

Abstract
Deleterious and intact mitochondrial DNA (mtDNA) mutations frequently co-exist in cells (heteroplasmy). Such mutations likely survive and are inherited due to complementation via the intra-cellular mitochondrial network. Hence, we hypothesized that compromised mitochondrial fusion would hamper such complementation, thereby affecting heteroplasmy inheritance. To test this hypothesis, we assessed heteroplasmic patterns in three Charcot-Marie-Tooth disease type 2A pedigrees, which carry a mutated mitofusin 2 (MFN2). We found reduced prevalence of a potentially functional mtDNA heteroplasmic mutation in these patients, as compared to healthy maternal relatives, while neutral heteroplasmic mutations fluctuated randomly. Secondly, we found that MFN2 dysfunction in a Caenorhabditis elegans model carrying a large heteroplasmic mtDNA deletion (ΔmtDNA) led to a severe developmental delay and embryonic lethality. Strikingly, these phenotypes were relieved during subsequent generations in association with complete ΔmtDNA removal. Such ΔmtDNA loss occurred during both gametogenesis and embryogenesis. Therefore, mitochondrial fusion is essential for inheritance of mtDNA heteroplasmy.
Introduction
Unlike the nuclear genome, mitochondrial DNA (mtDNA) is present in multiple copies per animal cell. For instance, human somatic cells contain an average of ∼1,000 mitochondria per cell, with each mitochondrion harboring 1-10 mtDNA copies (Schon and Gilkerson, 2010). Although this large intracellular mtDNA population is inherited from the maternal germline, and hence carries a single major haplotype, mtDNA molecules can differ in sequence (heteroplasmy) either due to inheritance of mutations from the ovum or due to the accumulation of changes over the lifetime of the individual (Avital et al., 2012). Some of these changes may have pathological consequences, as reflected in a variety of mitochondrial disorders, yet only upon crossing a threshold of prevalence in the cell (Hahn and Zuryn, 2019). Accordingly, the penetrance of disease-causing mutations ranges between 60-80%, depending on the symptoms and tissues that display the specific phenotype (Craven et al., 2017).
The repertoire of heteroplasmic mutations varies among cells and tissues of an individual, mainly due to replicative segregation (drift) of the mitochondria during cell division and mitochondrial bottlenecks that appear during embryo development (Floros et al., 2018). However, it has been suggested that heteroplasmy can be modulated by non-random factors, including selection (Avital et al., 2012; Burgstaller et al., 2014). Indeed, it has been shown that mitophagy, a mechanism of mitochondrial quality control, partially provides selection against defective mitochondria and maintains disease-causing mtDNAs below the threshold level both in human cells (Suen et al., 2010) and in a Caenorhabditis elegans model (Valenci et al., 2015). Mitophagy requires proper fission-fusion cycles of the mitochondrial network so as to allow removal of dysfunctional mitochondria (Mao et al., 2013; Twig et al., 2008). In agreement with this notion, reduction in heteroplasmy levels of potentially deleterious mtDNA mutations was observed when components of the fusion machinery were compromised in Drosophila models (Kandul et al., 2016). Furthermore, elevated heteroplasmy levels of pathological mtDNA were observed when the fission machinery was disrupted in cell culture (Malena et al., 2009). This suggest that the effect of disease-causing mutations is functionally compensated at sub-threshold levels of heteroplasmy by the intracellular mitochondrial network (Busch et al., 2014; Hahn and Zuryn, 2019; Schon and Gilkerson, 2010). This offers an appealing explanation for the relatively high abundance of low-level disease-causing heteroplasmic mutations in the general population (Rebolledo-Jaramillo et al., 2014; Ye et al., 2014). We thus hypothesized that interfering with the mitochondrial cellular network would prevent functional compensation of heteroplasmic mutations, and potentially lead to their removal. This question is of special interest in the case of human disorders in which the fusion machinery is compromised. A good example for such, is the mutated mitochondrial fusion protein MFN2, leading to the hereditary motor and sensory neuropathy Charcot-Marie-Tooth type 2A (CMT2A), which is characterized by progressive loss of muscle mass across the entire human body, and primary axonal neuropathies (Zuchner et al., 2004).
