

Abstract
Glioblastoma (GBM) is the most common malignant brain tumor, and particularly difficult to treat due to its inherent heterogeneity, which is promoted by a variety of genetic drivers. A lack of models that robustly recapitulate heterogeneity has been a major obstacle for research progress on this disease. Here we show that neural progenitor cells derived from human induced pluripotent stem cells, CRISPR/Cas9 engineered with different combinations of authentic GBM-related genetic drivers give rise to GBM models that recapitulate the pathobiology of this tumor, including inter- and intra-tumor heterogeneity, differential drug sensitivity, extrachromosomal DNA amplifications, and rapid clonal evolution. Different models established with this approach could serve as a platform for longitudinal assessment of drug treatment sensitivity governed by subtype-specific driver mutations.
One Sentence Summary hiPSC-derived GBMs recapitulate disease.
Main Text
GBM, the most common primary malignant tumor of the central nervous system (1), has been studied using different varieties of tumor models. Transgenic mouse models (2) and engineered human astrocyte-derived models (3, 4) have been utilized for decades, but application of exogenous viral factors such as SV40 T/t-Ag, HPV E6, and E7, or mutations of genes not commonly affected in GBM such as Src, K-ras, and H-ras have the potential to make these models dissimilar to the actual disease. Patient derived xenograft (PDX) models do overcome such limitations (5), but acquired passenger mutations make it difficult to study the effect of each driver mutation on different characteristics of this disease, and their direct effects on tumor initiation and evolution can only be inferred based on single cell sequencing and phylogenetic relationships.
We reasoned that introducing combinations of mutations identified by The Cancer Genome Atlas (6, 7) as GBM driver mutations into human neural progenitor cells (NPCs), potential cells of origin of GBM (8), could generate a model for studying different types of GBM in the context of an isogenic background. We first introduced two different combinations of driver mutations into human induced pluripotent stem cells (iPSCs) by CRISPR/Cas9 genome editing (9, 10) (Fig. 1, A and B). One combination of deletions targeted tumor suppressor genes PTEN and NF1, which are commonly altered together in mesenchymal subtype of GBM (6, 7). A second combination of deletions targeted TP53 and exon 8 and 9 of PDGFRA (PDGFRAΔ8-9). This creates a constitutively active truncating PDGFRA mutation observed in 40% of PDGFRA amplified GBM (11), resulting in a genotype common in the proneural subtype of isocitrate dehydrogenase-wildtype GBM (6, 7). The genetic modifications in single clones were confirmed by genotyping PCR (fig. S1) and RT-PCR (Fig. 1C). Edited iPSC clones with desired mutations were differentiated into NPCs, using a small molecule protocol (12) and differentiation status was confirmed by downregulation of pluripotency markers, Nanog and Oct4, and corresponding upregulation of NPC markers, Pax6, Nestin, and Sox1 (fig. S2). Confirmation of a neural progenitor state was illustrated by further differentiation into astrocytes, neurons, and oligodendrocytes (fig. S3).

(A) Schema of iGBM generation. (B) Designs for gene editing indicating placement of sgRNAs. (C) RT-qPCR evaluating designated edits. (D) H&E, GFAP, Olig2, and Ki-67 staining of tumors generated from engrafted PTEN−/−;NF1−/− NPCs and TP53−/−;PDGFRAwt/Δ8-9 NPCs.
