

Abstract
Microbial communities underpin earth’s biological and biogeochemical processes, but their complexity hampers understanding. Here we draw upon predictions from the theory of selfish genetic elements (SGEs), combined with approaches from experimental evolution, comparative metagenomics and biochemistry, and show how naturally occurring SGEs can be used to manipulate genes that underpin community function. Communities comprising hundreds of bacterial genera established from garden compost were propagated with bi-weekly transfer for one year in nitrogen-limited minimal medium with cellulose (paper) as sole carbon source. At each transfer, SGEs from one set of independent communities were collected, pooled and redistributed among this same set of communities (horizontal treatment). A control was included in which SGEs never moved beyond the community in which they were originally present (vertical treatment). SGEs along with genes of ecological relevance were rapidly amplified across horizontal communities and their dynamics tracked. Enrichment of genes implicated in nitrogen metabolism, and particularly ammonification, led to biochemical assays that showed a significant functional difference between communities subject to horizontal versus vertical treatments. This simple experimental strategy offers a powerful new approach for unravelling dynamical processes underpinning microbial community function.
Introduction
Microbial communities underpin all major biological and biogeochemical processes1. Appropriate functioning of communities has direct implications for health of people2, terrestrial3 and marine4 ecosystems and even global climate5. While the species composition of many communities have been exhaustively documented6, the link between genes and community function is poorly understood1.
Needed are new approaches to the study of communities that are process-focused. Ideally such approaches will provide knowledge on the complex interconnections between community members, their combined effects including feedbacks that shape the evolution of community members. Particularly desirable are approaches that provide a link between genes, their spatial and temporal dynamics, and community function, achieved using strategies that leave as far as possible the natural complexity of communities undisturbed. Here we achieve this via an experiment in which we combine theory governing the behaviour of selfish genetic elements7-9 (SGEs) with experimental evolution10,11, comparative metagenomics and functional assay.
Results
SGEs including bacteriophages and plasmids are abundant in microbial communities12. Typically capable of autonomous replication — or able to exploit mobilisation genes provided in trans — SGEs can disseminate across diverse hosts with movement often coincidental with genes of ecological significance to which, by chance, they become linked. Long-term persistence, requires occasional exposure to new hosts: frequent exposure to new hosts is expected to drive rapid spread and evolution of SGEs and linked genes13.
Recognising that SGEs are naturally adept at mobilising not only themselves, but also ecologically significant genes, and that their activity is fuelled by exposure to new hosts, we reasoned that promoting the activity of SGEs would bring about changes in the abundance and distribution of ecologically significant genes, allow tracking of their dynamics and even measurement of impacts on community function.
A set of ten independent experimental communities were established from 1 g samples of garden compost and propagated with bi-weekly transfer in static nitrogen-limited minimal M9 medium with cellulose (paper) as the sole carbon source. After one month, each of the 10 communities was used to found two new communities, giving a total of 20 paired communities. One of each pair was assigned to the “horizontal regime”, while the remainder were assigned to the “vertical regime” (Supplementary Data Fig. 1). The terms horizontal (H) and vertical (V) indicate the imposed transfer regime: the former is expected to fuel the life of SGEs, the latter to castrate.

Experimental setup. 1) Establish communities: Cellulose-degrading microbial communities (n=10) were established by placing 1 gram of fresh compost into 20 mL of M9 Minimal Media supplemented with cellulose paper (sole carbon source). 2) Vertical and Horizontal transfer regimes: Following four weeks of incubation the communities were homogenized and split into the vertical and horizontal transfer regimes. In the vertical transfer regime, 1 mL of homogenized community was transferred to 19 mL of M9 Minimal Media and provided with a fresh piece of cellulose paper. In the horizontal transfer regime, 1 mL of homogenized community was transferred using the same method as the vertical along with 1 mL of “SGE-cocktail”. The “SGE-cocktail” was prepared by mixing 1 mL of homogenized community (n=10) followed by centrifugation and filtration to remove microbial cells. 3) Incubate communities: Paired horizontal and vertical communities were incubated for two weeks and total DNA was extracted for metagenomic sequencing. At each transfer samples were collected for DNA sequencing and glycerol stocks of each community were created and stored at -80 °C.

