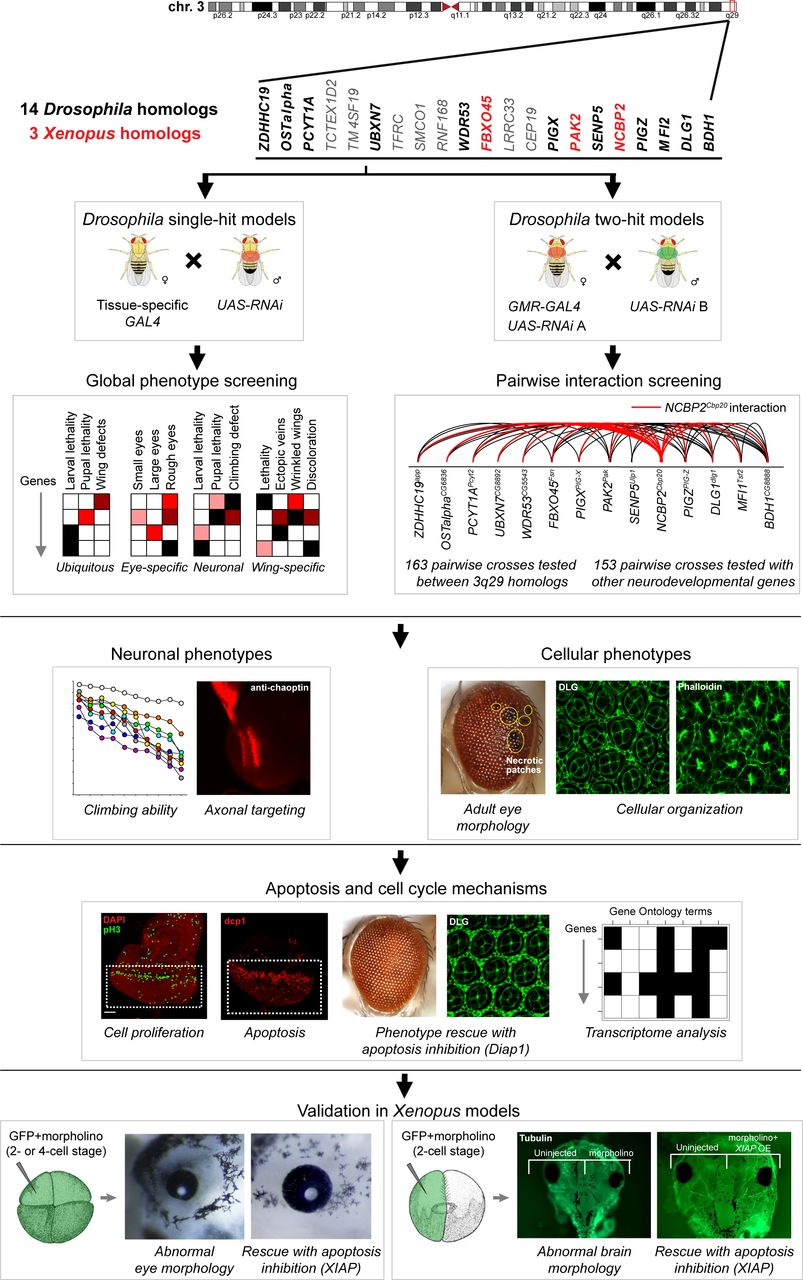
Strategy for identifying cellular phenotypes and genetic interactions of 3q29 homologs. We first knocked down individual or pairs of 14 homologs of 3q29 genes in Drosophila using tissue-specific RNAi. After screening for global phenotypes of single homologs, we tested 316 pairwise interactions using the fly eye system, and found that NCBP2Cbp20 enhanced the phenotypes of other 3q29 homologs and interacted with other neurodevelopmental genes outside of the 3q29 region. Next, we assayed for deeper cellular and neuronal phenotypes of single-hit and two-hit flies, and found cellular defects that identified apoptosis and cell cycle as candidate mechanisms for pathogenicity of the deletion. We confirmed our results by rescuing cellular phenotypes with overexpression of the apoptosis inhibitor Diap1 as well as by analyzing genes differentially expressed with knockdown of 3q29 homologs. Finally, we tested three 3q29 homologs in the Xenopus laevis vertebrate model system by injecting two- or four-cell stage embryos with GFP and 3q29 morpholinos (MOs) to observe abnormal eye morphology, as well as injecting one cell with GFP and 3q29 MOs at the two-cell stage to observe abnormal brain morphology. We found similar developmental defects to those observed in Drosophila, including increased apoptosis that was enhanced with pairwise knockdown of 3q29 homologs and rescued with overexpression of the apoptosis inhibitor XIAP. X. laevis embryo diagrams were produced by Nieuwkoop and Faber (94) and provided by Xenbase (96).

Neurodevelopmental defects in flies with knockdown of individual 3q29 homologs. (A) Percentage of 3q29 homologs with tissue-specific knockdown that manifest lethality or developmental phenotypes. (B) Eight 3q29 homologs with pan-neuronal knockdown showed defects in climbing ability over ten days (two-way ANOVA, p<2.2×10−16, df = 8, F = 33.962). Data represented show mean ± standard deviation of 10 independent groups of 10 flies for each homolog. (C) Representative brightfield adult eye images of single-hit flies with eye-specific GMR-GAL4;UAS-Dicer2 (scale bar = 100 µm) show rough eye phenotypes due to knockdown of 3q29 homologs. The boxplot shows Flynotyper-derived phenotypic scores for eyes with knockdown of 3q29 homologs (n = 10–14, *p < 0.05, one-tailed Mann–Whitney test). (D) Boxplot of adult eye area in 3q29 single-hit flies with GMR-GAL4 (n = 13–16, *p < 0.05, two-tailed Mann–Whitney test). (E) Confocal images of pupal eyes (scale bar = 5 µm) stained with anti-DLG (top) and larval eye discs (scale bar = 30 µm) stained with anti-pH3 (middle) and anti-dcp1 (bottom) illustrate cellular defects posterior to the morphogenetic furrow (white box) upon knockdown of select 3q29 homologs. Yellow circles in DLG images indicate cone cell defects, white circles indicate bristle cell defects, yellow arrows indicate rotation defects, and yellow arrowheads indicate secondary cell defects. (F) Boxplot of pH3-positive cells in larval eye discs of 3q29 knockdown flies (n = 9–12, *p < 0.05, two-tailed Mann–Whitney test). (G) Boxplot of dcp1-positive cells in larval eye discs of 3q29 knockdown flies (n = 11–12, *p < 0.05, two-tailed Mann–Whitney test). All boxplots indicate median (center line), 25th and 75th percentiles (bounds of box), and minimum and maximum (whiskers).
We next tested for defects in neuronal function, including climbing assays for motor defects and staining of larval brains for axonal targeting, with knockdown of 3q29 homologs. Interestingly, Elav-GAL4 mediated pan-neuronal knockdown caused partial larval or pupal lethality in DLG1dlg1, MFI2Tsf2 and WDR53CG5543 flies (Figure 2A), and about 30% of adult flies with knockdown of DLG1dlg1 did not survive beyond day 5 (Figure S1B), indicating an essential role for these genes in neuronal development. Furthermore, we found that flies with pan-neuronal knockdown of several 3q29 homologs, including DLG1dlg1 and NCBP2Cbp20, exhibited a strong reduction in climbing ability over ten days (Figure 2B, Video S1), suggesting that these genes could contribute to abnormalities in synaptic and motor functions (24). We next examined the axonal projections of photoreceptor cells into the optic lobe by staining third instar larval brains with anti-chaoptin. We found that GMR-GAL4 mediated eye-specific knockdown of NCBP2Cbp20, DLG1dlg1, FBXO45Fsn and PAK2Pak showed several axonal targeting defects (Figure S1C) similar to those identified in models of other schizophrenia-associated genes, such as DISC1 on chromosome 1q24.2 and ZDHHC8, located within the 22q11.2 CNV region (7, 25). Overall, our data show that multiple 3q29 homologs are important for Drosophila neurodevelopment, suggesting an oligogenic model for pathogenicity of the deletion.
Drosophila eye models of 3q29 homologs show cellular defects
The Drosophila compound eye has been classically used for performing high-throughput genetic screens and quantitative assays of neurodevelopmental defects (26). In fact, about two-thirds of all vital genes in the fly genome are predicted to be involved in fly eye development (27). For instance, the Drosophila eye model was recently used to screen a large set of intellectual disability genes (28), and genetic interaction studies using the fly eye have identified modifier genes for Rett syndrome, spinocerebellar ataxia type 3, and other conserved developmental processes (29–31). We used the developing fly eye as an in vivo system to quantify the effect of gene knockdown on adult eye morphology, cellular organization in the pupal eye, and cell proliferation and death in the larval imaginal eye disc. The wild-type adult Drosophila eye consists of about 750 ommatidia containing different cell types arranged in a regular hexagonal structure, which can be easily perturbed by genetic modifications (32, 33) (Figure S2). Because of this, we first performed eye-specific knockdown of 3q29 homologs using GMR-GAL4 and measured the rough eye phenotype of each homolog using Flynotyper, a quantitative tool that calculates a phenotypic score based on defects in ommatidial arrangement (34). We found that 8 out of 13 homologs showed significant external eye phenotypes compared with control flies, while eye-specific knockdown of MFI2Tsf2 caused lethality (Figure 2C, Figure S3). For example, knockdown of NCBP2Cbp20 resulted in a severe rough eye phenotype that was comparable to knockdown of other neurodevelopmental genes, such as SHANK3Prosap and CHD8kis (34).