Here, we took the first steps towards testing this hypothesis by assessing patterns of mtDNA heteroplasmy in three unrelated CMT2A pedigrees. In doing so, we found significantly reduced heteroplasmy levels only in patients carrying a potentially functional heteroplasmic mutation. Secondly, crossing C. elegans harboring a MFN (fzo-1) deletion to animals carrying a large heteroplasmic mtDNA deletion resulted in a developmental delay and embryonic lethality, which was alleviated in subsequent generations concomitant with a complete loss of the truncated mtDNA molecules. The mechanistic implications of these findings are discussed.
Results
MFN2 modulates heteroplasmic patterns in CMT2A patients
To assess the potential impact of defective MFN2 on the dynamics of mitochondrial heteroplasmy in humans, we employed massive parallel sequencing (MPS) of the entire mtDNA in CMT2A patients and healthy maternal relatives in three independent pedigrees (Fig. S1). High coverage per mtDNA nucleotide (>1000X) was attained at a mean of 16560 mtDNA positions, thus allowing both assignment of samples to specific mtDNA genetic backgrounds (haplogroups) and the detection of heteroplasmy levels >1% (Table S1).
In pedigree 1, a Caucasian-Cherokee American Indian family in which a Leu146Phe MFN2 mutation (Klein et al., 2011) has been segregating, five heteroplasmic mutations were identified, two of which (mtDNA positions 310 and 16172) are shared between the female patient (sample V17) and her healthy siblings (samples V13 and V14; Table 1). Interestingly, although the C-to-G transition at position 16172 was highly prevalent (∼95%) in the healthy maternal siblings, it became rare in the patient (∼5%). Notably, position 16172 maps within the mtDNA transcription termination site (TAS) (Barshad et al., 2018), thus suggesting potential functionality. In pedigree 2 (Caucasian family 1706, with the L76P MFN2 mutation (Zuchner et al., 2004)), a heteroplasmic transition at mtDNA position 13830 was identified (a synonymous mutation in the ND5 gene), which differentially segregated among patients and healthy maternal relatives. Nevertheless, the ratio between the two segregating alleles at this position was the opposite of what was seen between the proband (individual 106) and his maternal uncle, who was also a patient (individual 1007). This finding is consistent with random segregation of this heteroplasmic mutation. Finally, analysis of the entire mtDNA sequence in patients and healthy maternal relatives in pedigree 3 (an Arab-Israeli family presenting the Q386P MFN2 mutation (Verhoeven et al., 2006)) did not reveal any trustworthy heteroplasmic mutations. Thus, we found that only in the case where a heteroplasmic mutation was potentially functional was its level significantly declined exclusively in the CMT2A patient, suggesting that selective heteroplasmy removal had occurred. As such, we hypothesized that MFN2 likely modulates the inheritance of functional heteroplasmic mutations. To test this proposal, we examined the dynamics of a previously characterized heteroplasmic mtDNA deletion (Gitschlag et al., 2016; Liau et al., 2007; Tsang and Lemire, 2002) in an MFN2 C. elegans mutant.
Heteroplasmy in analyzed patient samples
A heteroplasmic deletion cannot be tolerated in a C. elegans mitofusin (fzo-1) mutant
The stable heteroplasmic C. elegans strain uaDf5/+ harbors a mixture of intact (+mtDNA) and ∼60% of a 3.1 kb mtDNA deletion (ΔmtDNA) (Gitschlag et al., 2016; Tsang and Lemire, 2002). Although lacking four essential genes (i.e., mt-ND1, mt-ATP6, mt-ND2 and mt-Cytb), this strain displays only mild mitochondrial dysfunction (Gitschlag et al., 2016; Liau et al., 2007; Tsang and Lemire, 2002). We showed that dysfunctional PDR-1, the worm orthologue of the key mitophagy factor Parkin, led to elevated levels of the truncated mtDNA, suggesting that mitochondrial quality control senses the presence of dysfunctional mitochondria (Valenci et al., 2015). In conjunction with this finding, RNAi knockdown of fzo-1, the C. elegans orthologue of MFN1/2, led to slight reduction in the levels of the heteroplasmic ΔmtDNA, although without any phenotypic consequences (Nargund et al., 2012). We, therefore, asked what would be the impact of the fzo-1(tm1133) deletion (hereafter designated as fzo-1(mut)) on the inheritance of the maternal ΔmtDNA.