Movement of genetic material between HCs and detection of ecological relevant genes within the HGT pool. To determine whether any SGE transfer occurred between communities, paired vertical and horizontal metagenomes were compared using a custom bioinformatic pipeline (Methods). First, each horizontal metagenome was compared to the founding T0 metagenome (before the horizontal vs. vertical split) and the paired vertical metagenome using BLASTn. Horizontal-T0 and horizontal-vertical sequence matches were removed, thus leaving a set of “unique” horizontal sequences that could not be identified in neither the founding T0-metagenome, nor the paired vertical-metagenome. Sequence matches were determined using a minimum e-value threshold of 1E-05, minimum alignment length of 100 base pairs, and a minimum percent identity of 90%. The “unique” horizontal sequences were then assembled, Open Reading Frames (ORFs) were extracted, and predicted protein domains were determined through comparison to the Conserved Domain Database (CDD). A. Movement of phage-like contigs between HCs. Unique horizontal contigs containing phage-specific domains were extracted and their distribution between horizontal and vertical bottles was determined. The Y-axis indicates the community from which a unique phage-like contig was assembled from while rows indicate the distribution of each unique phage-like contig in all HCs and VCs. The total number of reads recruited to each phage-like contig is indicated. See Supplementary Table 3 for full domain predictions and Extended Data Figure 2a for annotation of contig 35969 (upper panel). B. Assembly of “unique” Horizontal sequences identified predicted domains with ecological relevance including genes associated with cellulose degradation, defence, and metabolism. See Supplementary Table 4 for domain descriptions and abundances.
From the moment of the initial split (time point T0) and for the ensuing 48 weeks (24 transfers (T24)) 1 ml of each community, including the slurry of any remaining paper, was transferred to a fresh mesocosm containing 19 ml of M9 medium plus fresh paper. This ensured serial passage of microbes plus SGEs from each mesocosm. Half of these mesocosms (one of each pair), labelled vertical communities (VCs), received no further treatment other than continued serial passage on a bi-weekly basis.
The remaining ten horizontal communities (HCs) received — at the time of serial transfer — an aliquot from a “SGE cocktail” that was a pooled sample of SGEs derived from each horizontal mesocosm (Supplementary Data Fig. 1). The SGE cocktail was obtained by collecting the filtrate after passage of a 1 ml sample of the community (from each of the ten mesocosms) through a 0.22 µm filter. Such filtration removes bacteria, but allows passage of bacteriophages, naked DNA and potentially other unknown or unrecognised SGEs. After pooling the filtrate from each of the ten horizontal treatments a 1ml aliquot of the mixture was placed in each HC.
To determine whether the horizontal regime was promoting movement of SGEs as predicted, total DNA was extracted from each T0 community and each of the 20 T1 communities and sequenced (Supplementary Data Fig. 1 and Supplementary Table 1). Analysis was straightforward given the paired experimental design. For each set of T0 and descendant HCs and VCs (e.g., T0-1 and descendants T1V-1 and T1H-1) DNA sequence data were interrogated to identify reads found solely in (and therefore unique to) the horizontal mesocosm (e.g., T1H-1) (Methods). Such unique reads are likely to stem from SGEs, or be associated with SGE activity, originating from an allopatric community.
Analysis of the sequences unique to each horizontal community resulted in identification of ∼26 million unique reads from a total of ∼152 million reads across all ten communities, with an average of ∼2.6 million unique reads per community (Supplementary Table 2). Assembly of reads into contigs revealed an average of 3,352 contigs per HC greater than 1 kb in length. The mean maximum contig size per assembly was 82 kb (Supplementary Table 2).
After extracting Open Reading Frames (ORFs), interrogation of the Conserved Domain Database (CDD) showed 1,279 ORFs predicted to encode phage-associated proteins involved in capsid formation, baseplate assembly, and phage-induced lysis of bacterial cells (Supplementary Table 3). An example of a contig (T1H-1_35969) containing numerous genes characteristic of phages is shown in Supplementary Data Fig. 2a.

A. Characterization of T1H1 unique contig 35969. The ∼90kb contig assembled from the unique horizontal sequences of community-1 contains many phage-specific domains including ORFs predicted to encode proteins involved with capsid structure, assembly of phage particles, and bacterial cell lysis. B. An ∼80 kb contig assembled from the unique sequences of T1H7 (Time point 1, Horizontal community 7) containing a predicted cellulose-degrading domain as well as various domains associated with mobile elements. See Supplementary Tables 3 and 5 respectively for full domain annotations.