To examine the cellular mechanisms underlying the rough eye phenotypes observed with knockdown of 3q29 homologs, we first measured changes in area and ommatidial size of the adult eyes. We found a significant reduction in eye size with knockdown of BDH1CG8888 and NCBP2Cbp20, while the eyes of DLG1dlg1 and PIGZPIG-Z flies were significantly larger than controls (Figure 2D). Similarly, we observed increases in ommatidial diameter for DLG1dlg1 flies and decreases in diameter for NCBP2Cbp20 and BDH1CG8888 flies, suggesting that these genes also contribute to abnormal cell growth phenotypes (Figure S3B). We also assessed the cellular structure of 44 hour-old pupal eyes by staining the ommatidial and photoreceptor cells with anti-DLG, a septate junction marker, and Phalloidin, a marker for F-actin at cell boundaries (Figure S2). We found that knockdown of 11 out of 12 tested 3q29 homologs caused disorganization or loss of the photoreceptor neurons and ommatidial cells (Figure 2E, Figure S4A-B, Table S3). For example, knockdown of BDH1CG8888, DLG1dlg1, NCBP2Cbp20 and WDR53CG5543 all showed defects in cone cell orientation and ommatidial rotation compared with control flies. Furthermore, NCBP2Cbp20 and DLG1dlg1 knockdown flies showed hexagonal defects and severe disorganization of photoreceptor neurons, while NCBP2Cbp20 flies also showed fused secondary cells and DLG1dlg1 flies had a complete loss of bristle cells.
We next hypothesized that abnormal proliferation and apoptosis may contribute to the observed cellular defects of 3q29 homologs. To test this, we stained the third instar larval eye discs of select single-hit knockdowns with anti-pH3 (phospho-Histone H3 (Ser10)) and Drosophila caspase-1 (dcp1), markers for proliferating and apoptotic cells, and quantified the number of cells posterior to the morphogenetic furrow (Figure S2). We observed a significant decrease in pH3-positive cells for BDH1CG8888 knockdown flies and trends towards increased pH3-positive cells for PIGZPIG-Z and DLG1dlg1 flies (Figure 2E-F, Figure S4C), while DLG1dlg1 also showed significant increases in cells stained with bromodeoxyuridine (BrdU), a marker for replicating cells (Figure S4D-E). Both NCBP2Cbp20 and DLG1dlg1 flies also showed a significant increase in dcp1-positive cells compared with controls (Figure 2G), and we validated increases in apoptosis for these lines using TUNEL assays (Figure S4F). We further tested for proliferation and apoptosis in the third instar larval wing discs of flies with knockdown of 3q29 homologs, and found significant changes in both processes for DLG1dlg1, BDH1CG8888 and NCBP2Cbp20 flies (Figure S5). Knockdown of NCBP2Cbp20 in particular showed dcp1-positive staining across the entire wing pouch in the larval wing disc. These data suggest that multiple 3q29 homologs are involved in apoptosis and proliferation during early development, leading to defects in cell count and organization with reduced expression (Table 1).
Summary of phenotypes for select 3q29 knockdown experiments show widespread defects due to disruption of apoptosis and cell cycle functions.
Interactions between 3q29 genes enhance neurodevelopmental phenotypes
As knockdown fly models of multiple 3q29 homologs showed a variety of neuronal, developmental, and cellular defects, we next hypothesized that interactions between multiple genes in the region could contribute to the neurodevelopmental phenotypes of the entire deletion. We therefore generated GMR-GAL4 recombinant lines for nine 3q29 homologs, crossed these lines with multiple RNAi or mutant lines for other 3q29 homologs to generate 94 pairwise knockdowns with 163 two-hit crosses, and assessed changes in severity of eye phenotypes compared with single-hits using Flynotyper (Figure 1, Table S4). We found a significant enhancement in phenotypic severity for 44 two-hit models, which were validated with a second line when available, compared with single-gene knockdowns (Figure 3A, Figure S6-7). In fact, we found that 20 out of 21 pairwise interactions involving NCBP2Cbp20 as either a first or second-hit gene resulted in more severe eye phenotypes, suggesting that reduced expression of NCBP2Cbp20 drastically modifies the neurodevelopmental phenotypes of other 3q29 genes (Figure 3B-D). For further validation, we also compared pairs of reciprocal crosses (i.e. FBXO45Fsn/BDH1CG8888 versus BDH1CG8888/FBXO45Fsn) and confirmed concordant results for 21 out of 30 reciprocal interactions, including all 16 reciprocal interactions involving NCBP2Cbp20 (Table S4). As enhancements of the rough eye phenotype could be due to either additive effects or synergistic interactions between pairs of genes (35), we further compared Flynotyper scores of 31 two-hit crosses that showed enhancement with those of the individual recombined lines (Table S4). We found that 14 out of 16 two-hit crosses involving NCBP2Cbp20 were significantly more severe than the additive effects of the individual single-hit crosses, suggesting that NCBP2Cbp20 synergistically enhances the neurodevelopmental phenotypes of other 3q29 genes (Figure S8). Five other two-hit crosses also synergistically enhanced the single-hit phenotypes, including DLG1dlg1/OSTalphaCG6836 and ZDHHC19app/FBXO45Fsn, while 10 pairwise crosses that showed enhancement of the single-hit phenotype were additive in nature. We also found a non-significant increase in severity for DLG1dlg1/PAK2Pak two-hit flies using both RNAi and mutant lines, concordant with the enhanced neuromuscular synapse and circadian rhythm defects observed in mutant DLG1dlg1/PAK2Pak two-hit flies described by Grice and colleagues (36).

Screening for pairwise interactions of 3q29 homologs in the Drosophila eye and nervous system. (A) Heatmap showing average changes in phenotypic scores for pairwise interactions of 3q29 homologs in the adult eye, compared with single-hit recombined lines. Gray boxes indicate crosses without available data. Boxplots of phenotypic scores for pairwise knockdown of (B) NCBP2Cbp20 and (C) DLG1dlg1 with other 3q29 homologs are shown (n = 5–14, *p < 0.05, two-tailed Mann–Whitney test). Green arrows indicate a pair of reciprocal lines showing enhanced phenotypes compared with their respective single-hit controls. (D) Representative brightfield adult eye images of flies with pairwise knockdown of 3q29 homologs (scale bar = 100 µm) show enhancement (Enh.) of rough eye phenotypes compared with single-hit recombined lines. (E) Representative confocal images of larval eye discs stained with anti-chaoptin (scale bar = 30 µm) illustrate enhanced defects in axonal targeting from the retina to the optic lobes of the brain with eye-specific knockdown of NCBP2Cbp20/DLG1dlg1 and NCBP2Cbp20/FBXO45Fsn compared with NCBP2Cbp20 knockdown. (F) Flies with pan-neuronal pairwise knockdown of 3q29 homologs showed enhanced defects in climbing ability over ten days (two-way ANOVA, p<5.88×10−4, df = 2, F = 7.630) compared with NCBP2Cbp20 knockdown. Data represented show mean ± standard deviation of 10 independent groups of 10 flies for each line tested. All boxplots indicate median (center line), 25th and 75th percentiles (bounds of box), and minimum and maximum (whiskers).
As NCBP2Cbp20 knockdown enhanced the rough eye phenotypes of multiple 3q29 homologs, we next tested for enhancement of other neuronal defects among two-hit flies with knockdown of NCBP2Cbp20. We found that the simultaneous knockdown of NCBP2Cbp20 with DLG1dlg1 or FBXO45Fsn led to an increase in severity of axon targeting defects (Figure 3E). For instance, while only the R7 and R8 axons failed to project into the medulla with knockdown of NCBP2Cbp20, nearly all axons failed to project in NCBP2Cbp20/DLG1dlg1 and NCBP2Cbp20/FBXO45Fsn knockdown flies. We also tested pan-neuronal knockdown of select pairs of 3q29 homologs, and found that both NCBP2Cbp20/DLG1dlg1 and NCBP2Cbp20/ FBXO45Fsn significantly enhanced the severity of climbing defects observed with knockdown of NCBP2Cbp20 (Figure 3F, Video S2). Overall, these data suggest that NCBP2Cbp20 additively or synergistically interacts with other 3q29 genes to enhance the observed cellular and neuronal phenotypes, potentially accounting for the major features of the deletion (Table 1).
To further characterize the functional effects of 3q29 gene interactions, we analyzed changes in gene expression by performing RNA-sequencing of heads from select single-hit (NCBP2Cbp20, DLG1dlg1, FBXO45Fsn, PAK2Pak) and two-hit (NCBP2Cbp20/DLG1dlg1, NCBP2Cbp20/FBXO45Fsn) flies with pan-neuronal knockdown of 3q29 homologs. We identified differentially-expressed genes in each of the single and two-hit fly models compared with controls, and performed enrichment analysis after converting the gene sets to their human homologs (Table S5). We found that each of the single-hit flies showed enrichment for dysregulation of basic cellular and developmental processes (Figure S9A). For example, DLG1dlg1 and NCBP2Cbp20 flies showed enrichment for dysregulation of synaptic transmission genes, including NLGN1 and HTR3A. DLG1dlg1 flies were also enriched for differential expression of ion transport genes, while all four single-hit knockdowns were enriched for disruption of genes related to metabolism and cellular respiration functions. While the dysregulated genes in NCBP2Cbp20/DLG1dlg1 flies only showed enrichments for the same functions observed in the single-hit flies, NCBP2Cbp20/FBXO45Fsn flies were uniquely enriched for dysregulated cell cycle function genes, including AURKB, CDK1, CHEK2 and RB1 (Figure S9B-C). We similarly found 17 differentially-expressed apoptosis genes in NCBP2Cbp20/FBXO45Fsn flies, including the DNA fragmentation protein ENDOG (37) and the apoptosis signaling proteins RET and HSPA1A (38). Furthermore, we found a strong enrichment for genes preferentially expressed in early and mid-fetal brain tissues among genes differentially expressed in NCBP2Cbp20/FBXO45Fsn flies (Figure S9D). These data provide further evidence that NCBP2Cbp20 synergistically interacts with other 3q29 homologs to disrupt neuronal phenotypes through apoptosis and cell cycle pathways (Table 1).