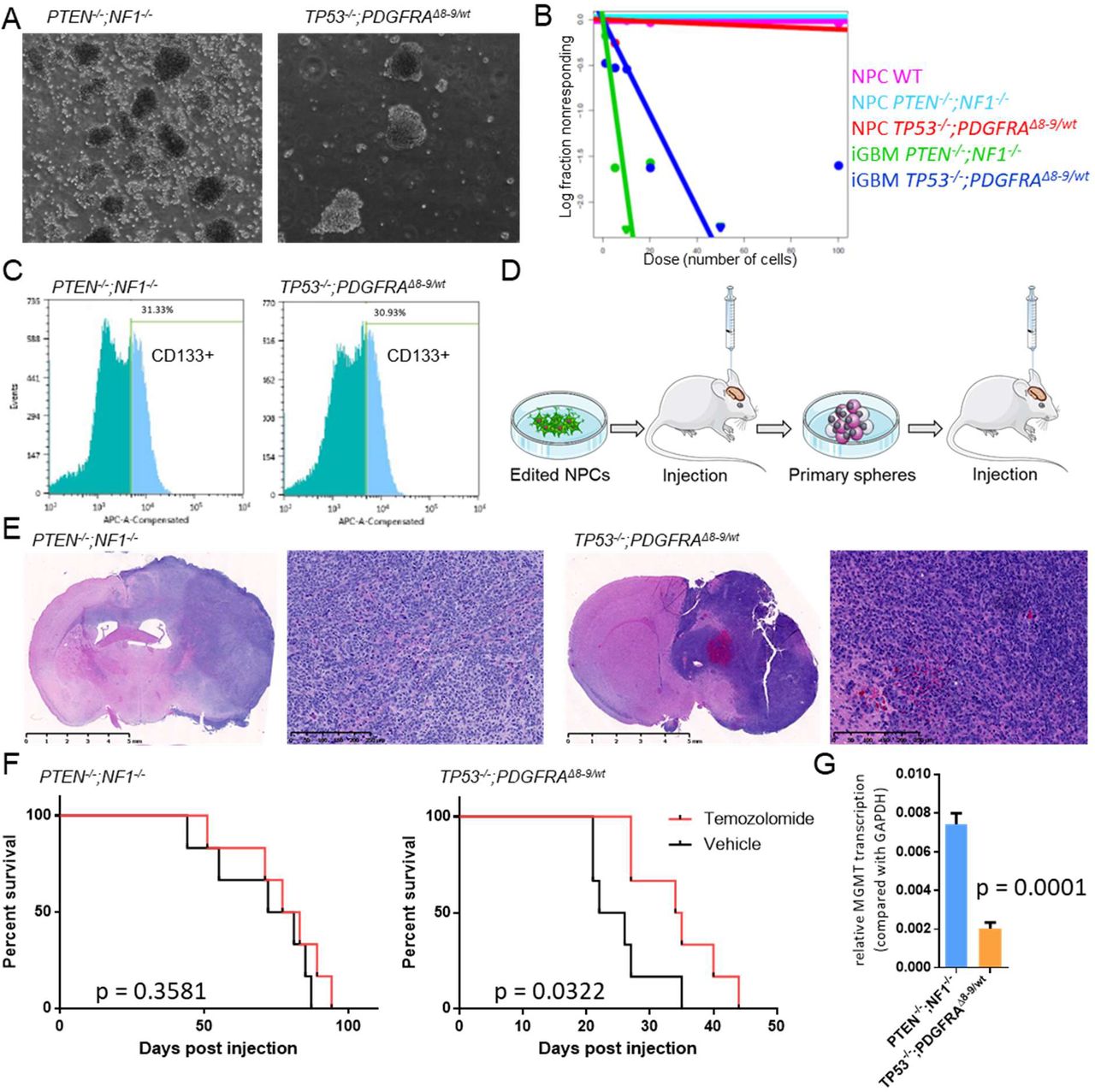
We next evaluated if these genetically modified NPCs were capable of forming orthotopic tumors in immunocompromised mice (Fig. 1A). When edited NPCs were engrafted in the brains of Nod/Scid mice, PTEN−/−;NF1−/− NPCs and TP53−/−;PDGFRAwt/Δ8-9 NPCs both formed brain tumors (Fig. 1D) with a latency of six to nine months. Pathological assessment of these tumors revealed regions of hypercellularity, positivity for GFAP and Olig2, and high expression of Ki-67, features consistent with aggressive tumors with GBM phenotype (Fig. 1D). In contrast, PTEN−/− and TP53−/− singly edited NPCs did not form tumors in the brain over the same time span (fig. S4, A and B), while unedited iPSCs formed teratoma-like tumors (fig. S5). Lack of teratomas after NPC injection suggests a high efficiency of differentiation to NPCs with the small molecule protocol. These results illustrate that small numbers of known driver mutations found in GBM are sufficient for phenotypic recapitulation of human tumors in this model system.
One of the benefits of using PDX models in cancer research is that they can be cultured in vitro and be re-engrafted in animals, thus enabling both in vitro and in vivo analyses (13). We evaluated if our induced GBM (iGBM) models could be used in a similar manner. Dissociated tumors obtained from the mouse brains were FACS sorted for human cells using a human MHC antibody, followed by propagation of isolated cells in the same neurosphere conditions used for GBM PDX spheres (14), which confirmed iGBM sphere formation capability (Fig. 2A). These iGBM spheres possessed the same genotypes as the corresponding input NPCs (fig. S6).
Extreme limiting dilution assays (15) showed that iGBM spheres had greater self-renewal capacity, a feature of cancer stem cells, when compared to pre-engraftment NPCs (Fig. 2B), again highlighting gain of cancerous phenotypes of iGBM cells compared to original input cells. The finding that only subpopulations of iGBM sphere cells expressed CD133, (Fig. 2C) a neural stem cell marker (16), while input NPCs were homogeneously positive for CD133 (fig. S7), further suggests intra-tumor heterogeneity in iGBM subpopulations displaying a cancer stem cell phenotype (17). We then evaluated if these iGBM-derived sphere cells maintained tumorigenic capacity by secondary orthotopic engraftment (Fig. 2D). When injected in the brains of Nod/Scid mice, iGBM-derived sphere cells formed tumors with a shortened latency period of one to two months (Fig. 2E). The cells obtained from these secondary iGBM tumors had even greater self-renewal capabilities compared with sphere cells obtained from the primary tumors (fig. S8), suggesting further malignant transformation through in vivo passage. We also tested if these models can be used for in vivo drug treatment experiments comparable to those applied to PDX lines by treating orthotopically engrafted animals with temozolomide (TMZ), a DNA-alkylating chemotherapeutic agent used for standard care treatment of GBM patients (18). TP53−/−;PDGFRAwt/Δ8-9 iGBMs proved to be more sensitive to TMZ compared to PTEN−/−;NF1−/− iGBMs (Fig. 2F). PTEN−/−;NF1−/− iGBMs were found to express higher levels of O6-methylguanine DNA methyl transferase (MGMT) (Fig. 2G), which is associated with resistance to TMZ in GBM patients (19), compared to TP53−/−;PDGFRAwt/Δ8-9 iGBMs, a possible explanation of this differential sensitivity, although MGMT-independent mechanisms in the context of TP53 alteration (20, 21) cannot be eliminated.

(A) iGBM spheres obtained by maintaining iGBM tumor cells in neurosphere culture conditions. (B) Extreme limiting dilution analysis of input NPCs and tumor-derived iGBM sphere cells. (C) CD133 staining of iGBM cells analyzed by flow cytometry. (D) Scheme of secondary iGBM xenograft tumor formation. (E) H&E staining of secondary xenograft tumors. (F) In vivo survival assays of mice orthotopically engrafted with primary iGBM sphere cells upon treatment either with vehicle or temozolomide. (G) MGMT expression levels in iGBM cells quantified by RT-qPCR.
We previously reported that extrachromosomal DNA (ecDNA) is prevalent in many cancer types, especially in GBM, and that ecDNA is associated with resistance to drug treatment and rapid evolution of tumor heterogeneity (22, 23). To determine if our iGBM models recapitulated the generation of ecDNA, we first investigated if the original input NPCs possessed karyotype abnormalities or traces of ecDNA. Based on DAPI staining of metaphase spreads and digital karyotyping, edited NPC were karyotypically normal (fig. S9 and S10) as were PTEN−/−;NF1−/− iGBM cells (Fig. 3A). In sharp contrast, metaphase spreads of cells obtained from TP53−/−;PDGFRAwt/Δ8-9 iGBMs showed small DAPI-stained dots adjacent to chromosomes, suggestive of ecDNA (Fig. 3B), consistent with our previous findings in GBM tumor samples (23). Furthermore, double minute-like structures became more apparent in the secondary tumors obtained by re-engraftment of the primary spheres (Fig. 3C). Those ecDNA incorporated EdU, suggesting replication of those extrachromosomal components (Fig. 3D). The TP53−/−;PDGFRAwt/Δ8-9 iGBMs also presented striking numerical and structural chromosome alterations (Fig. 3E). This is the first human stem cell-derived cancer model presenting with spontaneous extrachromosomal DNA amplifications, a frequently observed genomic characteristic of cancer (23). The involvement of TP53 in this model might have been crucial for the formation of ecDNA as has been seen in previous mouse model studies (24, 25).

(A) DAPI staining of PTEN−/−;NF1−/− primary iGBM cells. (B) DAPI staining of TP53−/−;PDGFRAwt/Δ8-9 primary iGBM cells. Red arrows indicate ecDNA. (C) DAPI staining of TP53−/−;PDGFRAwt/Δ8-9 secondary iGBM cells. Red arrows indicate ecDNA. (D) EdU labeling of chromosomes and ecDNA in a metaphase spread of TP53−/−;PDGFRAwt/Δ8-9 secondary iGBM. (E) Spectral karyotyping analysis of TP53−/−;PDGFRAwt/Δ8-9 iGBM cells.
We further investigated if these iGBMs showed inter- and intra-tumor heterogeneities, which are other notable hallmarks of GBM (26), and have not been recapitulated by previous models. First, the pattern of invasiveness was strikingly different between the two models, with PTEN−/−;NF1−/− iGBM showing more prominent diffuse invasion compared with TP53−/−;PDGFRAwt/Δ8-9 iGBM (fig. S11). These tumors were different as well in terms of transcriptomes. Single-cell RNA sequencing (scRNA-seq) was performed using primary iGBM spheres, secondary iGBM tumor cells obtained from orthotopic injection of the primary spheres, and secondary spheres obtained by in vitro culture of the secondary tumor cells (Fig. 4A). Transcriptome profiles varied between primary and secondary spheres of the same genotype as well as between spheres and tumors, but the greatest variation occurred between the two iGBM models of different genotypes (Fig. 4, B and C, fig. S12). This is suggestive of inter-tumor heterogeneity between iGBM models which wasnot apparent in pre-engraftment NPCs with different gene edits (fig. S13). These findings suggest that a small number of key driver mutations play an important role in developing such pathognomonic inter-tumor heterogeneity that arises through the process of transformation. When GBM subtype specific genes were analyzed, TP53−/−;PDGFRAwt/Δ8-9 iGBM showed upregulation of genes characteristic of the proneural subtype, while the PTEN−/−;NF1−/− iGBM showed a mesenchymal subtype signature (Fig. 4D, fig. S14, tables S1 to S4). However, when examined at single cell resolution, each sample shows intra-tumor heterogeneity with different populations of cells presenting signatures of different subtypes (Fig. 4E, fig. S14), as is observed in actual GBM patient samples (26). Within each iGBM model, there existed populations of cells with variations in cell cycle (Fig. 4F, fig. S15) and stemness signatures (Fig. 4G, fig. S16), resembling populations present in patient GBM tumors (26). Furthermore, TP53−/−;PDGFRAwt/Δ8-9 iGBM presented greater diversity compared to the PTEN−/−;NFl−/− model (Fig. 4C, fig. S17), both between samples from different tumor passages (primary vs secondary tumors and spheres) or replicates from different animals. This suggests a clonally unstable nature of the TP53−/−;PDGFRAwt/Δ8-9 model, where genomic instability or ecDNA could be driving dynamic clonal evolution (23, 27).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Scheme of single-cell RNA sequencing experiments. Primary spheres (S1), secondary tumors (T2.1-T2.3), and secondary spheres (S2.1-S2.3) from both of TP53−/−;PDGFRAwt/Δ8-9 (T) and PTEN−/−;NF1−/− (P) iGBMs were analyzed. Nomenclatures shown in quoted bold characters are used in subsequent panels of this figure. (B) Principal component analysis of all the sequenced samples. (C) Uniform Manifold Approximation and Projection (UMAP) analysis of all the sequenced samples. (D) GBM subtype analysis based on the average of individual cells in each sample. (E) GBM subtype analysis at single cell resolution. (F) UMAP showing populations with cell-cycle signatures and (G) stemness signatures.
In summary, we generated a robust system to model human glioblastoma. By introducing different combinations of essential genetic alterations into human iPSCs, resulting tumors faithfully recapitulated hallmarks of GBM pathobiology including histology, gene expression signatures, inter- and intra-tumor heterogeneity, and clonal evolution. Each model presents different characteristics depending on the background genetic alterations, and thus, we expect this to be a useful platform for examining phenotypes resulting from genetic alterations of GBM and other cancer types to derive effective treatments based on specific driver mutations.
Funding
This work was supported by National Institutes of Health R01NS080939 (F.B.F.), the Defeat GBM Research Collaborative, a subsidiary of the National Brain Tumor Society (F.B.F., and P.S.M.), Ruth L. Kirschstein Institutional National Research Award T32 GM008666 (A.D.P.), grants from National Institute of Neurological Disorders and Stroke NS73831 (P.S.M.), the Ben and Catherine Ivy Foundation (P.S.M.), and National Institutes of Health P30CA023100 (IGM Genomics Center).
Author contributions
T.K., Se.M., G.W.Y., and F.B.F conceived and designed the study. T.K., J.A.B., A.D.P., K.M.T., F.M.H., S.S., J.M., Sh.M., and A.D.B. conducted the experiments. I.A.C, N.D.N., and M.D.A. performed bioinformatics analyses. J.R., K.A.F., V.B., C.C.C., and P.S.M. supervised parts of the study. G.W.Y and F.B.F supervised all aspects of the study. T.K. and F.B.F. wrote the manuscript with inputs from other authors.
Competing interests
P.S.M. is co-founder of Pretzel Therapeutics, Inc. He has equity and serves as a consultant for the company. PSM also did a one-time consultation for Abide Therapeutics, Inc.
Data and materials availability
All data are available from the main text and supplementary materials.
Acknowledgments
We thank W.K. Cavenee, A.H. Thorne, C. Zanca, and Furnari lab members for discussions and helpful suggestions. We are grateful to the IGM Genomics Center, University of California San Diego for conducting RNA sequencing, digital karyotyping, single-cell RNA sequencing and whole genome sequencing. We thank the Center for Advanced Laboratory Medicine, University of California San Diego for assistance with immunohistochemistry. We thank M. F. Camargo for providing graphical work.
References and Notes