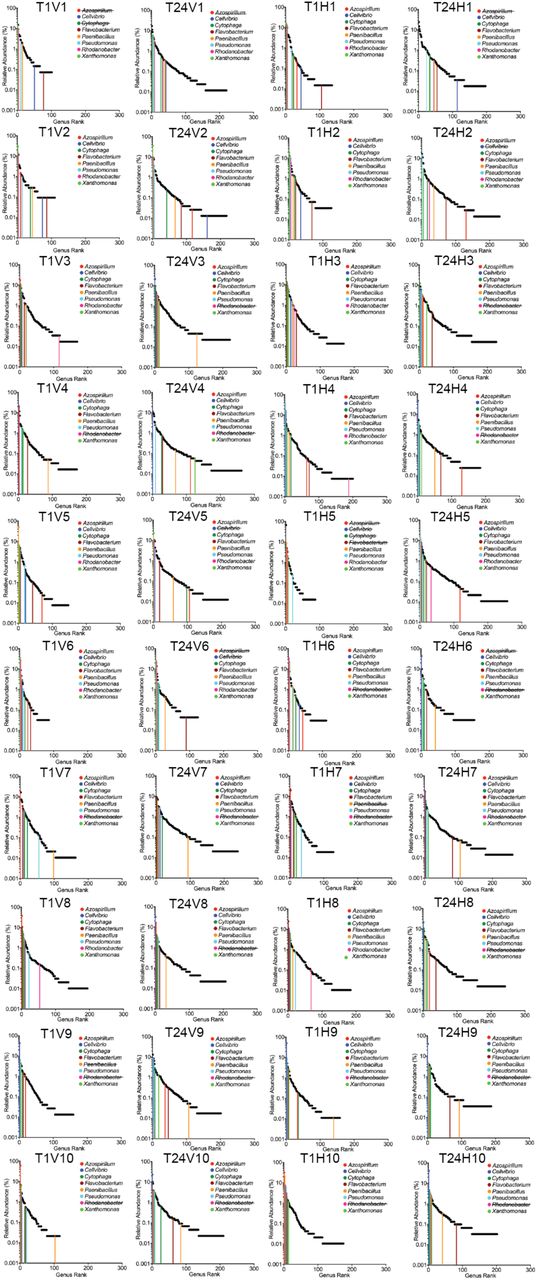
Genus relative abundance at T1 (2 weeks) and T24 (one year). HCs and VCs were combined at each time point due to lack of significant difference between treatments. Sequences annotated by MG-RAST as 16s rRNA were extracted, the abundance of each genus was determined, and all genera were ranked by percent abundance. See Supplementary Table 6 for genus abundance at all sequenced time points (T0, T1, T2, T4, T10, T16, T24) and Extended Data Figure 4 for rank abundance curves indicating the eight most abundant genera at T1 and their subsequent abundance at T24. Mean values are displayed with standard deviation indicated (n=20 for T1 and T24).
The distribution and abundance of contig T1H-1_35969 and 19 other representative phage-like entities (Supplementary Table 3b) across independent mesocosms was determined by mapping total reads from each horizontal and vertical metagenome onto each of the 20 phage-like contigs (Fig. 1a). The predominance of these contigs in HCs is evidence of rapid amplification and dissemination of genetic material. T1H-1_35969, originally assembled from the unique set of sequences from T1H-1, is also present in both T1H-2 and T1V-2 and was therefore likely present in the founding community (T0-2). Within two weeks this contig has amplified and spread to horizontal communities 1, 5 and 6 (Fig. 1a). Contig T1H2_39307 is below the level of detection in all vertical treatments, but two weeks after imposition of the horizontal treatment, this contig featured in seven of the independent HCs (Fig. 1a, Supplementary Table 3). From the community perspective, less than four of the 20 phage-like contigs were detected on average per VC, whereas the mean number of contigs per HCs was 11 (Fig. 1a).
In addition to phage-like contigs, assemblies of the unique sequences from HCs contained genes predicted to encode traits of likely ecological significance to life in the cellulose mesocosms (Fig. 1b). These include genes implicated in cellulose degradation, defence against SGE invasion and nitrogen metabolism (Table 4)14-16. Particularly notable is enrichment of reads mapping to β-glucosidase genes that define the rate-limiting step in cellulose degradation17. On occasion these ecological significant genes were linked to genes encoding features found on SGEs (Supplementary Table 5), but often these contigs lacked such features (Supplementary Data Fig. 2b). Nonetheless they were amplified and disseminated through the course of the evolution experiment with the same dynamic as expected of a mobile genetic element (see below).
Given evidence indicative of the amplification and dissemination of DNA via the horizontal treatment, combined with ability to detect such sequences, communities were allowed to evolve for a further year with, at each transfer, the HCs experiencing continual exposure to the SGE cocktail. Continuance was in part motivated by the expectation that movement of DNA — particularly that encoding genes of ecological relevance — mediated by SGEs would result in a measurable change in the function of HCs relative to VCs. To complement existing DNA sequence from T0 and T1, DNA was extracted from all HCs and VCs at time points T2, T3, T4, T10, T16, T20 and T24 yielding a total of 180 metagenomes (5.2 billion reads, each ∼150 bp).