Interactions between NCBP2Cbp20 and 3q29 homologs enhance apoptosis defects
Cell death and proliferation are two antagonistic forces that maintain an appropriate number of neurons during development (39). While cell cycle defects have been previously linked to multiple neurodevelopmental disorders (40–42), apoptosis in particular has been proposed as a candidate mechanism for schizophrenia (43, 44). For example, increased apoptosis has been observed among schizophrenia patient-derived cell lines (45, 46), and abnormal apoptosis has been proposed as a candidate mechanism for decreased brain volume (47) and synaptic pruning defects associated with schizophrenia (48). As the larval eye and wing discs of NCBP2Cbp20 flies showed strong increases in apoptosis, we hypothesized that apoptosis pathways could mediate the synergistic interactions observed between NCBP2Cbp20 and other 3q29 homologs. For example, we observed black necrotic patches on the ommatidia in NCBP2Cbp20/DLG1dlg1 and NCBP2Cbp20/FBXO45Fsn adult eyes, indicating an increase in cell death with these interactions (Figure 4A, Figure S10A). In fact, significantly larger regions of necrotic patches were observed in flies with concomitant homozygous knockdown of NCBP2Cbp20 and heterozygous knockdown of DLG1dlg1 (i.e. NCBP2Cbp20/NCBP2Cbp20; DLG1dlg1), suggesting that the knockdown of both genes contributes to ommatidial cell death (Figure 4A). Furthermore, we found an enhanced disruption of ommatidial cell organization and loss of photoreceptors in pupal flies with concomitant knockdown of NCBP2Cbp20 and DLG1dlg1, FBXO45Fsn or BDH1CG8888, emphasizing the role of these genes in maintaining cell count and organization (Figure 4B-C, Figure S10B, Table S6). Based on these observations, we assayed for apoptotic cells in the larval eye discs of two-hit knockdown flies involving NCBP2Cbp20 to determine whether the observed cellular defects were due to increased apoptosis. We observed significant increases in the number of apoptotic cells, as measured by dcp1 (Figure 4D-E) and TUNEL staining (Figure S10C-D), when NCBP2Cbp20 was knocked down with BDH1CG8888, DLG1dlg1, or FBXO45Fsn. NCBP2Cbp20/ BDH1CG8888 flies also showed a decreased number of pH3-positive cells, suggesting that both apoptosis and proliferation could mediate the interaction between these two genes (Figure 4F).
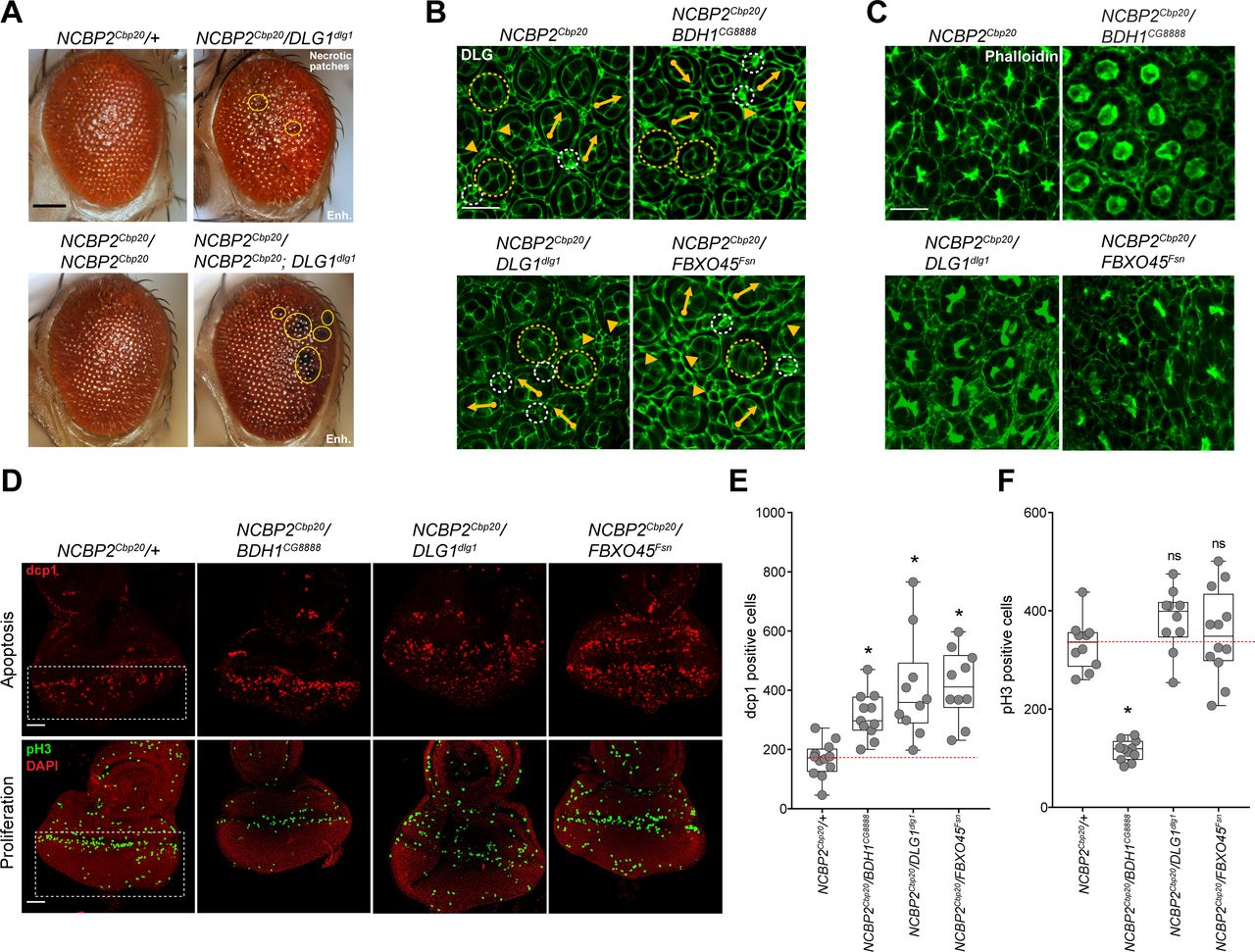
Cellular phenotypes with pairwise knockdown of 3q29 homologs. (A) Representative brightfield adult eye images (scale bar = 100 µm) show that knockdown of DLG1dlg1 enhanced the rough eye phenotype and necrotic patches (yellow circles) of both heterozygous and homozygous knockdowns of NCBP2Cbp20. (B) Representative confocal images of pupal eyes (scale bar = 5 µm) stained with anti-DLG illustrate enhanced defects in ommatidial organization upon concomitant knockdown of NCBP2Cbp20 with other 3q29 homologs compared with NCBP2Cbp20 knockdown. (C) Representative confocal images of pupal eyes (scale bar = 5 µm) stained with Phalloidin illustrate enhanced defects in photoreceptor cell count and organization upon concomitant knockdown of NCBP2Cbp20 and other 3q29 homologs compared with NCBP2Cbp20 knockdown. (D) Representative confocal images of larval eye discs (scale bar = 30 µm) stained with anti-dcp1 (top) and anti-pH3 (bottom) show enhanced defects in apoptosis and cell proliferation with pairwise knockdown of NCBP2Cbp20 and other 3q29 homologs compared with NCBP2Cbp20 knockdown. (E) Boxplot of dcp1-positive cells in the larval eye discs of 3q29 two-hit knockdown flies (n = 10–11, *p < 0.05, two-tailed Mann–Whitney test). (F) Boxplot of pH3-positive cells in the larval eye discs of 3q29 two-hit knockdown flies (n = 10–12, *p < 0.05, two-tailed Mann–Whitney test). All boxplots indicate median (center line), 25th and 75th percentiles (bounds of box), and minimum and maximum (whiskers).
To validate apoptosis as a mechanism for the cellular defects of flies with knockdown of 3q29 homologs, we crossed recombinant lines of NCBP2Cbp20 and DLG1dlg1 with flies overexpressing Diap1 (death-associated inhibitor of apoptosis). Diap1 is an E3 ubiquitin ligase that targets Dronc, the fly homolog of caspase-9, and prevents the subsequent activation of downstream caspases that lead to apoptosis (49). We found that overexpression of Diap1 rescued the adult rough eye phenotypes (Figure 5A-B) and increased the eye sizes of NCBP2Cbp20 and DLG1dlg1 flies (Figure S11A-B). These observations were corroborated by the reversal of cellular changes in the eye, including the rescue of ommatidial structure and cell count deficits observed in NCBP2Cbp20 and DLG1dlg1 flies with Diap1 overexpression (Figure S11C-D). Furthermore, overexpression of Diap1 led to significant reductions in the number of TUNEL and dcp1-positive cells in the larval eye discs of NCBP2Cbp20 and DLG1dlg1 flies, confirming the rescue of apoptosis defects in these flies (Figure 5E-F, Figure S11E-F). When we crossed NCBP2Cbp20/DLG1dlg1 and NCBP2Cbp20/FBXO45Fsn two-hit flies with flies overexpressing Diap1, we observed a similar rescue of the eye phenotypes, including the elimination of necrotic patches (Figure 5C-D) and a concomitant decrease in apoptotic cells (Figure 5G-H). Interestingly, Diap1 overexpression also corrected the photoreceptor axon targeting defects of NCBP2Cbp20 knockdown flies (Figure S11H), suggesting that the neuronal defects observed in these flies could be attributed to increased apoptosis. We further confirmed these mechanistic findings by observing increased severity in cellular phenotypes upon overexpression of Dronc in NCBP2Cbp20 and DLG1dlg1 knockdown flies. For example, we observed black necrotic patches (Figure 5A) and exaggerated apoptotic responses (Figure 5E-F, Figure S11F-G) in NCBP2Cbp20 knockdown flies with overexpression of Dronc. These results indicate that NCBP2Cbp20-mediated apoptosis is a primary driver of the cellular defects observed in both single and two-hit flies, suggesting that apoptosis is a candidate mechanism for pathogenicity of the deletion.

