

Abstract
Fecal IgA production depends on colonization by a gut microbiota. However, the bacterial strains that drive gut IgA production remain largely unknown. By accessing the IgA-inducing capacity of a diverse set of human gut microbial strains, we identified Bacteroides ovatus as the species that best induced gut IgA production. However, this induction varied biomodally across different B. ovatus strains. The high IgA-inducing B. ovatus strains preferentially elicited more IgA production in the large intestine through both T-cell-dependent and T-cell-independent B cell-activation pathways. Remarkably, a low-IgA phenotype in mice could be robustly and consistently converted into a high-IgA phenotype by transplanting a multiplex cocktail of high IgA-inducing B. ovatus strains but not individual ones. Thus, microbial strain specificity is essential for the optimal induction of high-IgA responses in the gut. Our results highlight the critical importance of microbial strains in driving phenotype variation in the mucosal immune system and provide a strategy to robustly modify a gut immune phenotype, including IgA production.
Introduction
Immunoglobulin A (IgA) is the most abundant mucosal antibody and plays an essential role in maintaining gut homeostasis as well as other physiological processes1-3. Secretory IgA, for example, can limit the access of bacteria and bacteria-derived toxins to intestinal epithelial cells4,5, facilitate the clearance of bacteria that have breached the mucosal barrier6-8 and regulate the colonization of bacteria in the mucosal lining9,10. In addition, IgA can also bind disease-associated gut microbiota11-13. Conversely, the gut microbiota and its metabolites drive the production of IgA as germ-free (GF) mice have an almost undetectable level of fecal IgA14. Upon bacteria colonization, even with a single bacterial strain15-17, B cells undergo class-switch to IgA+ cells in gut-associated lymphoid tissues (GALT), which include Peyer’s patches (PP), isolated lymphoid follicles (ILF) and mesenteric lymph nodes (MLN), and in the gut lamina propria (LP)8,18. Much of the intestinal IgA is bacteria-specific15,16,19, and the B-cell repertoire is highly influenced by the microbiota composition20. To date, a few murine derived bacterial species have been identified as being able to enhance or reduce intestinal IgA production21-25. However, key questions regarding the impact of microbiota in this process remain largely unanswered including the importance of colonization order, the contribution of individual bacterial species versus that of microbial communities, the potential to modulate IgA production by altering gut microbiota composition with commensal organisms, and the role of each microbial species in the development of IgA+ B cells in specific tissues8,26.
Apart from IgA-secreting cells, the gut microbiota has the capacity to influence numerous other immune cell populations including colonic regulatory T cells27-29, IL-17 producing T helper cells30, and macrophages31. Importantly, many of these responses seem to be bacterial strain-specific as communities with comparable species composition can drive gut immune responses characterized by largely different cell compositions32. These discoveries indicate that manipulation of the gut microbiota, with appropriate bacterial strains, represents a potential therapeutic pathway for the treatment of diseases including inflammatory bowel disease, rheumatoid arthritis and multiple sclerosis through shaping the host immune system33. Although the studies of microbiota-based therapeutics (MT) and fecal microbiota transplantation (FMT) have heavily focused on the engraftment of the transmitted microbiota and its influence on the composition of the recipient microbiota34-37, the clinical application of microbiota manipulation as an immunomodulatory strategy will require combinations of bacterial strains optimized for the induction of specific immune phenotypes that are robust to the interpersonal variation in the pre-existing microbiota of each recipient.
Here we demonstrate that, upon transfer into GF mice, human isolates of the Bacteroides ovatus species, one of the most common human gut commensals, are uniquely capable of inducing high mucosal IgA production compared with other common gut commensal species. This IgA-inducing capacity, however, was restricted to specific strains of B. ovatus that preferentially led to IgA production in the large intestine through both T-cell-dependent (TD) and T-cell-independent (TI) B cell-activation pathways. While no individual bacterial strain functioned as an effective enhancer of gut IgA production, we found that cocktails of these high IgA-inducing (IgAhigh) strains could serve as effective immunomodulators, that elicited higher fecal IgA levels upon administration to animals harboring a pre-existing microbiota with low IgA-inducing potential (IgAlow). Our work demonstrates the importance of strain-level variation in gut microbiota composition on mucosal immune responses. It also supports the potential utility of cultured multi-bacterial effector strain cocktails as a strategy to overcome phenotype transfer resistance in microbiota-based immunomodulation38.
Results
B. ovatus elicits robust gut IgA production
To determine if individual gut bacterial species have a distinct IgA-inducing potential, we monocolonized GF C57BL/6 mice with one of eight different human gut commensal bacteria (Supplementary Table 1) with representatives from the most prominent phyla of the human gut including Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria39,40. After three weeks of colonization to allow optimal steady-state gut IgA secretion (Supplementary Fig. 1a), we measured serum and fecal IgA levels in each group of gnotobiotic mice16. Although all tested species significantly increased IgA level relative to control GF mice, B. ovatus monocolonized mice secreted significantly more IgA in their feces compared with mice colonized with any of the other seven human gut bacteria (Fig. 1a; p < 0.001). Most species also increased serum IgA (Supplementary Fig. 1b). However, consistent with previous reports18, fecal IgA and serum IgA levels in these mice did not correlate significantly (Supplementary Fig. 1c; R2 = 0.226; p = 0.196). GF mice colonized with the cocktail of all eight bacterial species yielded as much fecal and serum IgA as mice monocolonized with B. ovatus.