The extraordinary flux of DNA detected within two weeks of establishing the horizontal treatment regime likely relies upon a minimum threshold of community-level diversity. Given propagation of communities on minimal medium with a single recalcitrant (cellulose) carbon source, we firstly interrogated the metagenomic data for sequences mapping to 16S rDNA to determine whether diversity was maintained for the duration of the experiment (Supplementary Table 6). Communities at T0 harboured on average ∼140 bacterial genera (SD + 23). At T24 the number of genera detected increased in all communities to an average of ∼200 (SD + 32), indicating an increase in abundance of rare types (Supplementary Table 6, Extend Data Fig. 3a). No significant difference was observed between HCs and VCs (Extend Data Fig. 3a-c). Rank abundance distributions from pooled HCs and VCs at T1 and T24 are shown in Fig. 2 and reveal the tail of the T24 distribution to be significantly flatter in both cases, consistent with an overall tendency for rare types to increase during the course of the year long selection. Rank abundance distributions for each T1 and T24 HC and VC, plus the rank order (and change therein) of the most common genera are shown in Supplementary Data Fig. 4. Overall communities differed markedly in genera composition. Among the most abundant genera, only two harbour species commonly associated with cellulolytic ability18 (Cytophaga and Cellvibrio), while others are better known for roles in ammonification19 (Azospirillum and Paenibacillus (nitrogen fixation); Rhodanobacter and Pseudomonas (dissimilatory nitrate reduction / denitrification)) (Supplementary Data Fig. 4). Single celled eukaryotes were also common features of all mesocosms with several evening maintaining populations of nematodes through the 48-week selection period. Because bacteria-specific protocols were used to extract DNA for sequencing, eukaryotes were not further considered.

Diversity indices over time. A. The total number of Genera based on the 16s rRNA subunit extracted from sequences annotated using the MG-RAST database. B. The number of Genera normalized to the total number of sequences per metagenome. C. Shannon-Weaver diversity index. For all panels mean values are displayed (n=10 for both Horizontal and Vertical) with standard deviation indicated.
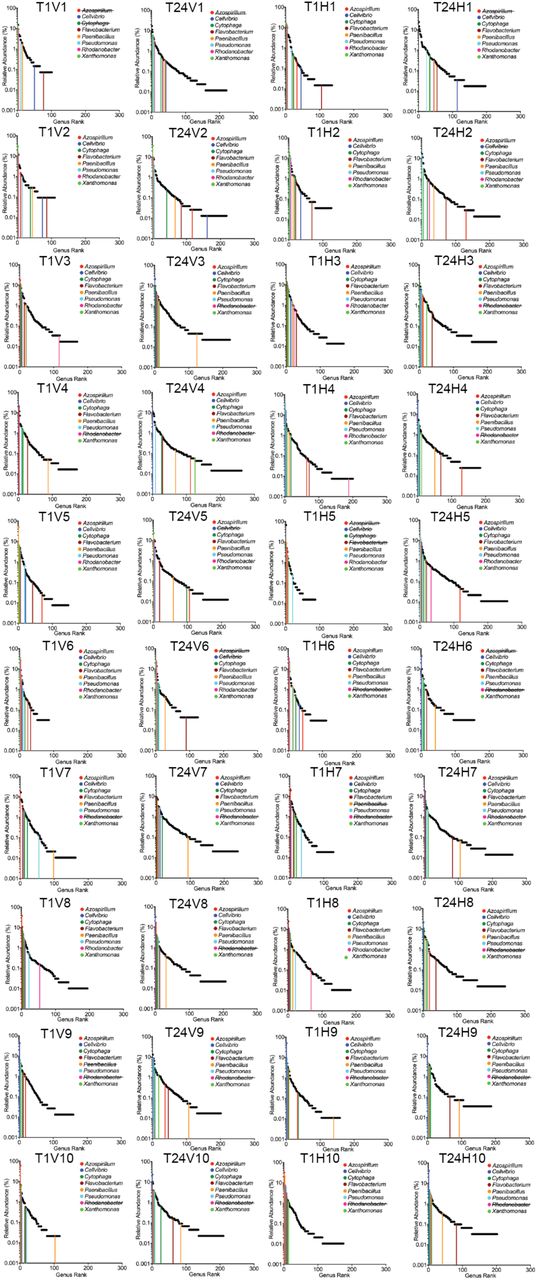
Relative abundance of representative genera over time. The most abundant genus from each T1 community was selected and the abundance of each genus was determined in all T1 and T24 communities.