To this end, we crossed ΔmtDNA heteroplasmic hermaphrodites with fzo-1(mut) heterozygote males (Fig. 1A). After self-cross of the F1 progeny, the distribution of the genotypes in the F2 heteroplasmic progeny did not deviate from the expected Mendelian ratios, namely 26% homozygous fzo-1(mut), 49% fzo-1 heterozygotes (ht) and 25% fzo-1(wt) (p=0.94, Chi square test; Table S2). No apparent phenotypic differences were observed among F2 animals (generation 1, G1) as well as in the non-heteroplasmic fzo-1(mut) animals. However, we noticed that only 13±5% of the second generation (G2m; i.e., the progeny of the self-crossed fzo-1(mut);ΔmtDNA worms) hatched, as compared to fzo-1(mut) animals (67±5%; Fig. 1B). Interestingly, the 13±5% G2m animals that did hatch were developmentally delayed, and no animals reached adulthood after six days. This was in contrast to ∼75% of the G1 animals that reached adulthood (Fig. 1C). These findings demonstrate that the interaction between the heteroplasmic ΔmtDNA and the nuclear DNA-encoded fzo-1 mutant led to a severe reduction in fitness. Therefore, the ΔmtDNA was not tolerated by mitofusin mutants.

(A) Schematic representation of experimental setup, Heteroplasmic hermaphrodites (ΔmtDNA) were crossed with fzo-1(mut) males. Cross progeny F1 were allowed to self-propagate and single F2 (generation 1; G1) animals were isolated, allowed to lay eggs and their genotypes were determined. We then monitored heteroplasmic mutant or wild type fzo-1 progeny over several generations (G2m-G4m and G2wt-G4wt, respectively). Heterozygous progeny was maintained to generate generation G1 without additional crosses. (B) The percent of hatched embryos of parental strains: N2(WT), ΔmtDNA and fzo-1(mut), of fzo-1(mut) mutant cross progeny across generations (G1m-G4m) and of the stable cross line (>20 generations). (C) The percent of gravid adults six days after egg laying of parental strains: N2(WT), ΔmtDNA and fzo-1(mut), and of mutant cross progeny across generations (G1m-G4m). (D) The percent of gravid adults of mutant cross progeny across generations (G1m-G4m) at the indicated times after egg laying. P values were calculated by comparison with fzo-1(mut) animals. (*) denotes P<0.05 and (**) denotes P<0.01.
Reversal of the adverse effects of the ΔmtDNA-fzo-1 interaction
To better characterize the phenotypic impact of interactions between ΔmtDNA and fzo-1(mut), we monitored the progeny of the self-crossed fzo-1(ht);ΔmtDNA worms. Specifically, we measured the duration of the larva-to-adulthood period during the course of development in the G1m-G4m generations (Fig. 1D). While ∼75% of the G1m generation reached adulthood after six days, the development of G2m animals was severely delayed, with 75% of the animals reaching adulthood only after nine days. To our surprise, the G3m animals showed significant improvement, with ∼60% of this population reaching adulthood after six days. Moreover, G4m animals showed a full reversal of ΔmtDNA-associated adverse effects (Fig. 1D). Thus, the adverse effects of the interaction between fzo-1(mut) and ΔmtDNA were reversed by the G4m generation.
We noted a similar pattern across generations when hatching was considered. In contrast to the ∼10% hatching seen among G2m embryos, 60±8% of the G3m embryos hatched. Strikingly, the hatching percentage of the G4m generation was indistinguishable from that of G1 animals and remained stable over subsequent generations (Fig. 1B). Finally, no phenotypic changes were observed across the G1-G4 generations while tracing the developmental pace and hatching percentage of fzo-1(wt);ΔmtDNA, G1wt-G4wt (Fig. S2). Taken together, our findings demonstrate a full reversal of the adverse effects of the interaction between the ΔmtDNA and the nuclear DNA-encoded mutant fzo-1 gene.
