

ABSTRACT
Under stressful conditions, bacterial RelA-SpoT Homologue (RSH) enzymes synthesise the alarmone (p)ppGpp, a nucleotide messenger. (p)ppGpp rewires bacterial transcription and metabolism to cope with stress, and at high concentrations inhibits the process of protein synthesis and bacterial growth to save and redirect resources until conditions improve. Single domain Small Alarmone Synthetases (SAS) are RSH family members that contain the (p)ppGpp synthesis (SYNTH) domain, but lack the hydrolysis (HD) domain and regulatory C-terminal domains of the long RSHs such as Rel, RelA and SpoT. We have discovered that multiple SAS subfamilies can be encoded in broadly distributed conserved bicistronic operon architectures in bacteria and bacteriophages that are reminiscent of those typically seen in toxin-antitoxin (TA) operons. We have validated five of these SASs as being toxic (toxSASs), and shown that the toxicity can be neutralised by six neighbouring antitoxin protein-coding genes. Thus, the ToxSAS-antiToxSAS system is a novel Type II TA paradigm comprising multiple different antitoxins, that exemplifies how ancient nucleotide-based signalling mechanisms can be repurposed as TA modules during evolution, potentially multiple times independently.
INTRODUCTION
Bacteria encounter a variety of different environment conditions during their life cycles, to which they need to respond and adapt in order to survive. This can include slowing down their growth and redirecting their metabolic resources during nutritional stress, until conditions improve and the growth rate can increase. One of the main signals that bacteria use for signalling stress are the alarmone nucleotides ppGpp and pppGpp, collectively referred to as (p)ppGpp (1). At high concentrations (p)ppGpp is a potent inhibitor of bacterial growth (2), targeting transcription (3), translation (4) and ribosome assembly (5). (p)ppGpp is produced and degraded by proteins of the RelA/SpoT homologue (RSH) superfamily, named after the two Escherichia coli representatives – multi-domain ‘long’ RSH factors RelA and SpoT (6). In addition to long RSHs, bacteria can encode single-domain RSHs: Small Alarmone Synthetases (SAS) (7-11) and Small Alarmone Hydrolases (SAH) (6,12).
It is currently unknown why some bacteria carry multiple SASs and SAHs, which can belong to many different subfamilies. Conservation of gene order through evolution can reveal potentially interacting proteins and shed light on the cellular role of proteins (13). Therefore, we developed a computational tool – FlaGs, standing for Flanking Genes (14) – for analysing the conservation of genomic neighbourhoods, and applied it to our updated database of RSH sequences classified into subfamilies. Surprisingly, we find that some subfamilies of SAS can be encoded in apparently bi- (and sometimes tri-) cistronic, often overlapping, conserved gene architectures that are reminiscent of toxin-antitoxin (TA) loci (15-18). The potential for SAS toxicity is supported by the observation that when (p)ppGpp is over-produced – for example, if synthesis by RelA is not balanced by hydrolysis by SpoT – the alarmone becomes toxic and inhibits growth (19).
The first direct evidence that RSH toxicity per se might be a bona fide function of some SASs was provided by Dedrick and colleagues (20). They showed that gp29, a SAS encoded by the mycobacterial Cluster N bacteriophage Phrann is exceedingly toxic to M. smegmatis. This toxicity is countered by co-expression of its neighbouring gene (gp30) – a proposed inhibitor of the SAS. Neither the molecular mechanism of gp29-mediated toxicity nor its neutralisation by gp30 are known. The gp29-mediated abrogation of growth is proposed to be a defence mechanism against co-infection by other bacteriophages, such as Tweety and Gaia (20).
The regulatory interplay between gp29 and gp30 is typical of that seen in toxin-antitoxin (TA) systems. When expressed, the protein toxin abolishes bacterial growth – and its toxicity can be efficiently countered by the protein or RNA antitoxin. Known toxins can act in a number of ways, commonly by targeting translation by cutting or modifying the ribosome (21), translation factors (22), tRNAs (23) or mRNAs (24). Similarly, antitoxins counteract the toxins through different mechanisms: through base-pairing of the antitoxin RNA with the toxin mRNA (type I TA systems (25)), direct protein-protein inhibition (type II (26)), inhibition of the toxin by the antitoxin RNA (type III (27)), or by indirect nullification of the toxicity (type IV (28)).