Rescue of cellular phenotypes of 3q29 knockdown flies with overexpression of the apoptosis inhibitor Diap1. (A) Representative brightfield adult eye images (scale bar = 100 µm) show suppression (Supp.) of rough eye phenotypes for NCBP2Cbp20 and DLG1dlg1 knockdown flies with overexpression of Diap1, as well as enhanced (Enh.) phenotypes with overexpression of caspase-9 homolog Dronc. (B) Boxplot of phenotypic scores for single-hit 3q29 knockdown flies with overexpression of Diap1 and Dronc (n = 8–9, *p < 0.05, two-tailed Mann–Whitney test). (C) Representative brightfield adult eye images (scale bar = 100 µm) show suppression (Supp.) of rough eye phenotypes and necrotic patches in NCBP2Cbp20/DLG1dlg1 and NCBP2Cbp20/FBXO45Fsn flies with Diap1 overexpression. (D) Boxplot of phenotypic scores in two-hit knockdown flies with overexpression of Diap1 (n = 5–6, *p < 0.05, two-tailed Mann–Whitney test). (E) Larval eye discs (scale bar = 30 µm) stained with anti-dcp1 show rescue of apoptosis phenotypes observed in NCBP2Cbp20 and DLG1dlg1 knockdown flies with Diap1 overexpression as well as enhanced phenotypes with Dronc overexpression. (F) Boxplot of dcp1-positive cells in the larval eye discs of 3q29 single-hit knockdown flies with Diap1 and Dronc overexpression (n = 9–18, *p < 0.05, two-tailed Mann–Whitney test). (G) Larval eye discs (scale bar = 30 µm) stained with anti-dcp1 show rescue of apoptosis phenotypes observed in two-hit knockdown flies with Diap1 overexpression. (H) Boxplot of dcp1-positive cells in the larval eye discs of 3q29 two-hit knockdown flies with Diap1 overexpression (n = 6–10, *p < 0.05, two-tailed Mann–Whitney test). All boxplots indicate median (center line), 25th and 75th percentiles (bounds of box), and minimum and maximum (whiskers).
3q29 genes interact with canonical neurodevelopmental genes
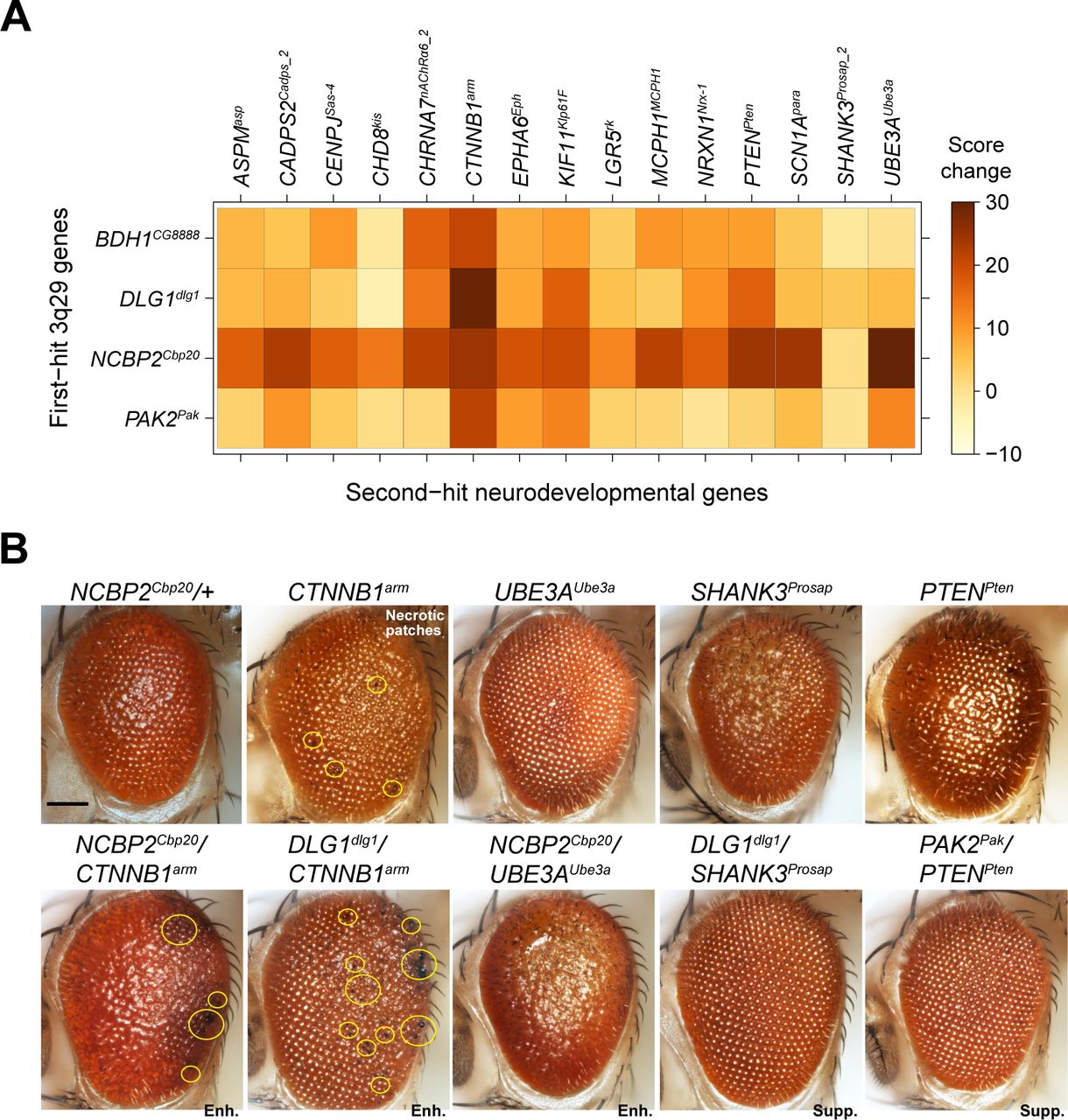
We further explored the role of 3q29 genes in neurodevelopmental pathways by screening four homologs with strong neurodevelopmental phenotypes (NCBP2Cbp20, DLG1dlg1, BDH1CG8888 and PAK2Pak) for interactions with 15 known neurodevelopmental genes, for a total of 60 pairwise interactions and 153 tested two-hit crosses (Figure 6A). We selected these neurodevelopmental genes for screening based on their association with developmental disorders in humans (34, 50), and included eight genes associated with apoptosis or cell cycle functions as well as four genes associated with microcephaly (51), a key phenotype observed in approximately 50% of 3q29 deletion carriers (8). We found that 34 pairwise interactions, validated with a second line when available, led to significant increases in eye phenotypes compared with single hits (Figure S12, Table S7). These interactions included 19 validated interactions of 3q29 genes with apoptosis or cell cycle genes as well as 10 interactions with microcephaly genes. We found that knockdown of NCBP2Cbp20 enhanced the phenotypes of 13 out of 15 neurodevelopmental genes, including all four microcephaly genes. In fact, four neurodevelopmental genes enhanced by NCBP2Cbp20 (SCN1Apara, UBE3AUbe3A, CADPS2Cadps and PTENPten) showed evidence of synergistic interactions (Figure S8), and knockdown of NCBP2Cbp20 also enhanced the ommatidial necrotic patches observed with knockdown of CTNNB1arm (Figure 6B). Interestingly, we also found that knockdown of BDH1CG8888 and DLG1dlg1 suppressed the rough eye phenotypes of SHANK3Prosap, while knockdown of PAK2Pak suppressed the phenotypes of both SHANK3Prosap and PTENPten (Figure 6B, Figure S8). Several of these interactions have been previously observed to modulate neuronal function in model systems. For example, SHANK3 interacts with DLG1 through the mediator protein DLGAP1 to influence post-synaptic density in mice (52) and binds to proteins in the Rac1 complex, including PAK2, to regulate synaptic structure (53, 54). These results suggest that 3q29 homologs interact with key developmental genes to modify neuronal phenotypes, providing a potential mechanism for variants outside of the 3q29 region to modulate the variably expressive CNV phenotypes (16).