MFN2 mutant selects against a large heteroplasmic truncation in C. elegans
We next asked how the deleterious interactions between fzo-1(mut) and ΔmtDNA were abrogated. We hypothesized that if the ΔmtDNA is not tolerated in the background of fzo-1(mut), then selection against the ΔmtDNA should occur. To test this prediction, we quantified the levels of ΔmtDNA by quantitative PCR (qPCR) across the G1m-G4m generations (at the adult stage) in both the fzo-1(mut);ΔmtDNA and the fzo-1(wt);ΔmtDNA strains. We found that ΔmtDNA levels declined two-fold in the G1m fzo-1(mut) animals, as compared to fzo-1 heterozygotes. This trend was enhanced, reaching a 10-fold reduction in ΔmtDNA levels in G2m animals. Values were below detection levels in most G3m (N=19/22) and all G4m (N=21) animals (Fig. 2A). In contrast, ΔmtDNA levels did not significantly change across the G1wt-G4wt generations of fzo-1(wt);ΔmtDNA animals (Fig. 2B).

(A) The percent of ΔmtDNA determined in individual animals (n ≥20) of the parental heteroplasmic strain ΔmtDNA, the fzo-1(mut) mutant cross-progeny strains (F1(ht), G1m-G4m) and the progeny of G4m animals crossed with fzo-1(wt), (G4m->G6-8wt). (B) The percent of ΔmtDNA determined in individual animals (n ≥20) of the fzo-1(wt) cross progeny strains (G1wt-G4wt). (C) The percent of ΔmtDNA determined in a population of animals of the stable cross lines (>20 generations). P values were calculated by comparison with fzo-1(wt);ΔmtDNA animals. (*) denotes P<0.05 and (**) denotes P<0.01.
These results suggest that the ΔmtDNA was completely lost during the G1m-G4m generations. To test this hypothesis, we crossed G4m hermaphrodites with wild type males to isolate fzo-1(wt) progeny (G6). Since G6 animals (and subsequent generations) did not display any ΔmtDNA (Fig. 2A and 2C), we concluded that disrupting fzo-1 function indeed resulted in a complete and specific loss of the deleterious heteroplasmic ΔmtDNA. Taken together, our results demonstrate, for the first time, that mitochondrial fusion is critical for maintaining heteroplasmic mutations.
ΔmtDNA levels are selected at two different stages in the C. elegans life cycle
We next asked at which point during the C. elegans life cycle was selection against ΔmtDNA realized. It was previously demonstrated that C. elegans mtDNA copy numbers started to increase significantly at the fourth larval stage (L4) and that this increase was associated with oocyte production (Bratic et al., 2009; Tsang and Lemire, 2002). Notably, the relative levels of ΔmtDNA are maintained during development (Tsang and Lemire, 2002). We, therefore, compared relative ΔmtDNA levels between embryos and adults in G2m animals. Our results indicate that relative ΔmtDNA levels were dramatically reduced (∼5-fold) during development of G2m animals but not in G2wt animals (Fig. 3A and 3B). This observation suggests that ΔmtDNA is most likely selected against during worm development. Secondly, ΔmtDNA levels became undetectable in the resultant embryos of G2m animals (i.e., in G3m animals; Fig. 3A). Hence, it is possible that selection against ΔmtDNA molecules had already occurred during gametogenesis. In support of this claim, we noted that upon isolation of the gonads of G2m animals and then comparing ΔmtDNA levels in gonads and somatic tissues, we found a two-fold decrease in ΔmtDNA levels in the gonads. In contrast, no such difference was observed when comparing gonads and somatic tissues of fzo-1(wt)/ΔmtDNA animals (Fig. 3C). Based on these observations, we suggest that selection against ΔmtDNA molecules occurs both during development and during gametogenesis.

(A) The percent of ΔmtDNA in embryos and adults of the G2m-G3m generations. (B) The percent of ΔmtDNA in embryos and adults of the G2wt-G3wt generations. (C) The ratio of ΔmtDNA or +mtDNA levels in the gonad and soma of generation G2m adults P values were calculated by comparison with fzo-1(mut) G2 embryos. (*) denotes P<0.05.