In this study we have uncovered the evolutionary diversity of SAS-based toxin (toxSAS) TA systems using sensitive in silico sequence searching and gene neighbourhood analysis. We have experimentally validated five SAS subfamilies as belonging to bona fide TA systems and demonstrated through mutagenesis that the toxicity of SASs is strictly dependent on a functional (p)ppGpp synthetase active site. Of our six identified antitoxins, five are strictly specific in counteracting only their cognate toxSAS and one other can universally neutralize all of the toxSASs. This antitoxin encodes a (p)ppGpp degrading enzyme – SAH – and acts as a type IV antitoxin degrading the molecular product of toxSAS synthetic activity.
MATERIAL AND METHODS
Identification and classification of RSH sequences across the tree of life
Predicted proteomes from 24072 genomes were downloaded from NCBI, selecting one representative genome per species for Archaea, Bacteria, Eukaryotes and all Viruses. The sequences from our previous RSH database (6) were extracted on a subfamily basis, aligned with MAFFT v7.313 (29) and hidden Markov models (HMMs) were made with HMMer v3.1b2 (30). All genomes were scanned with the HMMs to identify RSH family members and classify them into subfamilies with E value cut-off thresholds that were previously determined (6): E-4 and E-5 for SYNTH and HD domains, respectively. HMMs of the HD and SYNTH domains were used to determine the (p)ppGpp synthesising and hydrolysing domains present in the 35615 identified sequences. Sequences, taxonomy of the source organism, domain composition and subfamily memberships were stored in a MySQL database. To update the classifications with novel subfamilies, phylogenetic analysis was carried out of RSHs from representative genomes based on taxonomy (one representative per genus). Three data sets of sequences (long RSHs, SASs and SAHs) were extracted and aligned as above. Phylogenetic analysis was carried out with FastTree v2.1.10 (31) after removing positions containing more than 50% gaps, and any extremely divergent proteins that could not be confidently aligned. The three resulting trees and alignments were examined by eye with FigTree v1.4.2 and Aliview v1.2.0 (32) to identify groups that are appear to be distinct, that is, are comprised of mostly orthologues, have a distinct domain structure, and, ideally, have strong support for monophyly. Eight of our subfamilies are paraphyletic in that they contain one or monophyletic groups nested within their diversity: MixSpo, AaRel, CapRel, FpRel, FunRel, MixRel, PRel3, and Rel. To further resolve relationships among subfamilies, trees were then made with RaxML v8.2.10 (33) and IQ-TREE v1.6.6 (34) on the Cipres Science Gateway v 3.3 portal (35), excluding divergent sequences that could not be assigned to subfamilies in the FastTree tree. For Maximum Likelihood phylogenetic analyses, we used the LG model of substitution, which is the best-fit model for our dataset, as predicted by IQ-TREE. RAxML was run with 100 bootstrap replicates to give a value (maximum likelihood boostrap, MLB percentage) for how much of the input alignment supports a particular branch in the tree topology. In the case of IQ-TREE, the ultrafast bootstrapping (UFB) approximation method was used to compute support values for branches out of 1000 replicates. Trees from RAxML and IQ-TREE were visualized with FigTree and subfamilies were inspected for whether they contain mostly orthologues with at least moderate (>60%) bootstrap support. Overall, we could classify sequences into 13 subfamilies of long RSHs, 30 subfamilies of SASs and 11 subfamilies of SAHs. The sequences of each subfamily were aligned and used to make HMMs, as above. The 35615 sequences in the MySQL database were re-scanned with the updated subfamily HMMs and the database was updated, as reproduced here as two Excel files of all sequences (Supplementary Table S1), and subfamily distribution across taxonomy (Supplementary Table S2).
Phylogenetic analysis of RSH subfamily representatives
To make the representative trees of Figure 1, we selected taxa from the RSH database to sample broadly across the tree of life, and cover all subfamilies of RSHs. We used a Python script to select 15 representatives per SAS or SAH subfamily, 145 representatives for the almost universal bacterial protein Rel, and 80 representatives for RelA and SpoT, based on taxonomy of the RSH-encoding organism. The script calculates the total number of unique names on each taxonomic level (e.g., phylum, class, order, genus and species) and optimises selection of representative sequences accordingly, in order to sample taxonomy as broadly as possible within that subfamily. The (p)ppGpp hydrolase (HD) domain-containing dataset and the (p)ppGpp synthetase (SYNTH) domain-containing dataset were separately aligned with MAFFT with the L-ins-i strategy, and alignment positions with >50% gaps were removed. After alignment curation, our HD domain-containing alignment contained 698 amino acid positions from 519 sequences, and the SYNTH domain-containing alignment contained 699 amino acid positions from 722 sequences (Supplementary Text S1). The two domain alignments were used for Maximum Likelihood phylogenetic analysis using RaxML and IQ-TREE as described above.