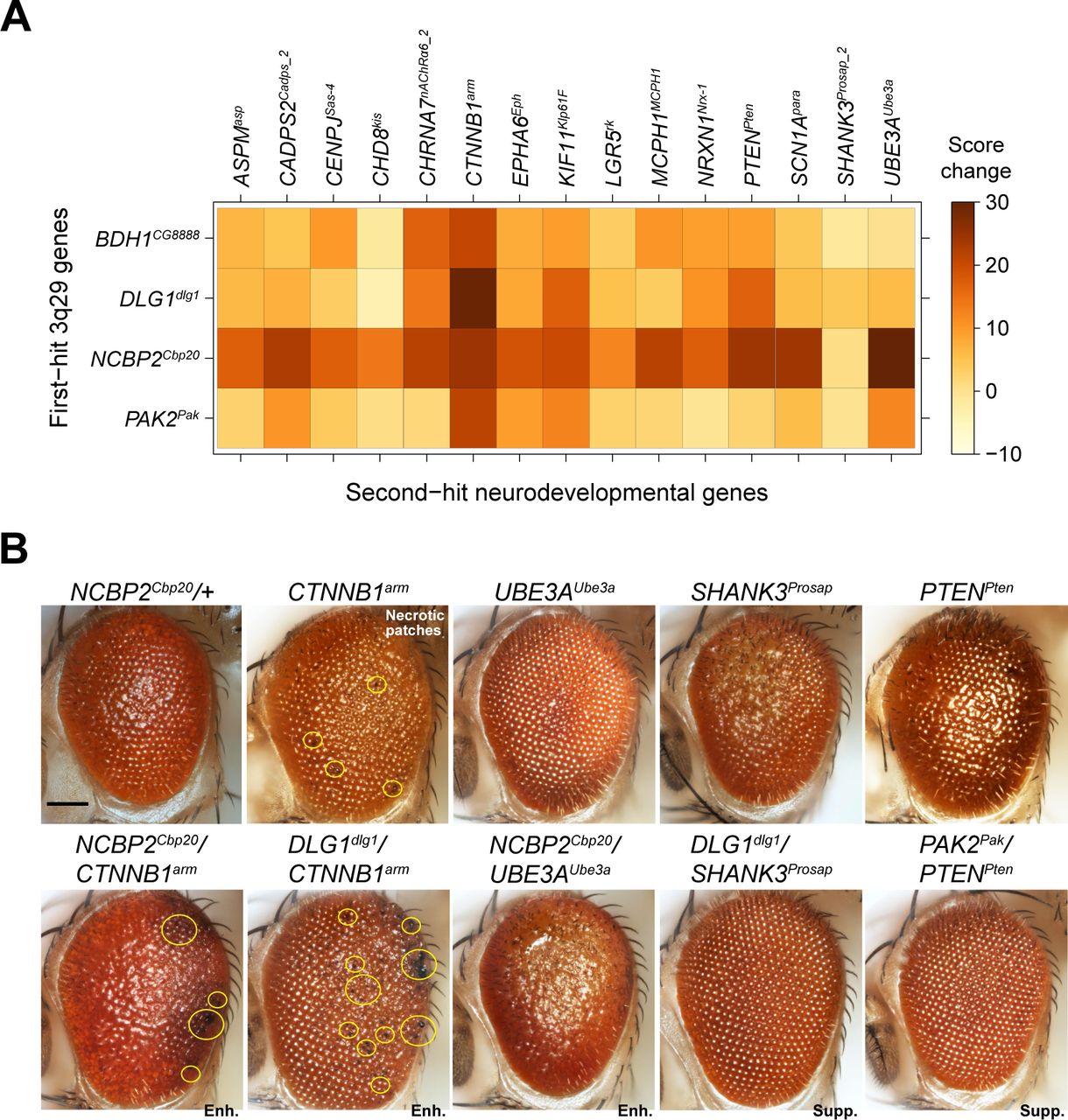
Interactions between 3q29 homologs and other neurodevelopmental genes. (A) Heatmap showing the average changes in phenotypic scores for the pairwise knockdown of 3q29 homologs in the adult eye and other neurodevelopmental genes, compared with single-hit recombined lines. (B) Representative brightfield adult eye images of flies with pairwise knockdown of 3q29 homologs and other neurodevelopmental genes (scale bar = 100 µm) show enhancement (Enh.) or suppression (Supp.) of rough eye phenotypes and necrotic patches compared with the single-hit genes.
Reduction of 3q29 gene expression causes developmental defects in Xenopus laevis
After identifying a wide range of neurodevelopmental defects due to knockdown of 3q29 fly homologs, we sought to gain further insight into the conserved functions of these genes in vertebrate embryonic brain development using the Xenopus laevis model system. We examined the effect of targeted knockdown of NCBP2, FBXO45, and PAK2, as these genes displayed multiple severe phenotypes with reduced gene expression in flies. Knockdown of each gene was accomplished using antisense morpholino oligonucleotides (MOs) targeted to early splice sites of each gene (Figure 1). X. laevis embryos were injected at either the two- or four-cell stage with various concentrations of each 3q29 gene MO or a standard control MO, and were validated using RT-PCR (Figure S13A-B). As reduction of NCBP2Cbp20, FBXO45Fsn, and PAK2Pak each resulted in neuronal defects in Drosophila, we first examined the effects of knockdown of these genes on X. laevis brain development at stage 47. To test this, we knocked down each gene in half of the embryo at the two-cell stage, and left the other half uninjected to create a side-by-side comparison of brain morphology (Figure 7A). We performed whole-mount immunostaining with anti-alpha tubulin and found that reduction of NCBP2, FBXO45, and PAK2 each resulted in smaller forebrain and midbrain size compared with controls (Figures 7A-C). We also found that simultaneous knockdown of NCBP2 with FBXO45 caused a significant decrease in forebrain size and a trend towards decreased midbrain size compared with NCBP2 knockdown (Figure 7A-C). Knockdown of PAK2 with NCBP2 showed a similar trend towards decreased forebrain size. We further examined the effect of knocking down 3q29 homologs on X. laevis eye development at stage 42, and found that knockdown of these genes caused irregular shapes and decreased size compared with controls (Figure S14A-B). The reductions in eye size were rescued to control levels when mRNA was co-injected along with MO for each gene (Figure S14C). Together, these data show that individual and pairwise knockdown of 3q29 genes in X. laevis leads to abnormal brain and eye morphology, confirming the conserved role of these genes during vertebrate development.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Developmental phenotypes observed with knockdown of 3q29 homologs in X. laevis models. (A) To study brain morphology upon knockdown of 3q29 homologs, one cell in a two-cell embryo was injected with MO for the 3q29 homolog(s) while the other cell remained uninjected. Representative images of stage 47 X. laevis tadpoles with MO knockdown of NCBP2 (scale bar = 500 µm) show morphological defects and decreased size compared with control tadpoles. Pairwise knockdown of FBXO45 and NCBP2 enhanced these phenotypes, which were also rescued with overexpression of XIAP. (B) Box plot of forebrain area in X. laevis models with knockdown of 3q29 homologs, normalized to controls (n = 30–63, *p < 0.05, two-tailed Welch’s T-test). (C) Box plot of midbrain area in X. laevis models with knockdown of 3q29 homologs, normalized to controls (n = 30–63, *p < 0.05, two-tailed Welch’s T-test). (D) Western blot analysis of X. laevis whole embryos show increased levels of cleaved caspase-3 with knockdown of 3q29 homologs, including enhanced caspase-3 levels with two-hit knockdown of 3q29 homologs and rescued levels with XIAP overexpression. β-actin was used as a loading control on the same blot. Western blot images shown are cropped; the full blots are provided in Figure S13C. (E) Quantification of Western blot band intensity for caspase-3 levels, normalized to the loading control. All boxplots indicate median (center line), 25th and 75th percentiles (bounds of box), and minimum and maximum (whiskers). X. laevis embryo diagrams were produced by Nieuwkoop and Farber (94) and provided by Xenbase (96).
To determine if the knockdown of 3q29 homologs also disrupted apoptotic processes in X. laevis, we tested whether overexpression of the X-linked inhibitor of apoptosis gene (XIAP) could rescue the observed developmental defects. We found that overexpression of XIAP rescued the midbrain and forebrain size deficits observed with NCBP2 knockdown to control levels (Figure 7A-C). Similarly, we found that the decreased eye sizes and morphological defects observed with knockdown of NCBP2 were rescued with XIAP1 overexpression (Figure S14A-B). To further validate these findings, we performed a Western blot following knockdown of FBXO45 and NCBP2 using anti-cleaved caspase-3 (Asp175) as a marker for apoptosis (Figure 7D, Figure S13C). We found that reduction of FBXO45 and NCBP2 expression each led to an increase in cleaved caspase-3 levels compared with controls, which were restored to control levels with concomitant overexpression of XIAP (Figure 7E). Caspase-3 levels were also enhanced when FBXO45 and NCBP2 were knocked down together (Figure 7E), suggesting that these two genes both contribute to developmental phenotypes by increasing apoptosis. Overall, these results suggest a role for apoptosis towards the developmental phenotypes observed with knockdown of 3q29 homologs in a vertebrate model, confirming that these findings are not specific to Drosophila (Table 1).
DISCUSSION
Using complementary Drosophila and X. laevis models, we analyzed the molecular functions and genetic interactions of 3q29 homologs towards neurodevelopmental phenotypes. While we did not examine the entire deletion in these model systems, assaying individual 3q29 genes and their interactions allowed us to identify candidate genes and cellular mechanisms potentially responsible for pathogenicity of the deletion. We found that multiple genes within the 3q29 region contribute to the observed neurodevelopmental defects, and that apoptosis pathways play a large role in mediating interactions among 3q29 homologs to determine the ultimate phenotypic trajectory of the CNV. Several themes emerged from our study that exemplify the genetic and mechanistic complexity of the 3q29 deletion, potentially leading to a better biological understanding of the disorder for its diagnosis and treatment.
First, our analysis of developmental phenotypes upon individual and pairwise knockdowns of 3q29 homologs showed that a single gene within the region is not likely to be solely responsible for the clinical features of the deletion. In fact, we found that 12 out of 14 homologs showed developmental defects in Drosophila on their own, while every homolog showed an enhanced rough eye phenotype when knocked down along with at least one other homolog. These data provide evidence towards a multi-genic model of pathogenicity for the deletion, suggesting that complex interactions among the genes in the region are responsible for the observed developmental defects (16). Furthermore, we identified 34 interactions between 3q29 genes and canonical neurodevelopmental genes, suggesting that variants in genes outside of the CNV region could modulate the pathogenicity of the entire CNV. While our study is limited to examining conserved cellular phenotypes of 3q29 homologs in Drosophila and X. laevis, evidence from other model organisms also supports an oligogenic model for the deletion. For example, a recent study found that a mouse model of the entire 3q29 deletion showed several behavioral defects, including decreased social interaction and increased startle response, that were not observed in mice with knockdown of Dlg1+/− on its own (15). In fact, knockout mouse models for several 3q29 genes have exhibited developmental phenotypes, including axonal and synaptic defects with knockout of Fbxo45−/− and embryonic lethality in Pak2−/− and Pcyt1a−/− mice (55–57) (Table S8). Furthermore, several 3q29 genes including PAK2, DLG1, PCYT1A, and UBXN7 are under evolutionary constraint in humans based on gene pathogenicity metrics (Table S8). These data, combined with our findings in Drosophila and X. laevis, implicate multiple genes towards the pathogenicity caused by deletion of the entire 3q29 region.