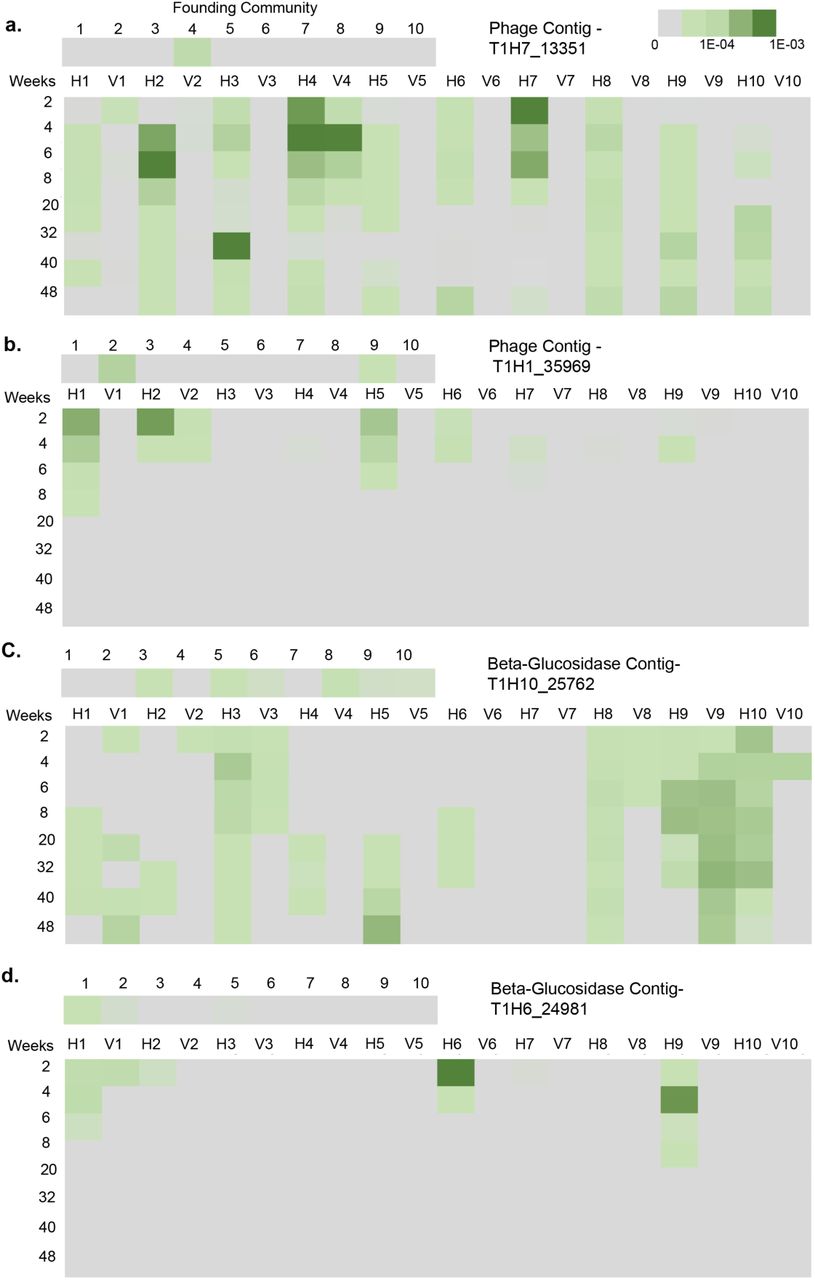
Experimental design provides the opportunity to observe HGT dynamics in complex microbial communities. A. Survival of a phage-like contig assembled from the unique horizontal sequences from time point-1 of HC-7 (T1H7). B. Extinction of the phage-like contig depicted in Extended Figure 2a. C. Survival of a contig containing a predicted Beta-glucosidase domain assembled from the unique horizontal sequences from time point-1 of HC-10 (T1H10). D. Extinction of the Beta-glucosidase contig depicted in Extended Figure 2b. Shade of green indicates the number of sequences recruited to each contig normalized by the total number of sequences in each metagenome. Upper panel displays contig abundance in the founding T0-communities indicating a potential source community for each contig. Rows indicate the number of weeks after the Vertical vs. Horizontal split while columns indicate the abundance of each contig in a community over time (H1 = HC-1 V1 = VC-1 etc.) A minimum alignment length of 100 base pairs and percent identity of 90% was used to determine sequence matches.

Enrichment of nitrogen metabolism genes over time and between HCs vs. VCs. A. Relative abundance of “Nitrogen Metabolism” gene categories based on sequence annotation using the MG-RAST database. See Supplementary Table 7 for the relative abundances of all gene categories in the T0 (founding) and T24 (one year) communities (* = p-value < 0.05, ** = p-value <0.01, Unpaired T-Test). The mean of T0 (n=10), T24V (n=10), and T24H (n=10) is displayed with minimum and maximum values indicated. B. Total number of Open Reading Frames (ORFs) predicted to encode specific domains associated with Ammonification, regulation of nitrogen metabolism, and other associated nitrogen metabolism genes in the assemblies of unique vertical and unique horizontal sequences. Horizontal metagenomes were compared to their paired vertical metagenomes at eight separate eight time points (T1, T2, T3, T4, T10, T16, T20, T24) as well as the metagenome of their founding T0 community using BLASTn. Horizontal sequences that matched vertical or T0 metagenomes were removed, leaving a set of unique horizontal sequences that could not be identified in any of the paired vertical or founding T0 communities. Unique horizontal sequences were assembled, Open Reading Frames (ORFs) extracted, and analyzed using the Conserved Domain Database (CDD). As a control the same analysis was performed using the T24 Vertical metagenomes to identify “unique” Vertical sequences (Methods). The mean of T24V (n=10), and T24H (n=10) is displayed with minimum and maximum values indicated. 1 = NifS, 2 = NifE, 3 = NapA, 4 = Nitrilase, 5 = NtrA, 6 = NifR3, 7 = NtrY, 8 = NifL, 9 = PtsN, 10 = NtrB, 11 = NtrC, 12 = NAC, 13 = FixI, 14 = FixG, 15 = Nitroreductase, 16 = Nitroreductase, 17 = Arginase, 18 = Cytochrome D1 (Supplementary Table 9).