Trees were generated from RaxML and IQ-TREE analyses of alignments of representatives across the RSH family with (A) the (p)ppGpp hydrolase (HD) domain-containing dataset (698 amino acid positions, 519 sequences), and (B) the ppGpp synthetase (SYNTH) domain-containing dataset (699 amino acid positions, 722 sequences). Shading behind the branches indicates the boundary between multi-domain type (long) RSHs and single domain (small) RSHs. The long RSH groups also contain members that seem to have secondarily lost domains through evolution to become single domain (members of the RickSpo and RickRel groups). The inset box shows the legend for subfamily and intersubfamily support, support values within subfamilies, and those that are less that 60% MLB are not shown. Branch length is proportional to the number of substitutions per site (see scale bar). The red skull and crossbones symbol indicates those subfamilies of SASs that we have confirmed with toxicity neutralisation assays to contain toxSASs. The SAH group PbcSpo that we have found contains an antitoxin is indicated with a green plus sign. Alignments used for phylogenetic analysis, and trees with all branch support values are available in Supplementary Text 1.
Analysis of gene neighbourhood conservation
To find conserved genomic architectures we used our Python tool FlaGs (14), with the NCBI accession numbers of representative RSH subfamily members (one per genus) as input. The legend files for the gene cluster numbers in all conservation figures in this paper are found in Supplementary Table S3.
Prediction of prophage-encoded RSHs
To detect if bacterial SAS or SAH genes are located in bacteriophage-like sequence regions, we used the PHASTER URLAPI (36). To create the input nucleotide data sets, we made a pipeline that takes the nucleotide sequence containing the four up and downstream genes around each SAS or SAH genes that are present in our RSH Database. The resulting PHASTER predictions are found in Supplementary Table S4.
Construction of plasmids
All bacterial strains and plasmids used in the study are listed in Supplementary Table S5. Oligonucleotides and synthetic genes were synthesized by Eurofins. Toxin ORFs were amplified using primers containing SacI and HindIII restriction sites and cloned in pBAD33 vector. To make the constructs with a strong Shine-Dalgarno motif, the 5’-AGGAGG-3’ sequence was incorporated into the pBAD33 vector. The full start codon context including the Shine-Dalgarno motif and intervening sequence was therefore 5’-AGGAGGAATTAAATG-3’. Antitoxin ORFs were amplified using primers containing EcoRI and HindIII restriction sites and cloned in a pKK223-3 vector. Ligation mixes were transformed by heat-shock in E. coli DH5α. PCR amplifications were carried out using Phusion polymerase, purchased from ThermoScientific along with restriction enzymes and T4 ligase. Point mutations were introduced using QuikChangeKit (Agilent). All final constructs were re-sequenced by Eurofins.
Toxicity neutralisation assays
Toxicity-neutralisation assays were performed on LB medium (Lennox) plates (VWR). E. coli BW25113 strains transformed with pBAD33 (encoding toxins) and pKK223-3 (encoding antitoxins) were grown in liquid LB medium (BD) supplemented with 100 µg/ml carbenicillin (AppliChem) and 20 µg/ml chloramphenicol (AppliChem) as well as 1% glucose (repression conditions). Serial ten-fold dilutions were spotted (5 µl per spot) on solid LB plates containing carbenicillin and chloramphenicol in addition to either 1% glucose (repressive conditions), or 0.2% arabinose combined with 1mM IPTG (induction conditions). Plates were scored after an overnight incubation at 37°C.
Growth assays
Growth assays were performed in liquid MOPS minimal medium (1x MOPS mixture (AppliChem), 0.132 M K2HPO4 (VWR Lifesciences), 1mg/ml thiamine (Sigma), 0.1% casamino acids (VWR Lifesciences) and the carbon source – either 0.5% glycerol (VWR Chemicals) or 1% glucose). The media was supplemented with carbenicillin and chloramphenicol. Overnight cultures were grown in MOPS medium supplemented with 1% glucose at 37°C. The cultures were diluted to a final OD600 of 0.01 in MOPS medium supplemented with 0.5% glycerol, 0.2% arabinose and 1mM IPTG. Growth was then monitored using a Bioscreen C Analyzer (Oy Growth Curves Ab Ltd) at 37°C for 10 hours.