Second, our screening of interactions between pairs of Drosophila 3q29 homologs identified 44 interactions with enhanced rough eye phenotypes compared with single-hit flies. Strikingly, NCBP2Cbp20 enhanced the phenotypes of 11 out of 12 other 3q29 homologs, suggesting that NCBP2 is a key modulator of the deletion phenotype. In fact, several two-hit flies involving NBCP2Cbp20 knockdown were more severe than the additive effects of the individual 3q29 homologs. While additive interactions are expected with the knockdown of any two genes affecting the same phenotype, these synergistic interactions indicate that NCBP2Cbp20 enhances the effects of other genes through involvement in common functional pathways (35). Previous studies of the 3q29 region have focused on other genes as candidates for neurodevelopmental features based on biological function and sequencing studies (58). For example, DLG1 organizes the synaptic structure at neuromuscular junctions (59) and is enriched for mutations among individuals with schizophrenia (13). In contrast, NCBP2 encodes a subunit of the nuclear cap-binding complex (CBC), which binds to the 5’ end of mRNA and microRNA in the nucleus (60). Given the role of the CBC in post-transcriptional regulatory mechanisms such as nonsense-mediated decay, alternative splicing and mRNA transport (61, 62), it is possible that disruption of this complex could result in changes to a broad set of genes and biological processes. In fact, our analysis of differentially-expressed genes in NCBP2Cbp20 knockdown flies showed disruption of synaptic transmission, cellular respiration and several metabolic pathways. Interestingly, NBCP2 is not predicted to be pathogenic on its own in humans (Table S8) and has not been identified with deleterious mutations in sequencing studies of neurodevelopmental disease cohorts so far, indicating its potential role as a modifier of the other candidate genes in the region. Furthermore, our results complement previous reports of synergistic interactions among 3q29 homologs in the nervous system (36), representing another hallmark of an oligogenic model for the deletion. As these genetic interactions may vary across different species, developmental time-points, and tissues, the role of NCBP2 towards deletion phenotypes and its interactions with other 3q29 genes should be more deeply explored using mouse or human cell culture models.
Third, through cellular phenotyping in Drosophila and X. laevis models, we identified apoptosis as a key mechanism for pathogenicity of the 3q29 deletion. In fact, our data suggest that apoptosis is not only affected by knockdown of single 3q29 genes, but can also be synergistically disrupted with knockdown of multiple 3q29 genes (Table 1). For example, simultaneous knockdown of NCBP2Cbp20 with FBXO45Fsn leads to enhanced cellular disorganization and altered expression of apoptosis genes in Drosophila, as well as enhanced morphological defects and increased caspase-3 levels in X. laevis. We further found that overexpression of the apoptosis inhibitor Diap1/XIAP rescued the cellular and neuronal phenotypes observed with knockdown of 3q29 homologs, providing important validations for the involvement of apoptosis towards the deletion phenotypes (Table 1). Several 3q29 homologs have been previously associated with apoptosis or cell cycle regulation functions (Table S8). For example, DLG1 is a tumor suppressor gene whose knockdown in Drosophila leads to neoplasms in the developing brain and eye disc (63, 64), while PAK2 is a key downstream mediator of the ERK signaling pathway for neuronal extension and is activated by caspases during apoptosis (56, 65, 66). Additionally, our work provides evidence that NCBP2 regulates apoptosis during neurodevelopment, and suggests that this pathway contributes to the enhanced phenotypes observed with interactions of NCBP2 and other 3q29 genes. Our data further implicates knockdown of several 3q29 genes, including DLG1 and BDH1, towards abnormal cell proliferation during development. Similar to our previous findings of disrupted cell proliferation with knockdown of genes in the 16p11.2 deletion (67), these results emphasize the importance of cell cycle regulation towards developmental phenotypes (40, 42).
More broadly, our results provide further evidence for apoptosis as a cellular mechanism associated with schizophrenia (44). Previous functional studies have associated abnormal apoptosis with several schizophrenia-associated processes during development (47, 48), and mutations within apoptotic engulfment pathways have also been identified in sequencing studies of schizophrenia cohorts (68, 69). Importantly, our study provides direct evidence for the role of apoptosis towards the etiology of a schizophrenia-associated genetic disorder. We found further validation for this association by identifying a significant enrichment (empirical p=0.0136) for genes associated with apoptotic processes among candidate schizophrenia genes (13) (Figure S15). Out of the 268 schizophrenia genes that were also involved in apoptosis, 13 genes were present within schizophrenia-associated CNV regions (70), including CORO1A, MAPK3 and TAOK2 in the 16p11.2 microduplication (71) and AATF and HNF1B in the 17q12 microdeletion (72) (Table S9). These genes can be further investigated using functional studies to determine the role of apoptosis towards the developmental phenotypes of these CNV disorders. In addition, our data also suggests that abnormal apoptosis could be responsible for the microcephaly phenotypes observed in 3q29 deletion carriers. Apoptosis has been previously implicated towards decreased brain size in animal models (73) and syndromic forms of microcephaly in humans (74). In fact, knockdown mouse models of the primary microcephaly genes CENPJ and STIL also showed increased apoptosis (75). Furthermore, a mouse model of the Nijmegen breakage syndrome gene NBN exhibited increased neuronal apoptosis leading to microcephaly and decreased body mass (76), suggesting that the reduction of body size observed in 3q29 deletion mice could also be due to apoptosis during early development (15). These findings highlight the importance of apoptosis towards modulating neurodevelopmental features.
Overall, our results show that interactions between NCBP2 and other 3q29 genes disrupt apoptosis mechanisms to contribute towards schizophrenia and other clinical features of the deletion. The use of Drosophila and X. laevis models, both of which are amenable to high-throughput screening of developmental phenotypes, allowed us to systematically examine the conserved cellular and mechanistic roles of 3q29 homologs and their interactions. Follow-up studies in more evolutionarily advanced systems, such as mouse or human cell lines, will be useful to overcome limitations of the Drosophila and X. laevis models, including testing the neurodevelopmental phenotypes and interactions of 3q29 genes without Drosophila homologs. Collectively, these results emphasize the utility of quantitative functional assays for identifying conserved pathways associated with neurodevelopmental disorders, which will hopefully allow for future discoveries of treatments for these disorders.
MATERIALS AND METHODS
Fly stocks and genetics
Using reciprocal BLAST searches and ortholog predictions from the DIOPT v.7.1 database (77), we identified 15 fly orthologs for the 21 human genes within the chromosome 3q29 region (Table S1). No fly homologs were present for six genes, including LRRC33, CEP19, RNF168, SMCO1, TFRC, and TM4SF19. We used a similar strategy to identify homologs for other neurodevelopmental genes tested for interactions in this study. RNAi lines for fly homologs were obtained from the Vienna Drosophila Resource Centre (78) (VDRC), including both KK and GD lines, and the Bloomington Drosophila Stock Centre (BDSC) (NIH P40OD018537). A list of fly RNAi lines used in this study is provided in Table S10 for 3q29 homologs and Table S11 for other neurodevelopmental and apoptosis genes. All 3q29 lines were tested for gene knockdown with qualitative real-time PCR, and select KK lines were also tested for overexpression of tiptop due to RNAi insertion at the 5’UTR of the gene (79) (Table S2). As the available lines for TCTEX1D2CG5359 failed to achieve the desired gene knockdown and showed overexpression of tiptop, these lines were excluded and a total of 14 fly homologs were tested in this study. Microarray data and modENCODE Anatomy RNA-Seq from FlyBase (80, 81) showed that all of the 14 tested homologs were expressed in the fly central nervous system and eye tissues (Table S1).
All fly stocks and crosses were cultured on conventional cornmeal-sucrose-dextrose-yeast medium at 25°C, unless otherwise indicated. RNAi lines were crossed with a series of GAL4 driver lines to achieve tissue-specific knockdown of genes, including da-GAL4 (Scott Selleck, Penn State) for ubiquitous, GMR-GAL4 (Zhi-Chun Lai, Penn State) and GMR-GAL4;UAS-Dicer2 (Claire Thomas, Penn State) for eye-specific, beadexMS1096-GAL4;UAS-Dicer2 (Zhi-Chun Lai, Penn State) for wing-specific, and Elav-GAL4 (Mike Groteweil, VCU) for pan-neuronal knockdown of gene expression. To perform interaction studies, we generated recombinant stock lines of GMR-GAL4 with reduced expression of nine select 3q29 homologs (Table S4). Females from these stocks with constitutively reduced gene expression for each of these genes were crossed with RNAi lines of other homologs to achieve simultaneous knockdown of two genes (Figure 1). We previously demonstrated that these two-hit crosses had adequate GAL4 to bind to two independent UAS-RNAi constructs (67).