The 180 metagenomic data sets (nine time points, 20 mesocosms per time point) spread across 48 weeks allows the abundance and distribution of SGEs and associate genes to be determined by read mapping. The spatial and temporal dynamics of two phage-like contigs and two contigs encoding β-glucosidases are shown in Fig. 3.
To determine whether the horizontal transfer regime affected the abundance and distribution of functional genes, the MG-RAST database was interrogated with sequence reads from all T0 and all T24 HCs and VCs (Supplementary Table 7). The relative abundance of reads assigned to 13 of 28 functional categories at Subsystems Level 1 (REF), including genes involved in virulence, motility and nitrogen metabolism, changed significantly during the course of the year (Supplementary Data Fig. 5a). HCs and VCs did not always change to the same extent, or in same direction (Supplementary Data Fig. 5a, b). No significant change occurred in the overall category of carbohydrate metabolism (Supplementary Data Fig. 5b), but a significant increase in β-glucosidase metabolism — the rate limiting step in cellulose degradation — was detected in both HCs and VCs (Supplementary Data Fig. 5c)

Differences in functional gene categories over time. A. Relative abundances of specific gene categories that were significantly different in either vertical (n=10) or Horizontal (n=10) communities compared to the T0 founding communities (before vertical/horizontal split). * = p-value < 0.05, Unpaired T-Test B. Relative abundances of specific gene categories that were not significantly different in either Vertical (n=10) or horizontal (n=10) communities compared to the T0 founding communities (before vertical/horizontal split). C. Relative abundance of genes associated with Beta-glucosidase metabolism in T0 (n=10), T24V (n=10), and T24H (n=10). Mean values are displayed with SEM indicated.

Manipulation of SGEs enhances ammonia production. To directly test whether the Horizontal transfer regime had a functional impact on ammonification the concentrations of nitrate, nitrite, and ammonia / ammonium were determined in T24 HCs and VCs revived from stored glycerol stocks. A. The nitrate (NO3-), nitrite (NO2-), and ammonia / ammonium (NH3 + NH4+) concentrations were determined at the end of the two-week incubation period for T24 HCs and T24 VCs. ** = p-value 0.0019, Unpaired T-test (Supplementary Table 10). B. Ammonia + ammonium concentrations were measured at specific time points during the two-week incubation period for HCs and VCs. * = p-value < 0.05, unpaired T-test. (Supplementary Table 11). Mean values for T24 horizontal (n=10) and T24 vertical (n=10) with minimum and maximum values are displayed in both panels.
The nitrogen-limited nature of the culture medium, combined with evidence of selection favouring genes involved in nitrogen metabolism, led to closer focus on this essential resource. The MG-RAST database was again interrogated, but this time at Subsystems Level 2, which provides information on functional categories within the broader category of nitrogen metabolism (Fig. 4a). Significant changes were observed in the relative abundance of reads mapping to genes involved in both ammonia assimilation and dissimilation (ammonification). Such changes are consistent with the nature of the selective environment. This prompted a query of the extent to which these changes may have been influenced by movement of genes via SGEs.
A modified version of the bioinformatic pipeline developed to identify sequences unique to HCs (Methods) was applied to the T24 metagenomes. T24 HC metagenomes were compared to their paired T24 VC metagenomes as well as to all metagenomes from VCs from earlier time points (Supplementary Data Fig. 6a, Supplementary Table 8). This showed a greater number of unique sequences in the HCs, but also showed a fraction of unique sequences present in VCs. The latter is to be expect given that T0 communities must contain rare sequences that through the course of the selection experiment become common — and is consistent with data on changes in abundance of genera (Fig. 2). To see whether it was possible to reduce this signal, genomic DNA samples from T0 were sequenced on the HiSeq platform resulting in additional 320 million reads per community. The effect of increasing depth of sequence made minimal impact on detection of unique sequences (Supplementary Data Fig. 6b). Nonetheless, the “deep” T0 metagenomes were used in all subsequent analyses.

Examination of potential artifacts associated with the “unique” analysis pipeline A. The percentage of “unique” Horizontal and Vertical sequences after applying the “unique” analysis pipeline with mean (n=10) and SEM indicated. B. To determine the impact of increasing the sequencing depth of the T0 communities on the final percentage of “unique” sequences the HiSeq T0 metagenomes were split into eight metagenomes each consisting of about ∼40 million sequences. The “unique” analysis pipeline was then applied using the first T0 metagenome, any sequence matches were removed, and the “unique” analysis pipeline was then repeated using the second T0 metagenome. The process was repeated with all eight T0 metagenomes while removing any sequence matches with each iteration of the
To determine the fraction of unique reads that mapped to genes involved in nitrogen metabolism, the final sets of unique reads were obtained from HCs and VCs; they were assembled, ORFs identified and functionally categorised using CDD. HCs contained significantly more unique ORFs predicted to be involved in nitrogen metabolism compared to VCs (Supplementary Data Figure S7). HCs were enriched in several functional classes of gene, especially those with predicted roles in regulation and ammonification (Fig. 4b).