Discussion
Cell culture experiments revealed that a mixture of mtDNA molecules differing in sequence in the same cell can complement each other by the diffusion of products via the mitochondrial network, which in turn leads to a restoration of mitochondrial function (Gilkerson et al., 2008; Schon and Gilkerson, 2010). This very same mechanism offers an explanation for the survival of mtDNA disease-causing mutations in cells and in turn, their transmission to the next generation. Therefore, we hypothesized that interfering with the intracellular mitochondrial network by compromising the fission-fusion cycle would hamper mitochondrial functional complementation.
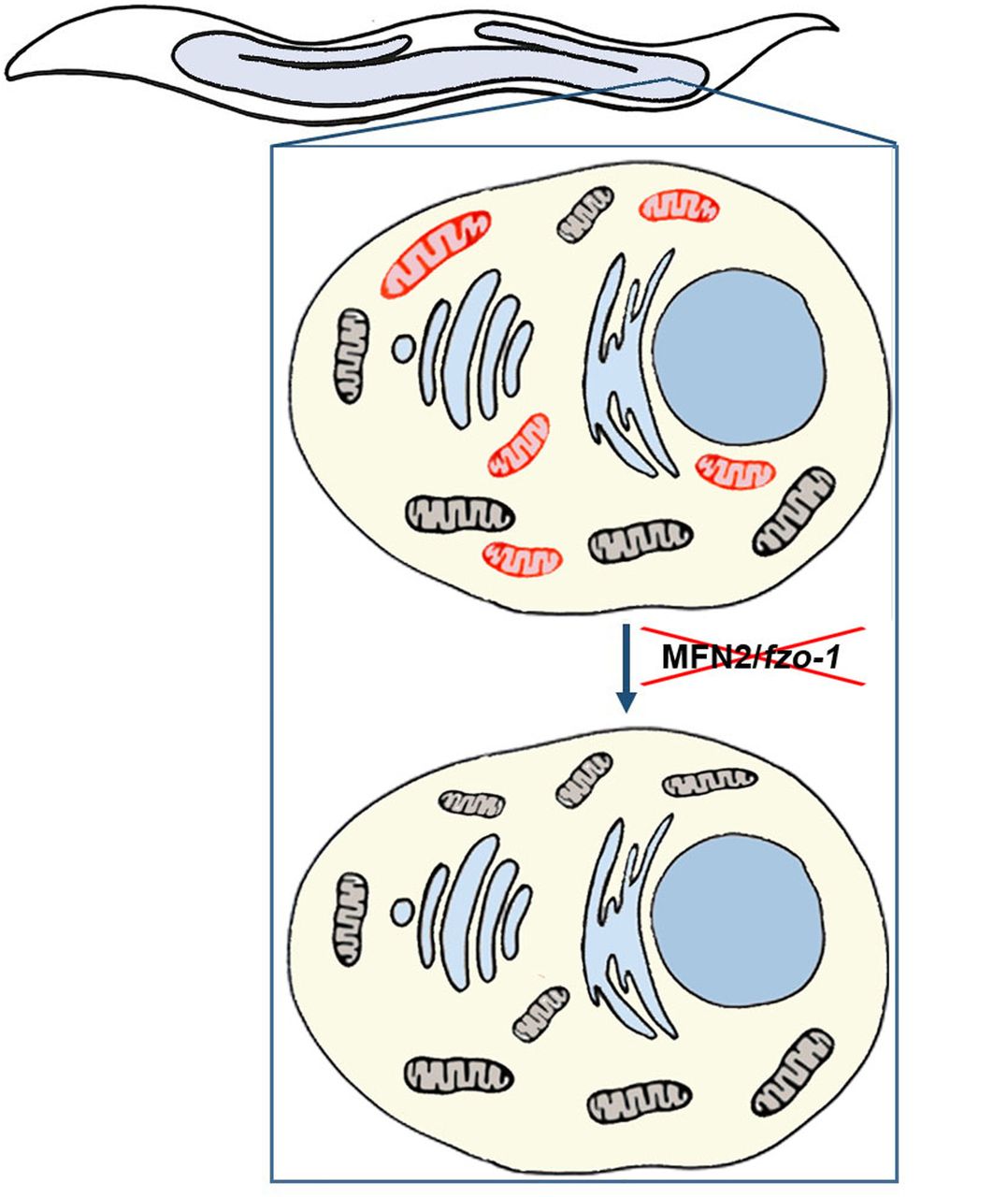
As a first step in testing this hypothesis, we investigated patterns of mtDNA heteroplasmy in three independent pedigrees of Charcot-Marie-Tooth type 2A characterized by mutated MFN2, a major mitochondrial fusion component. We found that the heteroplasmy level of a potentially functional mutation in the mtDNA transcriptional-associated sequence was strongly reduced in patients, as compared to healthy maternal relatives. In contrast, the levels of apparently neutral mtDNA mutations fluctuated without any clear correlation to phenotype. Although offering support to our working hypothesis, the impact of a compromised fusion machinery on heteroplasmy required assessment in a more controlled system. Thus, we turned to a well-characterized C. elegans heteroplasmic ΔmtDNA model (Tsang and Lemire, 2002) into which we introduced a mutation in MFN2 (fzo-1) (Kanazawa et al., 2008). We found that fzo-1 mutant worms experienced significant developmental delays and high rates of embryonic lethality, which were relieved within two generations. Strikingly, this improvement was accompanied by complete and selective removal of the ΔmtDNA molecules (Fig. 4). These observations strongly argue for the essentiality of a functional intracellular mitochondrial network for the survival of potentially deleterious heteroplasmic deletions.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
ΔmtDNA (red), intact mtDNA (black).
Our findings provide experimental support for the hypothesis that functional complementation among mitochondria in the intracellular network likely enables the survival and prevalence of deleterious heteroplasmic mtDNA mutations in the population (Rebolledo-Jaramillo et al., 2014; Ye et al., 2014). However, in contrast to our findings, either in vitro loss of mitochondrial fusion or in vivo conditional deletion of both MFN1 and MFN2 in