Measurement of cellular (p)ppGpp levels by thin layer chromatography (TLC)
Overnight cultures were pre-grown at 37°C in liquid MOPS medium supplemented with carbenicillin, chloramphenicol and 1% glucose, diluted to a final OD600 of 0.05 in MOPS medium supplemented with 0.5% glycerol and antibiotics. The cultures were grown at 37°C until an OD600 of 0.5, at which point they were re-diluted to an OD600 of 0.05, and 500 µl were transferred to 2 ml Eppendorf tubes and spiked with 2.5 µCi of 32P-orthophosphoric acid (Perkin Elmer). The cultures were grown for two generations (OD600 0.2) and expression of the toxins and antitoxins was induced by addition of 0.2% arabinose and 1 mM IPTG (final concentrations), respectively. At 0, 5, 15 and 30 minutes post-induction, 50 µl samples were transferred to 1.5 mL Eppendorf tubes containing 10 µl of 2 M formic acid and pelleted for 2 minutes at 14,000 rpm 4°C. 10 µl of the resultant supernatant was spotted on PEI Cellulose TLC plates (Merck). The nucleotides were resolved in 1.5 M KH2PO4, pH 3.4 (VWR Chemicals). Plates were dried and imaged on Phosphorimager Typhoon FLA 9500 (GE Healthcare).
RESULTS
Updated RSH phylogeny across the tree of life
Our previous evolutionary analysis of the RSH protein family applied high-throughput sensitive sequence searching of 1072 genomes from across the tree of life (6). Since the number of available genomes has grown dramatically in the last decade (37), we revisited the evolution of RSHs, taking advantage of our new computational tool, FlaGs to analyse the conservation of gene neighbourhoods that might be indicative of functional associations (14). FlaGs clusters neighbourhood-encoded proteins into homologous groups and outputs a graphical visualisation of the gene neighbourhood and its conservation along with a phylogenetic tree annotated with flanking gene conservation. We identified and classified all the RSHs in 24072 genomes from across the tree of life using our previous Hidden Markov Model (HMM)-based method. We then carried out phylogenetic analysis to identify new subfamilies, generated new HMMs and updated the classification in our database (Supplementary Tables S1 and S2). We have identified 30 subfamilies of SASs, 11 subfamilies of SAHs, and 13 subfamilies of long RSHs (Figure 1).
Putative toxSAS TA modules are widespread in Actinobacteria, Firmicutes and Proteobacteria
We ran FlaGs on each of all the subfamilies and discovered that Small Alarmone Synthetase (SAS) genes can be frequently found in conserved bicistronic (sometimes overlapping) loci that are characteristic of toxin-antitoxin (TA) loci. Five SAS subfamilies displaying particularly well conserved TA-like arrangements: FaRel (which is actually tricistronic), FaRel2, PhRel, PhRel2 and CapRel (Figure 2, Supplementary File S1 and Supplementary Table S3) were selected for further investigation. Among bacteria, PhRel (standing for Phage Rel, the group to which Gp29 (20) belongs) and FaRel are found in multiple species of Firmicutes and Actinobacteria (hence the “Fa” prefix), along with representatives of various Proteobacteria; FaRel2 is found in multiple Actinobacteria, and Firmicutes, while PhRel2 is found in firmicutes in addition to bacillus phages. CapRel as a subfamily can be found in a wide diversity of bacteria (including Cyanobacteria, Actinobacteria and Proteobacteria), hence the “Cap” prefix. The putative antitoxins are non-homologous among cognate groups, with the exception of PhRel and CapRel, which share a homologous putative antitoxin (Figure 2). PhRel and CapRel are sister groups in the RSH phylogeny with medium support (81% MLB RAxML, 96% UFB IQ-TREE, Figure 1 and Supplementary Text S1), suggesting the TA arrangement has been conserved during the diversification of these groups from a common ancestor.

Genes that encode proteins belonging to a homologous cluster in more than one genomic neighbourhood are coloured and numbered (see Supplementary Table S3 for the identity of clusters with flanking gene accession numbers). The SAS gene is shown in black, and non-conserved genes are uncoloured. Validated TAs are boxed in red, and have red taxon names. SASs that we have tested and are non-toxic have purple taxon names. Purple- and green-outlined grey genes are pseudogenes and RNA genes, respectively. Bacteriophage names are in bold. Numbers on branches are percentage bootstrap support from 100 replicates.