Nitrogen-related genes are enriched in “unique” horizontal sequences. A. 136 nitrogen related domains were extracted from the Conserved Domain Database (CDD) based on their presence in at least one of the twenty “unique” assemblies and the abundance of all domains was determined. Mean values (n=10 for horizontal and vertical) are indicated with SEM. P-value = 0.047, Unpaired T test. See Supplementary Table 9 for domain descriptions and abundance values.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Supplementary Data – Methods
Taking this one final step we asked whether there was evidence of an effect of the horizontal treatment on community function that might be associated with movement via SGEs. To this end, and with focus on the process of ammonification (production of ammonia through either fixation, or reduction of nitrate / nitrite) the concentration of nitrate, nitrite, and ammonia were determined in the T24 HCs and VCs at the end of the two-week period immediately prior serial passage. The concentration of ammonia was significantly higher in HCs (Fig. 5a). To check the robustness of this finding the concentration of ammonia was measure during the course of a two-week period. Despite substantive variability in composition of the independent communities, a significantly greater concentration of ammonia was detected in HCs at each one of nine sampling occasions throughout the two week period (Fig. 5b).
Discussion
Microbial communities are dynamical systems defined by complex ecological interactions and evolutionary feedbacks1. The challenges associated with understanding this complexity are immense. Here we have demonstrated the utility of an altogether new approach in which we have used naturally occurring SGEs to manipulate genes of ecological significance, shown that the dynamics of these genes can be followed, and established that the magnitude of effect is sufficient to allow links to community function to be made. Central to the strategy is a simple manipulation that promotes the evolutionary success of SGEs. That this involves nothing more than frequent exposure of SGEs to new hosts, combined with routine metagenomic analysis, means it is readily transferable to a wide range of microbial communities, from experimental constructs as here, through microbiomes of plants and animals, to naturally occurring marine and terrestrial communities.
The work described here delivered numerous surprises. Among these, ability to maintain exceptionally diverse communities through the course of a year-long selection experiment, was particularly significant. The transfer regime, involving passage of 1/20th of the two-week culture into fresh medium, meant possibility for resource partitioning and cross-feeding to become established, but to witness diversity in all replicate mesocosms increase over the course of the experiment was unexpected20. Although barely touched in our analysis so far, cycles for replenishing all major nutrients were established within each mesocosm, with that involving nitrogen being most evident. Each community harboured a single dominant free-living nitrogen-fixing bacterium, but the identity — even at the genus level — differed between mesocosms. Attempts to establish such complex communities by choosing key players before the fact, would almost certainly fail. That such diversity and stability can arise from a single gram of compost speaks to an abundance of redundancy in these natural systems.
A second surprise was the speed and magnitude of the response of communities to the mixing regime. Within just two weeks of implementation, ∼17 % of DNA sequence reads within HCs were derived from allopatric communities. That these reads could be assembled into substantive contigs containing genes of ecological relevance including those involved in cellulose degradation emphasises the dynamism of processes driven by SGEs. Ability to so clearly detect movement of this DNA via comparative genomic approaches was an additional, but critical, bonus.
The association established between genes involved in nitrogen metabolism and enhanced activity of SGEs in HCs is correlative, but is nonetheless linked to horizontally transferred sequences that map to genes with predicted roles in ammonification. The association is further linked to data that demonstrate a significant effect of the experimental treatment on concentrations of ammonia in HCs (Fig. 5). Additionally, increased ammonia in the HCs is consistent with the prediction that horizontal movement of DNA will increase the rate at which community function improves.
The two processes that generate ammonia: nitrogen fixation and nitrate ammonification, both require environments devoid of oxygen (or mostly so in the case of nitrate ammonification)19, 21. It is possible that enhanced metabolic activity associated with cellulose degradation causes lower oxygen conditions at the paper surface (where community members grow as biofilms) compared to the VCs, leading to enhanced production of ammonia. Intriguingly, the cattle industry promotes addition of ammonia to feed because of beneficial effects on digestibility of plant matter in the rumen22. This raises an alternate possibility, which is that elevated ammonia levels are similarly beneficial to digestion of cellulose in the experimental mesocosms, and reflect a more rapid response to selection in the HCs.