mouse skeletal muscle led to a strong reduction in mtDNA levels, which was accompanied by the accumulation of mtDNA point mutations and deletions (Chen et al., 2007; Chen et al., 2010). This apparent contradiction could stem from the different outcome of manipulating the fusion machinery in the germ line (our study) versus somatic cells. We thus argue that unlike what occurs with somatic cells, manipulating fusion in the germline leads to strong negative selection against deleterious mtDNA mutations. Hence, the complete loss of ΔmtDNA across C. elegans generations suggests that the fusion machinery represents an attractive candidate target for future treatment of mitochondrial disorders. Furthermore, partial inhibition of the fusion machinery might offer a safer route to selectively remove disease-causing heteroplasmic mutations. Consistent with this suggestion, it has been demonstrated that knock-down of mitochondrial fusion components in both flies and worms reduced the levels of heteroplasmic deleterious mutations (Kandul et al., 2016; Nargund et al., 2012). While such a course of action may not completely remove the deleterious heteroplasmic mutation, selective reduction in heteroplasmy level below the functional threshold in cells could provide an attractive therapeutic approach (Bacman et al., 2018; Gammage et al., 2018).
Our results indicate that there are two stages during the nematode life cycle in which ΔmtDNA was lost, i.e., during the transition from larva to adult, and during embryogenesis. The relative decline of ΔmtDNA levels during late larval stages occurred concomitantly with mtDNA replication, which is part of gametogenesis (Ahier et al., 2018; Bratic et al., 2009). Close inspection of the data revealed that while intact mtDNA levels increased as expected, ΔmtDNA levels increased only some two-fold (Fig. S3B). Moreover, since ΔmtDNA levels dropped specifically in the gonad, ΔmtDNA molecules could either have been actively removed or selectively not replicated during gametogenesis. Secondly, our observed total loss of ΔmtDNA in the worm G3m generation (in contrast to its presence in the germline of the G2m generation) suggests another step of selection against ΔmtDNA during embryogenesis. Indeed, changes in mitochondrial morphology and function associated with lysosomal activation upon fertilization were demonstrated (Bohnert and Kenyon, 2017), which might also play a role in mitochondrial clearance. Taken together, our results suggest the existence of specific checkpoints of mtDNA heteroplasmy during development.