Quantitative real-time polymerase chain reaction
Levels of gene expression knockdown were confirmed using quantitative real-time PCR (RT-PCR) on RNA isolated from fly heads (Table S2). Briefly, each RNAi line was crossed with Elav-GAL4 at 25°C to achieve pan-neuronal knockdown of the fly homolog. Adult fly heads at day 3 were separated by vortexing, and total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA, USA). cDNA was prepared using the qScript cDNA synthesis kit (Quantabio, Beverly, MA, USA). Quantitative real-time PCR (qPCR) was performed using an Applied Biosystems Fast 7500 system with SYBR Green PCR master mix (Quantabio) to estimate the level of gene expression. Primers were designed using NCBI Primer-BLAST (82), with primer pairs separated by an intron in the corresponding genomic DNA. All experiments were performed using three biological replicates. A list of primers used in the experiments is provided in Table S2. The delta-delta Ct value method was used to obtain the relative expression of fly homologs in the RNAi lines compared with controls (83).
Climbing assay
We set up fly crosses at 25°C with Elav-GAL4 to obtain pan-neuronal knockdown of the tested 3q29 homologs. For each genotype, groups of ten female flies were first allowed to adjust at room temperature for 30 minutes and then transferred to a climbing apparatus, made by joining two vials, and allowed to adjust for 5 minutes. The flies were tapped down to the bottom, and the number of flies climbing past the 8 cm mark measured from the bottom of the apparatus in 10 seconds was then counted. This assay was repeated nine additional times for each group, with a one-minute rest between trials. The sets of 10 trials for each group were repeated daily for ten days, capturing data from 100 replicates from day 1 until day 10, starting the experiments with 1-2-day old flies. All experiments were performed during the same time of the day for consistency of results. Two-way ANOVA and pairwise two-tailed T tests were used to determine significance for each genotype and day of experiment (Table S12). All statistical analysis in the manuscript was performed using R v.3.4.2 (R Foundation for Statistical Computing, Vienna, Austria).
Imaging of adult fly eyes and wings
We crossed RNAi lines with GMR-GAL4 and reared at 29°C for eye-specific knockdown and beadexMS1096-GAL4 at 25°C for wing-specific knockdown. For eye imaging, adult 2-3-day old female progenies from the crosses were collected, immobilized by freezing at −80°C, mounted on Blu-tac (Bostik Inc, Wauwatosa, WI, USA), and imaged using an Olympus BX53 compound microscope with LMPLan N 20X air objective using a DP73 c-mount camera at 0.5X magnification and a z-step size of 12.1μm. (Olympus Corporation, Tokyo, Japan). We used CellSens Dimension software (Olympus Corporation, Tokyo, Japan) to capture the images, and stacked the image slices using Zerene Stacker (Zerene Systems LLC, Richland, WA, USA). Eye images for Diap1 rescue of two-hit models were captured with a Leica Z16APO microscope using a PLAN APO 2X objective lens, and stacked using Leica Application Suite v.4.1 (Leica Microsystems, Buffalo Grove, IL, USA). All eye images presented in this study are maximum projections of 20 consecutive optical z-sections. Adult wings were plucked from 2-5 day old female flies, mounted on a glass slide, covered with a coverslip and sealed with clear nail polish. The wings were imaged using a Zeiss Discovery V20 stereoscope (Zeiss, Thornwood, NY, USA) with ProgRes Speed XT Core 3 camera (Jenoptik AG, Jena, Germany) using a 40X objective, and images were captured with ProgRes CapturePro v.2.8.8.
Quantitative phenotyping of fly eyes using Flynotyper
We used a computational method called Flynotyper (http://flynotyper.sourceforge.net/) to measure the degree of roughness of the adult eyes (34). The software uses an algorithm to detect the center of each ommatidium, and calculates a phenotypic score based on the number of ommatidia detected, the lengths of six local vectors with direction pointing from each ommatidium to the neighboring ommatidia, and the angle between these six local vectors (Figure S2). Using Flynotyper, we obtained quantitative measures for roughness of the fly eye with single gene or pairwise gene knockdown. These results were compared to controls using one-tailed or two-tailed Mann-Whitney tests (Table S12), and two-way ANOVA tests were used to identify synergistic enhancement or suppression of the respective single-hit phenotypes (Figure S8).
Immunohistochemistry of eye and wing discs
Third instar larval and 44-hour-old pupal eye discs, reared at 29°C, and third instar larval wing discs, reared at 25°C, were dissected in 1X phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde for 20 minutes. The eye and wing discs were then washed thrice in PBT for 10 minutes each, treated with 1% blocking solution for 30 minutes, and then incubated overnight with primary antibodies at 4°C. Rabbit anti-cleaved Drosophila dcp1 (Asp216) (1:100; 9578S, Cell Signaling Technology, Danvers, MA, USA), a marker for cells undergoing apoptosis, and Mouse anti-phospho-Histone H3 (S10) antibody (1:100; 9706L, Cell Signaling Technology), a mitotic marker for measuring proliferating cells, were used to assay cell cycle and apoptosis defects in larval eye and wing discs. Mouse anti-DLG (1:200; 4F3, DSHB, Iowa City, Iowa, USA), a septate junction marker, and Rhodamine Phalloidin (1:200; R415, Invitrogen Molecular Probes, Carlsbad, CA, USA), an F-actin marker, were used to visualize and count ommatidial cells and photoreceptor cells in pupal eyes. Mouse anti-chaoptin (1:200; 24B10, DSHB) was used to visualize retinal axon projections. Preparations were then washed for 10 minutes thrice with PBT, and incubated for two hours with fluorophore-conjugated secondary antibodies (Alexa fluor 568 goat anti-mouse (1:200) (A11031), Alexa fluor 488 goat anti-mouse (1:200) (A11029), Alexa fluor 647 goat anti-rabbit (1:200) (A21245), and Alexa fluor 647 goat anti-mouse (1:200) (A21236), Invitrogen Molecular Probes, Carlsbad, CA, USA)) with gentle shaking. Final washes were performed in PBS for 10 minutes, and the tissues were then mounted in Prolong Gold antifade mounting media with DAPI (P36930, Thermo Fisher Scientific, Waltham, MA, USA) or Vectashield hard set mounting media with DAPI (H-1500, Vector Laboratories, Burlingame, CA, USA) for imaging.
Bromouridine staining
Third instar larval eye discs were dissected in PBS and immediately transferred to Schneider’s Insect Media (Sigma-Aldrich, St. Louis, MO). The tissues were then incubated in 10 µM BrdU (Sigma-Aldrich) at 25°C for one hour with constant agitation to allow for incorporation of BrdU into DNA of replicating cells during the S-phase of cell cycle. The samples were washed thrice with PBS for five minutes each and fixed in 4% paraformaldehyde for 20 minutes. To denature DNA, the tissues were acid-treated in 2N HCl for 20 minutes, neutralized in 100 mM Borax solution for 2 minutes, washed thrice in 10X PBT (PBS+0.1% Tween-20) for 10 minutes, and treated with blocking solution (PBS, 0.2% Triton X-100, 5% Normal Goat serum) for one hour. The tissues were then incubated in mouse anti-BrdU (1:200; G3G4, DSHB, Iowa City, Iowa, USA) diluted in blocking solution overnight at 4°C. The next day, the tissues were washed thrice in PBT for 20 minutes each and incubated in Alexa fluor 568 goat anti-mouse (1:200, Invitrogen Molecular Probes, Carlsbad, CA, USA) for two hours with constant agitation. Finally, the samples were mounted in Prolong Gold antifade reagent with DAPI (Thermo Fisher Scientific, Waltham, MA, USA) for imaging.
Terminal deoxynucleotidyl transferase (TUNEL) Assay
The levels of cell death in the developing eye were evaluated by staining using the In Situ Cell Death Detection Kit, TMR Red (Roche, Basel, Switzerland). The third instar larval eye discs were dissected in PBS and fixed in 4% paraformaldehyde for 20 minutes at room temperature. The dissected tissues were permeabilized by treating with 20 µg/ml proteinase K (Sigma-Aldrich, St. Louis, MO, USA) for two minutes, washed in PBT for 30 minutes, and fixed again in 4% paraformaldehyde for 15 minutes. The tissues were then incubated overnight with TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) reaction mixture at 4°C per the manufacturer’s instructions, and washed five times in PBT for 15 minutes each. Finally, tissues were mounted in Prolong-gold antifade containing DAPI (Thermo Fisher Scientific, Waltham, MA, USA) for imaging.
Confocal imaging and analysis
Confocal images of larval and pupal eye discs were captured using an Olympus Fluoview FV1000 laser scanning confocal microscope (Olympus America, Lake Success, NY). Maximum projections of all optical sections were generated for display. Acquisition and processing of images was performed with the Fluoview software (Olympus Corporation, Tokyo, Japan), and the z-stacks of images were merged using ImageJ (84). Images for experiments using Diap1 overexpression for rescue of two-hit models were captured using a Nikon A1RMPsi-STORM (4.0) confocal microscope with a PLAN APO 20X lens (Nikon Instruments Inc., Tokyo, Japan), and were stacked using NIS Element AR Analysis v.4.51.00. The number of pH3, BrdU, TUNEL, and dcp1-positive cells from larval eye discs were counted using two ImageJ plugins, AnalyzeParticles and Image-based Tool for Counting Nuclei (ITCN). As we found a strong correlation (Pearson correlation, r=0.736, p<2.2×10−16) between the two methods (Figure S2D), all cell counts displayed for eye data were derived from ITCN analysis. Proliferating cells in larval wing discs stained with pH3 were counted using AnalyzeParticles, and apoptotic cells in wing discs stained with dcp1 were analyzed using manual counting. For statistical analysis, we compared the tested genotypes with controls using two-tailed Mann-Whitney tests (Table S12).