The potential antitoxins are named with an ‘AT’ prefix to the SAS name. ATfaRel is a predicted SAH of the PbcSpo family (Figure 1), and ATphRel2 is a GepA (Genetic Element protein A) family homologue. GepA proteins, which carry the DUF4065 domain have previously been associated with TA loci (38-40), and are related to the proteolysis-promoting SocA antitoxin of the SocB toxin (41). The other potential antitoxins (ATcapRel, ATfaRel2, AT2faRel and ATPhRel) have no detectable homology to proteins or domains of known function.
toxSAS-anti-toxSAS operons encode bona fide type II and type IV TAs
We tested whether SASs encoded in conserved TA-like architectures act as bona fide TA systems using a toxicity neutralisation assay in E. coli strain BW25113 (42). Putative toxSAS and antitoxin genes were expressed under the control of arabinose- and IPTG-inducible promoters, respectively (42). We have verified five toxSASs as toxic components of bona fide TA systems: Bacillus subtilis la1a PhRel2 (Figure 3A), Coprobacillus sp. D7 FaRel2 (Figure 3B), Mycobacterium phage Phrann PhRel (gp29) (Figure 3C) and Cellulomonas marina FaRel (Figure 3D). Importantly, co-expression of putative antitoxins restored bacterial viability in all of the cases. FaRel is encoded as the central gene in a tricistronic architecture (Figure 2), and its toxic effect can be neutralised by expression of either the upstream or, to a lesser extent, the downstream flanking gene (Figure 3D). Despite the well-conserved bicistronic organisation, Mycobacterium tuberculosis AB308 CapRel (Figure 3E) initially displayed no detectable toxicity. Thus, we added a strong Shine-Dalgarno motif (5’-AGGAGG-3’) to increase the translation initiation efficiency in order to drive up its expression levels. In the case of Mycobacterium sp. AB308 CapRel, the protein became toxic. Importantly, this toxicity is readily counteracted by the antitoxin ATcapRel (Figure 3E). Mycobacterium phage Squirty PhRel (20) did not display significant toxicity even when the expression was driven with a strong Shine-Dalgarno sequence (Supplementary Figure S1A). The reason for this seems to be a large deletion in the synthetase active site in Squirty PhRel (Supplementary Figure S2). We also tested well-studied bacterial SASs that are not encoded in TA-like arrangements (Staphylococcus aureus RelP (43,44) and Enterococcus faecalis RelQ (45,46)). We detected no toxicity, even when the expression is driven by a strong Shine-Dalgarno sequence (Supplementary Figure S1B).

Representatives of groups of bi and tri-cistronically encoded toxSAS are validated as TAs: (A) Bacillus subtilis la1a phRel2:aTphRel2, (B) Coprobacillus sp. D7 faRel2:aTfaRel2, (C) Mycobacterium phage Phrann phRel:aTphRel (gp29:gp30) (D) Cellulomonas marina faRel:aTfaRel and faRel:aT2faRel and (E) Mycobacterium sp. AB308 capRel:aTcapRel. To perform the toxicity neutralisation assays on LB plates, overnight cultures of E. coli strains transformed with pBAD33 and pKK223-3 vectors or derivatives expressing putative toxSAS toxins and antitoxins, correspondingly, were serially diluted from 101 to 108-fold and spotted on LB medium supplemented with appropriate antibiotics as well as either 1% glucose (repression conditions, left) or 0.2% arabinose and 1 mM IPTG (induction conditions, right). To assay the toxicity in liquid media, bacteria were grown at 37°C in MOPS minimal media supplemented with 0.5% glycerol, 0.2% arabinose and 1mM IPTG (induction conditions). The growth curves represent the geometric mean of three biological replicates and shading represents the standard error, µ2 is the growth rate (±standard error) either upon induction of the toxin (in red) or in the absence of the toxin (in black, vector control).
The validated toxSAS toxins differ in the strength of the toxic effect in our system (Figure 3A-E): i) FaRel2 and PhRel2 are exceedingly potent and no bacterial growth is detected upon expression of these toxins from the original pBAD33 vector, ii) FaRel and PhRel are significantly weaker and small colonies are readily visible and iii) CapRel is weaker still, with toxicity requiring the introduction of a strong Shine-Dalgarno sequence in the pBAD33 vector. We have validated the observed toxicity by following bacterial growth in liquid culture (Figure 3).