In summary, we have provided clear evidence showing that compromising the mitochondrial fusion machinery relieves the protective communal ‘shelter’ for deleterious heteroplasmic mutations, resulting in their complete and selective removal. The occurrence of such a process during embryogenesis and gametogenesis further underlines the importance of fission-fusion cycles in maintaining the integrity of mtDNA during transmission from generation to generation. Since reduced expression of MFN2 is frequently observed in the placenta after miscarriages of unexplained etiology (Pang et al., 2013), one can speculate that the combination of mtDNA disease-causing hetereoplasmic mutations along with MFN2 dysfunction is negatively selected. Finally, our findings may offer previously overlooked avenues to actively reduce levels of mtDNA heteroplasmy as an approach to treat mitochondrial disorders.
Materials and methods
Human DNA samples
Patients diagnosed with Charcot-Marie-Tooth type 2a (CMT2A) and healthy maternal relatives from three unrelated pedigrees were considered in the current study. Total blood DNA was extracted from available patients and healthy individuals of all three sibs. Pedigree 1, of English-native American admixed origin, harbors patients heterozygous for the dominant L146F MFN2 mutation (Klein et al., 2011). Pedigree 2, of North American Caucasian origin, harbors patients previously identified as heterozygous for the L76P MFN2 dominant mutation (Zuchner et al., 2004). In pedigree 3 of Israeli Arabian origin, the patient was identified as a compound heterozygote for A1157C and T1158G mRNA nucleotide position mutations, both leading to the recessive Q386P mutation (Verhoeven et al., 2006). In Fig. S1, arrows point to the tested individuals in each pedigree.
Massive parallel sequencing of the entire human mtDNA and bioinformatics analysis
The entire mtDNA of available patients and controls (Fig. S1) was amplified in three fragments using three sets of primers, as previously described (Avital et al., 2012). For each of the analyzed samples, the amplified fragments were mixed in equimolar ratios and sent for library construction and sequencing utilizing the Illumina MiSeq platform (Technion Genome Center, Israel). Paired-end reads were trimmed using TrimGalore (version 0.4.5)) F., 2015 (and then mapped to the mtDNA reference sequence (rCRS) using BWA mem (version 0.7.16) with default parameters (Li and Durbin, 2009). Alignment files (SAM format) were compressed to their binary form (BAM format) using SAMtools (version 1.3) with default parameters [view –hb] and sorted using the [sort] function (Li et al., 2009). True heteroplasmic mutations were annotated using an in-house script that followed the logic of the previously established MitoBamAnnotator (Cohen et al., 2016). In brief, a pileup of the mtDNA-mapped reads was generated using the SAMtools [mpileup] function with the [-Q 30] parameter to only consider reads with a phred score higher than 30 as a measure of quality control. Next, the mapped bases were counted and the most frequent nucleotide in each mtDNA position was considered the major allele only if that nucleotide position had a minimal coverage of 10X. To avoid strand bias, the secondary mutation at a given nucleotide position was recorded only if it was represented by at least two reads per strand. mtDNA sequences were then haplotyped using HaploGrep2.0 (Kloss-Brandstatter et al., 2011).
Sanger sequencing and cloning
Sanger sequencing was employed to verify the identified heteroplasmic mutations at position 16172 (pedigree 1) and position 13830 (pedigree 2). Primers used for this amplification reactions are listed in Table S3.
NUMT exclusion
To control for possible contamination by nuclear mitochondrial DNA pseudogenes (NUMTs) in the MPS reads, mtDNA sequence contigs encompassing each of the identified mutations were BLAST screened as previously described (Blumberg et al., 2014). In brief, the highest scored nuclear DNA BLAST hits encompassing each of the identified mutations were aligned against the mtDNA sequence reads of the relevant sample and the number of reads harboring the exact identified mutation was counted using IGV viewer (Robinson et al., 2011). Notably, as the mtDNA is maternally inherited as a single locus, variants appear in linkage. Hence, combinations of variants that were present in nuclear DNA BLAST hits but which were not identified in sequence reads of the relevant samples were not considered as corresponding to NUMTs.