Differential expression analysis of transcriptome data
We performed RNA sequencing (RNA-Seq) of samples isolated from fly heads of Elav-GAL4 crosses for four single-hit (DLG1dlg1, NCBP2Cbp20, PAK2Pak, FBXO45Fsn) and two two-hit (NCBP2Cbp20/DLG1dlg1 and NCBP2Cbp20/FBXO45Fsn) knockdowns of 3q29 homologs, and compared gene expression levels to VDRC control flies carrying the same genetic background (GD or KK). We prepared cDNA libraries for three biological replicates per genotype using TruSeq Stranded mRNA LT Sample Prep Kit (Illumina, San Diego, CA), and performed single-end sequencing using Illumina HiSeq 2000 at the Penn State Genomics Core Facility to obtain 100 bp reads at an average coverage of 36.0 million aligned reads/sample. We used Trimmomatic v.0.36 (85) for quality control assessment, TopHat2 v.2.1.1 (86) to align the raw sequencing data to the reference fly genome and transcriptome (build 6.08), and HTSeq-Count v.0.6.1 (87) to calculate raw read counts for each gene. edgeR v.3.20.1 (88) (generalized linear model option) was used to perform differential expression analysis, and genes with log2-fold changes >1 or <-1 and corrected false-discovery rates less than 0.05 were considered to be differentially expressed (Table S12). Human homologs of differentially-expressed fly genes (top matches for each fly gene, excluding matches with “low” rank) were identified using DIOPT (77). Enrichment analysis of Panther GO-Slim Biological Process terms among the differentially-expressed genes was performed using the PantherDB Gene List Analysis tool (89). Enrichments for genes preferentially expressed in the developing brain were calculated using the Cell-type Specific Expression Analysis tool (90) based on expression data from the BrainSpan Atlas (91).
X. laevis embryos
Eggs collected from female X. laevis frogs were fertilized in vitro, dejellied, and cultured following standard methods (92, 93). Embryos were staged according to Nieuwkoop and Faber (94). All experiments were approved by the Boston College Institutional Animal Care and Use Committee and were performed according to national regulatory standards.
Morpholino and RNA constructs
Morpholinos (MOs) were targeted to early splice sites of X. laevis NCBP2, FBXO45, PAK2, or standard control MO, purchased from Gene Tools LLC (Philomath, OR, USA). MO sequences are listed in Table S13. For knockdown experiments, all MOs were injected at either the 2-cell or 4-cell stage, with embryos receiving injections two or four times total in 0.1X MMR containing 5% Ficoll. FBXO45 and control MOs were injected at 10ng/embryo, NCBP2 and control MOs were injected at 20ng/embryo, and PAK2 and control MOs were injected at 50ng/embryo. For rescue experiments, the same amounts of MOs used in the KD experiments were injected along with gene-specific mRNA tagged with GFP (800pg/embryo for XIAP-GFP; 1000pg/embryo for NCBP2-GFP and FBXO45-GFP, and 300pg/embryo for PAK2-GFP) in the same injection solution. Capped mRNAs were transcribed in vitro using SP6 or T7 mMessage mMachine Kit (Thermo Fisher Scientific, Waltham, MA, USA). RNA was purified with LiCl precipitation. X. laevis NCBP2, FBXO45, PAK2, and XIAP ORFs obtained from the European Xenopus Resource Center (EXRC, Portsmouth, UK) were gateway-cloned into pCSf107mT-GATEWAY-3’GFP destination vectors. Constructs used included NCBP2-GFP, FBXO45-GFP, PAK2-GFP, XIAP-GFP, and GFP in pCS2+. Embryos either at the 2-cell or 4-cell stage received four injections in 0.1X MMR containing 5% Ficoll with the following total mRNA amount per embryo: 300pg of GFP, 800pg of XIAP-GFP, 1000pg NCBP2-GFP, 1000pg of FBXO45-GFP, and 300pg of PAK2-GFP.
RT-PCR for morpholino validation and knockdown
Morpholino validation and knockdown was assessed using RT-PCR. Total RNA was extracted using TRIzol reagent (Life Technologies, Grand Island, NY, USA), followed by chloroform extraction and ethanol precipitation from 2-day old embryos injected with increasing concentrations of MO targeted to each tested 3q29 gene. cDNA synthesis was performed with SuperScript II Reverse Transcriptase (Life Technologies, Grand Island, NY, USA) and random hexamers. PCR primers are listed in Table S14. RT-PCR was performed in triplicate (Figure S13A), with band intensities quantified by densitometry in ImageJ and normalized to the uninjected control mean relative to ODC1, which was used as a housekeeping control.
Brain and eye morphology assays
In brain morphology experiments, all embryos received two injections at the 2-cell stage in 0.1X MMR containing 5% Ficoll. One cell was left uninjected and the other cell was injected with either control MO or MO targeted to the tested 3q29 gene, along with 300pg of GFP mRNA in the same injection solution. Stage 47 tadpoles were fixed in 4% PFA diluted in PBS for one hour, rinsed in PBS and gutted to reduce autofluorescence. Embryos were incubated in 3% bovine serum albumin and 1% Triton-X 100 in PBS for two hours, and then incubated in anti-acetylated tubulin primary antibody (1:500, monoclonal, clone 6-11B-1, AB24610, Abcam, Cambridge, UK) and goat anti-mouse Alexa fluor 488 conjugate secondary antibody (1:1000, polyclonal, A11029, Invitrogen Life Technologies, Carlsbad, CA). Embryos were then rinsed in 1% PBS-Tween and imaged in PBS. Skin dorsal to the brain was removed if the brain was not clearly visible due to pigment. For eye phenotype experiments, all embryos received four injections at the 2-cell or 4-cell stage in 0.1X MMR containing 5% Ficoll with either the control MO or MOs targeted to each 3q29 gene. Stage 42 tadpoles were fixed in 4% PFA diluted in PBS. Tadpoles were washed three times in 1% PBS-Tween for one hour at room temperature before imaging.
X. laevis image acquisition and analysis
Lateral view images of stage 42 tadpoles for eye experiments and dorsal view images of state 47 tadpoles for brain experiments were each collected on a SteREO Discovery.V8 microscope using a Zeiss 5X objective and Axiocam 512 color camera (Zeiss, Thornwood, NY, USA). Areas of the left and right eye, forebrain, and midbrain were determined from raw images using the polygon area function in ImageJ. Eye size was quantified by taking the average area of both the left and right eye, while forebrain and midbrain area were quantified by taking the ratio between the injected side versus the uninjected side for each sample. For statistical analysis, we compared the tested genotypes with controls using unpaired two-tailed t-tests (Table S12).
Western blot for apoptosis
Embryos at stages 20-22 were lysed in buffer (50mM Tris pH 7.5, 1% NP40, 150mM NaCl, 1mM PMSF, 0.5 mM EDTA) supplemented with cOmplete Mini EDTA-free Protease Inhibitor Cocktail (Sigma-Aldrich, Basel, Switzerland). Blotting was carried out using rabbit polyclonal antibody to cleaved caspase-3 (1:500, 9661S, Cell Signaling Technology, Danvers, MA, USA), with mouse anti-beta actin (1:2500, AB8224, Abcam, Cambridge, UK) as a loading control. Chemiluminescence detection was performed using Amersham ECL Western blot reagent (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA). Band intensities were quantified by densitometry in ImageJ and normalized to the control mean relative to beta-actin.
Overlap between schizophrenia and apoptosis gene sets
We obtained a set of 1,794 genes annotated with the Gene Ontology term for apoptotic processes (GO:0006915) or children terms from the Gene Ontology Consortium (95) (AmiGO v.2.4.26), and compared this gene set to a set of 2,546 candidate schizophrenia genes curated by Purcell and colleagues (13). We found 268 genes that overlapped across the two sets (Table S12). To determine the statistical significance of this overlap, we performed 100,000 simulations to identify the number of apoptosis genes in randomly selected groups of 2,546 genes (Figure S15), and found that the 268 overlapping genes were in the top 98.64% of the simulation results (empirical p=0.0136).
Reproducibility
Drosophila negative geotaxis, immunohistochemistry/confocal microscopy and key eye imaging experiments were performed on multiple independent occasions to ensure reproducibility of our results; all Drosophila data shown were derived from single experimental runs containing multiple samples. X. laevis experiments were performed on at least three independent occasions to ensure reproducibility, with the data representing consistent findings from these multiple replicates.
Data availability
Gene expression data for the six Drosophila single-gene and two-hit models of 3q29 homologs are deposited in the GEO (Gene Expression Omnibus) database with accession code GSE128094, and the raw RNA Sequencing files are deposited in the SRA (Sequence Read Archive) with BioProject accession PRJNA526450. All unique biological materials described in the manuscript, such as recombinant fly stocks, are readily available from the authors upon request.
Code availability
All source code and datasets for generating genomic data (RNA-Seq and schizophrenia/apoptosis gene overlap) are available on the Girirajan lab GitHub page at https://github.com/girirajanlab/3q29_project.
AUTHOR CONTRIBUTIONS
M.D.S., M.J., and S.G. designed the study. M.D.S, E.H., T.Y., L.P., B.L., I.P., A.K., S.Y., J.I. and D.E.R.L. performed the Drosophila experiments, and M.L. and L.A.L. designed and performed the X. laevis experiments. M.D.S., M.J., T.Y., J.I., M.L., and S.G. analyzed the data. M.D.S., M.J., and S.G. wrote the manuscript with input from all authors.
COMPETING INTERESTS
The authors declare that they have no competing interests.
ACKNOWLEDGMENTS
We thank V. Faundez for useful discussions and critical reading of the manuscript, and J. Tiber for technical assistance with the X. laevis experiments. This work was supported by a Basil O’Connor Award from the March of Dimes Foundation (#5-FY14-66), NIH R01-GM121907, a NARSAD Young Investigator Grant from the Brain and Behavior Research Foundation (22535), and resources from the Huck Institutes of the Life Sciences to S.G., NIH T32-GM102057 to M.J., and NIH R01-MH109651 to L.A.L.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵