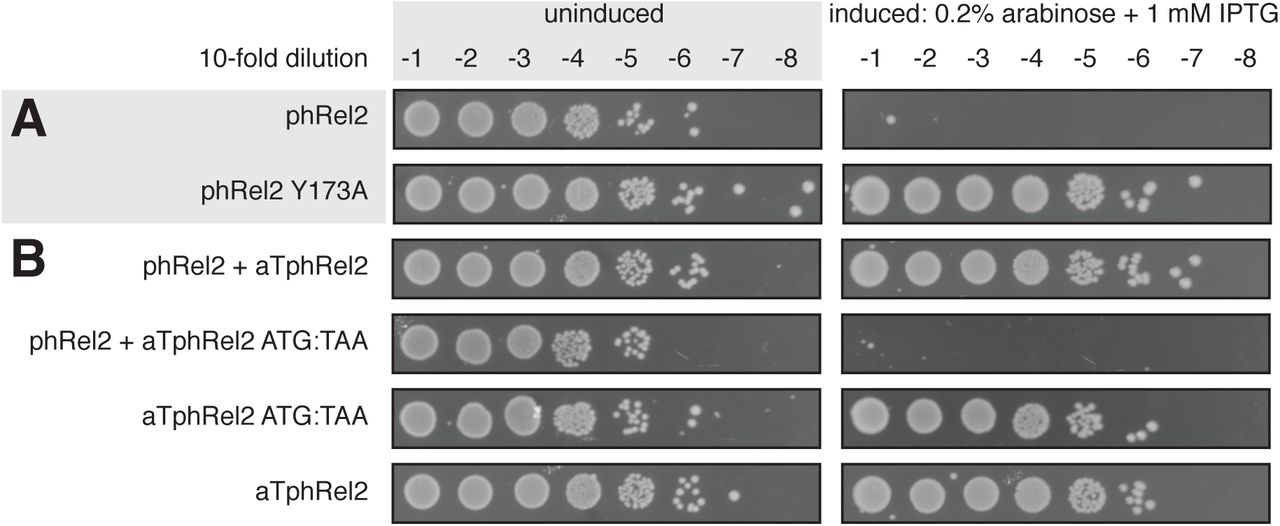
Next we tested whether enzymatic activity is responsible for the toxicity of toxSASs. To do so, we substituted a conserved tyrosine in the so-called G-loop for alanine (Supplementary Figure S3). This residue is critical for binding the nucleotide substrate and is highly conserved in (p)ppGpp synthetases (11). All of the tested mutants – PhRel2 Y173A (Figure 4A), FaRel2 Y128A, PhRel Y143A and FaRel Y175A – were non-toxic (Supplementary Figure S3). Therefore, we conclude that production of a toxic alarmone is, indeed, the universal causative agent of growth inhibition by toxSASs.

(A) Active site mutation Y173A renders phRel2 toxSAS non-toxic. Analogous experiments with all other identified toxSAS support the essentiality of the enzyme function for toxicity (Supplementary Figure S3). (B) Mutation of the start codon to stop renders the aTphRel2 antitoxin ORF unable to protect from the phRel2 toxin. Equivalent experiments of other toxSASs are presented in Supplementary Figure S4.
We then investigated whether toxSAS antitoxins inhibit toxSASs on the level of RNA (as in type I and III TA systems) or protein (as in type II and IV TA systems). The former scenario is theoretically possible, since, as we have shown earlier, E. faecalis SAS RelQ binds single-strand RNA and is inhibited in a sequence-specific manner (45). To discriminate between the two alternatives, we mutated the start codon of the aTphRel2, aTfaRel2 and aTphRel antitoxin ORFs to a stop codon, TAA. Since all of these mutants fail to protect from the cognate toxSAS (Figure 4B and Supplementary Figure S4), we conclude that they act as proteins, that is, are type II or IV antitoxins.