Nematodes and growth conditions
A list of strains used in this work and name abbreviations is found in Table S4. All strains were outcrosses to our N2 stock at least four times. Nematodes were grown on Nematode Growth Medium (NGM) plates seeded with the Escherichia coli OP50-1 strain at 15°C. Experiments were repeated at least three times. P values were calculated using the Wilcoxon Mann-Whitney rank sum test to compare two independent populations.
Crosses
Mutant fzo-1(tm1133) animals (strain CU5991) are very poor in mating and, therefore, were first crossed with males expressing a yellow fluorescent protein marker (unc-54p::YFP). Heteroplasmic (uaDf5/+) hermaphrodites were then crossed with fzo-1(tm1133); unc-54p::YFP heterozygote males to ensure maternal inheritance of uaDf5 ΔmtDNA and to establish independent heteroplasmic lines carrying the wild type or fzo-1(tm1133) mutation. F2 progeny were screened for the fzo-1(tm1133) mutation using a single worm PCR Phire Animal Tissue Direct PCR Kit (Thermo Scientific) with fzo-1 MUT primers (Table S5), followed by gel electrophoresis of the PCR products. Heteroplasmic (uaDf5/+) animals that were heterozygotes for fzo-1(tm1133) were maintained to easily produce G1 animals.
Gonad dissection
G2 wild type or mutant animals were placed in a drop of ultra-pure water on a coverslip slide and a 25 gauge needle was used to remove the gonads from the body of the animals. 5-10 worms were used for each biological repeat. Gonads or the remaining carcasses were then transferred to DNA extraction buffer.
DNA purification and extraction
Total DNA was extracted using a QuickExtract kit (Lucigen). Unless otherwise indicated, DNA was extracted from a single worm (n ≥20). When populations were examined, ∼5 animals were collected. For embryos, DNA was extracted from ∼30 embryos. For gonad and soma analysis, gonads were dissected from ∼5 animals per biological repeat. DNA was extracted separately from the gonads and soma.
Assessment of mtDNA copy numbers
mtDNA levels were measured by qPCR performed on a C1000 Thermal Cycler (Bio-Rad) with KAPA SYBRFAST qPCR Master Mix (KAPA Biosystems). Analysis of the results was performed using CFX Manager software (Bio-Rad). To quantify the different mtDNA molecules, three set of primers were used for truncated, intact and total mtDNA molecules (Table S5). The average CT (threshold cycle) of triplicate values obtained for these mtDNA molecules was normalized to a nuclear DNA marker using the  method (Livak and Schmittgen, 2001). At least four independent experiments were used to determine the normalized CT values of each strain or generation. Truncated/total ratio was defined as the ratio of the normalized CT values of truncated to total mtDNA for a given strain.
method (Livak and Schmittgen, 2001). At least four independent experiments were used to determine the normalized CT values of each strain or generation. Truncated/total ratio was defined as the ratio of the normalized CT values of truncated to total mtDNA for a given strain.
Embryo hatching
Gravid animals were moved to fresh plate for 2-12 hours and then removed from the plates. Embryos were allowed to develop and hatching was examined after 48 hours. Experiments were repeated independently at least four times.
Developmental timing
Single embryos were placed on fresh plates and allowed to grow at 15°C. The animals’ developmental stage was examined every day and the number of animals reaching reproductive adulthood on each day was recorded. Developmentally arrested animals were excluded. Experiments were repeated independently at least three times.
Author Contributions
Conceptualization, A.B. and D.M.; Methodology, L.M., D.B. and I.V.; Investigation, L.M., D.B., D.K., M.K. and S.D.; Formal Analysis, D.B and T.C; Resources, C.J.K., J.M.V., Y.N. and S.Z; Writing – Original Draft, A.B. and D.M.; Writing – Review & Editing, A.B. and D.M.; Supervision, A.B. and D.M.; Funding Acquisition, A.B. and D.M.
Acknowledgements
This study was funded by the Israel Science Foundation (ISF) grant 278/18 to ABZ and grant 372/17 to DM and by Israel Ministry of Science and Technology grant 3-14337 to ABZ.
References