The genomic era has done much to resolve controversy surrounding the importance of horizontal gene transfer as a driver of evolutionary change23-25. Direct evidence of gene transfer events — many facilitated by SGEs — from one organism to another are common26, 27, but the possibility that the process assumes far greater evolutionary significance when at play within communities, has, to the best of our knowledge, been little considered, let alone directly studied. That the vast diversity of DNA sequence encompassed within complex microbial communities might exist in fluid association with SGEs generating permutations of effects significantly beyond those observed through study of individual SGEs and their hosts, is both interesting and plausible.
Materials and Methods
Establishment of cellulose-degrading microbial communities
Microbial communities were initially established by placing 10 independent 1-gram samples of fresh compost into 20 mL of M9 minimal media supplemented with a 1 cm2 piece of cellulose paper (Whatman cellulose filter paper). All incubations were performed in 140 mL sterile glass bottles with loosened screw caps allowing for gas exchange between the community and the environment. Compost was sampled from the Square Theodore-Monod compost heap (Paris, France) in February 2016. During the primary incubation period of 2 weeks the piece of cellulose was suspended by a wire from the screw cap into the media allowing for paper colonization. After two weeks, the community was transferred by moving the wire-suspended cellulose paper into 20 mL of fresh M9 minimal media that also contained a new 1 cm2 piece of cellulose paper in suspension. Two more weeks of incubation provided the opportunity for colonization of the new piece of suspended cellulose paper thus eliminating the need for a wire-suspended community for each transfer. All community transfers were performed by vortexing each bottle at maximum speed until the cellulose fibers were dissolved into a cellulose-microbial slurry.
Horizontal and vertical transfer regimes
Vertical and horizontal transfers were performed at the exact same time every two weeks. Before each transfer aliquots of the community were taken for glycerol stocks, DNA extraction, and phage storage. Glycerol stocks were created by mixing 500 µl of cellulose-microbial slurry with 500 µl of 80% glycerol and stored at -80 °C. For DNA extractions 2 mL of cellulose-microbial slurry was centrifuged at 13,000 g for 10 min and the pellet was stored at -80 °C for later processing. DNA was extracted from the founding T0 communities (before horizontal vs. vertical split) as well as transfers 1, 2, 3, 4, 10, 16, 20, 24 (T1, T2, T3, T4, T10, T16, T20, T24) using the soil DNA extraction kit (Norgen Biotek). DNA sequencing was performed using the MiSeq and NextSeq platforms for all time points and T0 communities with additional deep sequencing of the T0 communities using the HiSeq platform.
Bioinformatic analysis
DNA sequences were demultiplexed using bcl2fastq, paired ends were joined using FLASh [31] and initial fastq files were generated. Preprocessing of fastq files was performed using PrinSeq [32] with a minimum length of 100 base pairs, minimum quality score of 25, and a maximum percentage of N’s of 10%. All metagenomes were uploaded to the MG-RAST metagenomic analysis server and are publicly available (Table 1). Gene category abundances (Supplementary Data Fig. 5) and genus identifications (Table 6) were determined using the MG-RAST pipeline [33]. Sequence matches were determined using BLASTn [34] with a minimum e-value threshold of 1E−05, minimum alignment length of 100 base pairs, and a minimum percentage identity of 90%. “Unique” Horizontal and Vertical sequences were assembled using ME-GAHIT [35], ORFs with a minimum length of 100 amino acids were extracted using getORF [36], compared to the Conserved Domain Database (CDD) [37] using RPS-BLAST, and top domain hits were extracted (Supplementary Table 4). Phage-like contigs were classified based on phage-specific protein domains (Supplementary Table 3) and their distribution was determined using Fr-Hit [38]. Sequences matches to phage-like contig were based on a minimum alignment length of 100 base pairs with a percentage identity of at least 90%. Nitrogen-related genes were based on gene function classifications and descriptions with a total of 136 domains (Supplementary Table 9).
Nitrogen assays
Vertical and Horizontal communities from the one-year time point (24 transfers, T24) were re-established by placing 100 µl of glycerol stock into 20 mL of M9 minimal media supplemented with a 1 cm2 piece of cellulose paper and incubated for two weeks. 1 mL of the cellulose-community slurry was transferred to 19 mL of M9 Minimal Media with a fresh piece of cellulose and incubated for an additional two weeks. 1 mL of the community was then transferred to a new bottle and 100 µl of surrounding media was sampled at various time points during the two-week incubation period to determine Ammonia+Ammonium, Nitrate, and Nitrite concentrations. The concentrations of all three nitrogen species were determined using fluorometric assay kits according the manufacturers protocol (Ammonia Assay Kit, Sigma-Alrdrich, and Nitrate/Nitrite Fluorometric assay kit) (Figure 5 and Table 11).
Accompanying data
Links to be provided (available from authors on request)
Acknowledgements
We thank members of the Rainey lab at ESPCI and the MPI for Evolutionary Biology for valuable discussion, Boris Shraiman and Daniel Fisher for fuelling interest in the intractability of microbial communities, and Sven Kuenzel for DNA sequencing. S.Q. acknowledges receipt of funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 747527.
References