C. marina ATfaRel efficiently degrades (p)ppGpp and cross-inhibits all identified toxSAS SASs
The antitoxin ATfaRel is a member of the PbcSpo subfamily of SAH hydrolases (Figure 1A). This suggests it acts via degradation of the alarmone nucleotide produced by the toxSAS (and thus as a type IV TA system that does not require direct physical interaction of the TA pair). Therefore, we hypothesised that ATfaRel is able to mitigate the toxicity of all of the identified toxSAS classes through alarmone degradation. This is indeed the case (Figure 5A). To test if the hydrolysis activity is strictly necessary for antitoxin function, we generated a point mutant of ATfaRel (D54A). Mutation of the homologous active site residue of Rel from Streptococcus dysgalactiae subsp. equisimilis (RelSeq) abolishes (p)ppGpp hydrolysis (47). As expected, the D54A mutant is unable to counteract the toxicity from FaRel (Figure 5B). We used metabolic labelling with 32P-orthophosphoric acid combined with TLC separation and autoradiography to assess the accumulation and degradation of nucleotide alarmones upon expression of the C. marina FaRel toxSAS and ATfaRel SAH. The expression of FaRel results in accumulation of 32P-ppGpp, which is counteracted by wild type – but not D54A substituted – ATfaRel (Figure 5C). The location of the SAS immediately downstream of the SAH raises the question of whether this gene pair has evolved from fission of a long RSH. However, if this was the case, we would see FaRel and ATfaRel in the long RSH part of the phylogeny, which we do not see (Figure 1).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) C. marina aTfaRel neutralises the toxicity of all identified toxSAS toxins. (B) Toxicity neutralisation by C. marina aTfaRel is abolished by the D54A mutation that inactivates the hydrolytic activity of aTfaRel. (C) The expression of C. marina faRel leads to the accumulation of the alarmone ppGpp. Alarmone accumulation is efficiently counteracted by wild type aTfaRel but not its enzymatically-inactive D54A mutant. Autoradiograms of a representative TLC plate and a biological replicate are presented (Figure 5C and Supplementary Figure S7).
Class II antitoxins protect only from cognate toxSAS toxins
The gp29-mediated abrogation of growth is employed by the Phrann phage as a defence mechanism against super-infection by other phages (20). This raises the question of cross-inhibition between toxSAS TA systems: do all of the identified antitoxins inhibit all of the toxSASs (similarly to how the type IV antitoxin SAH ATfaRel protects from all of the tested toxSASs, see Figure 5A and Table 1) or is the inhibition specific to toxSAS subfamilies TAs? Therefore, we exhaustively tested pairwise combinations of all of the toxSASs with all of the antitoxins (Table 1 and Supplementary Figure S5). ATphRel2, ATfaRel2, ATphRel, ATcapRel and AT2faRel antitoxins could not counteract their non-cognate toxSASs (Table 1 and Supplementary Figure S5), demonstrating that different classes provide specific discrimination of self from non-self.
Toxicity neutralisation assays are presented in (Figure 5A and Supplementary Figures S5 and S6). Plus (+) and minus (–) symbols indicate the ability and inability of the antitoxin to neutralise toxicity, respectively.
Numerous SASs and SAHs are encoded in prophage-derived regions of bacterial genomes
Our initial search has identified 13 SASs in bacteriophage genomes, five of which we have confirmed as toxSASs (Figures 2 and 3). However, this is likely to be an underestimate for two reasons. Firstly, the currently sequenced phage genomes are a small sample of their entire diversity (48), and secondly, as prophages reside in bacterial genomes, their genes may not be identified as phage in origin. To detect SAS genes that may be phage in origin but reside in bacterial genomes, we used the tool PHASTER (PHAge Search Tool Enhanced Release (36)), taking a region of DNA equivalent to four upstream and four downstream genes around each SAS and SAH gene (one representative strain per bacterial species). In addition to the already identified phage-encoded CapRel, PhRel and PhRel2, we find 18 prophage regions around representatives in groups belonging to CapRel, FpRel, FpRel2, FunRel and RelP (Supplementary Table S4). It is notable that of RelP and RelQ (the two most broadly distributed SASs), RelP but not RelQ can be phage-associated. An evolutionary history that includes transduction may be part of the reason why the various operon structures of RelP are less well conserved across genera compared with RelQ (Supplementary File S1). SAHs are found in many more prophages and prophage-like regions than SASs (90 versus 63 instances, Supplementary Table S4). We have tested SAHs encoded by Salmonella phages PVP-SE1 (49) (PbcSpo subfamily) and SSU5 (50) (PaSpo subfamily) in toxicity neutralisation assays against validated toxSASs. Like the C. marina SAH ATfaRel, both of these stand-alone phage-encoded SAHs efficiently mitigate the toxicity of all the tested toxSASs (Table 1 and Supplementary Figure S6).
DISCUSSION
Using our tool FlaGs, we have made the striking and surprising discovery that multiple SAS subfamilies can be encoded in TA-like genetic architectures. Through subsequent experimental validation, we have found that the organisation of SAS genes into conserved TA-like bi- (and in one case tri-) cistronic arrangements is a strong indicator of toxicity. Identification of bicistronic architectures has previously been used as a starting point for prediction of TAs (51,52). However, these studies focussed on species that do not encode toxSASs, and therefore these TA systems were not detected. By being associated with novel antitoxins, toxSASs have also avoided identification in “guilt by association” analysis of thousands of genomes (53). This long-term obscurity is despite toxSAS-containing subfamilies being broadly distributed, present in 239 genera of 15 Gram-positive and -negative phyla of bacterial genomes sampled in this study. Thus, it is likely that there are other previously unknown TA systems to be found that are identifiable through searching for conservation of gene neighbourhoods across disparate lineages, as we have done with FlaGs.
The RSH protein family is widespread; most likely being present in the last common ancestor of bacteria. Thus, for billions of years, these proteins have been used by bacteria to regulate their growth rate in response to their environment by synthesising and hydrolysing nucleotide alarmones. Paradoxically, the very ability of an alarmone to downregulate growth for continued survival is also what gives it toxic potential. We have identified 30 subfamilies of SASs, five of which we have validated as containing toxins, and two of which we have validated as non-toxic (RelP and RelQ). It is likely that SASs exist on a continuum in terms of toxicity, with an antitoxin only being required at a certain level of toxicity. This is supported by the observation that not all toxSASs have the same level of toxicity, with one (M. tuberculosis AB308 CapRel) requiring a strong Shine-Dalgarno in order to observe any toxicity in our system. For our five validated toxSAS systems, there are five different homologous groups of antitoxins. This – and the lack of a multi-subfamily toxSAS-specific clade in phylogenetic analysis – suggests toxic SASs could have evolved independently multiple times from non-toxic SASs. In the evolution of a ToxSAS-antiToxSAS module from a non-toxic SAS, it is unlikely that the toxic component evolved before the regulatory antitoxin, as this would be detrimental to fitness. Rather, it is more likely that a SAS became regulated by a neighbouring gene, which relaxed enzymatic constraints on the SAS, allowing it to evolve increased alarmone synthesis rates.
Thus, we suggest that it may be useful to consider Type II TAs in the broadest sense as two (or three)-gene regulatory systems of enzyme activity rather than the toxicity necessarily being the main function in itself; especially as toxicity is usually defined using overexpression in a heterologous system (54). Nevertheless, some toxSASs do seem to have a specific role that depends on their toxicity: inhibition of superinfection in the case of the phage PhRel-ATphRel (Gp29-Gp30) toxSAS TA pair (20). In this system, PhRel encoded by a prophage protects Mycobacteria from infection by a second phage. Phage infection has previously been linked to alarmone accumulation and stringent response in bacteria (55-57). Presumably this is an example of a so-called abortive infection mechanism (58), where infected hosts are metabolically restricted, but the larger population is protected. A corollary of alarmone-mediated phage inhibition is that incoming phages could bypass this defence system by encoding alarmone hydrolases. Indeed, we have found a variety of different SAHs in different phage genomes and prophage-like regions of bacterial genomes, suggesting there could be cross-talk between ToxSASs and SAHs during infection and superinfection.
DATA AVAILABILITY
FlaGs is an open source Python application available in the GitHub repository (https://github.com/GCA-VH-lab/FlaGs).
SUPPLEMENTARY DATA
Supplementary Data are available online.
FUNDING
This work was supported by the funds from European Regional Development Fund through the Centre of Excellence for Molecular Cell Engineering to VH; support from Molecular Infection Medicine Sweden (MIMS) to VH; Estonian Science Foundation grants [IUT2-22 to VH]; Swedish Research council [grant numbers 2017-03783 to VH, 2015-04746 to GCA]; Ragnar Söderbergs Stiftelse to VH; Carl Tryggers Stiftelse för Vetenskaplig Forskning [grant numbers CTS 14:34, CTS 15:35 to GCA]; Kempestiftelserna [grant numbers JCK-1627 to GCA]; Jeanssons Stiftelser grant to GCA; Umeå Universitet Insamlingsstiftelsen för medicinsk forskning to GCA and VH; Umeå Centre for Microbial Research (UCMR) gender policy programme grant to GCA. Funding for open access charge: Swedish Research council [grant number 2015-04746 to GCA]; AGP would like to acknowledge support from the Fonds National de Recherche Scientifique [FRFS-WELBIO CR-2017S-03, FNRS CDR J.0068.19 and FNRS-PDR T.0066.18].
Conflict of interest statement
None declared.
ACKNOWLEDGMENTS
We are grateful to the Protein Expertise Platform (PEP) Umeå University and Mikael Lindberg for constructing the plasmids used in this work, and to Anaïs Poirier for help with experiments.
REFERENCES