(a) Fecal IgA level in C57BL/6 gnotobiotic mice colonized with individual or a cocktail of human gut commensal bacteria for three weeks. (b-e) The concentration of fecal IgA (b and d) and proportion of each bacterial strain (c and e) in stool of gnotobiotic mice that were colonized sequentially with individual or combined bacterial communities starting from E. coli (b and c) or B. ovatus (d and e). Feces were harvested before addition of new bacteria to the same mice. C.C.R.: cocktail of C. bolteae, C. aerofaciens and R. gnavus; B.B.B.: cocktail of B. caccae, B. theta. and B. vulgatus (f) Quantification of fecal IgA in gnotobiotic mice upon colonization with an individual strain of B. ovatus for three weeks. Unique strains of B. ovatus were isolated from the stools of different human donors. Dotted line separates high- and low-IgA phenotypes. (g) Dendrogram clustering of different B. ovatus strains basing on the dissimilarity of bacterial genomic DNA sequences. (h) Correlation of stool IgA levels between single B. ovatus strain monocolonized mice versus mice colonized with a microbiota arrayed culture collection that included that single B. ovatus strain. Both single B. ovatus strains and arrayed culture collections were isolated from the same donor. (i) Correlation of fecal IgA concentration between single B. ovatus strain colonized mice versus human donor. Data shown are mean ± standard error of the mean. Each dot in a, b, d and f represents a biological replicate. The average fecal IgA concentration from 4-10 mice was used for correlation in h and i. Detailed strain information is listed in Supplementary Tables 1 and 2. p-values with statistical significance (assessed by two-tailed Student’s t test or one-way ANOVA) are indicated: ***p < 0.001; ns, not significant.
To address if the order of bacterial colonization could influence fecal IgA secretion, GF mice were sequentially colonized every three weeks with individual species or small cocktails of the same eight bacterial species. We first assayed fecal IgA level in mice sequentially colonized with low IgA inducers (e.g. E. coli) to high IgA inducers (e.g. B. ovatus). Fecal IgA increased gradually with the colonization of additional bacterial species. However, the more striking (>2-fold) increase in IgA occurred after colonization with B. ovatus (Fig. 1b). Metagenomic sequencing of fecal microbiota in these mice revealed gut colonization by each bacterial species, albeit with different proportions (Fig. 1c). We then reversed the order of colonization from high IgA inducers (e.g. B. ovatus) to low IgA inducers (Fig. 1d). Once again, B. ovatus elicited the largest increase of fecal IgA production, while the other species led to smaller increases (Fig. 1e). Remarkably, the relative abundance of each organism at the end of the colonization was very similar, regardless of the order of colonization (Fig. 1c,e). These results demonstrate that B. ovatus is a uniquely potent gut IgA inducer and that the species composition of the gut microbiota impacts IgA production more than the order of bacterial colonization.
To test the role of bacterial viability in the induction of gut IgA by B. ovatus15,41,42, GF mice were administered heat-killed B. ovatus or B. ovatus metabolites (i.e. filtered growth medium from stationary phase of B. ovatus cultures) for three weeks. Neither approach was capable of enhancing fecal IgA above the level detected in GF mice (Supplementary Fig. 1d). To ensure the above result was not due to the underdeveloped mucosal immune system of GF mice, we performed similar experiments by first colonizing GF mice with E. coli for three weeks and subsequently treated these mice with heat-killed B. ovatus for an additional three weeks. Again, we found no significant fecal IgA increase (Supplementary Fig. 1d). Thus, neither dead B. ovatus nor its metabolites triggered efficient gut IgA responses in the murine intestine. All together, live B. ovatus species elicited more gut IgA production than other tested gut commensal bacterial species in GF mice.
B. ovatus-driven gut IgA production is strain-specific
Given the remarkable microbial strain variation across individuals43-46, we wondered whether all B. ovatus strains within this common bacterial species induced comparably high fecal IgA. GF mice monocolonized for three weeks with one of 19 B. ovatus strains isolated from 19 different individuals (Supplementary Table 2) showed a strain-specific gut IgA response (Fig. 1f; p < 0.0001 one-way ANOVA). In contrast to the large variability of fecal IgA levels, serum IgA levels were comparable across mice monocolonized with different B. ovatus strains (Supplementary Fig. 1e). Similarly, the colonization density was also comparable across mice harboring different B. ovatus strains (Supplementary Fig. 1f). This observation suggests that the global density of each individual strain was not implicated in the genesis of strain-specific differences of gut IgA responses. Of note, the distribution pattern of IgA induction across multiple B. ovatus strains was bimodal (Supplementary Fig. 1g; p = 0.0481 Hartigans’ Dip Test), allowing these strains to be categorized as IgAhigh or IgAlow. The genomic similarity of B. ovatus strains was not a significant predictor of their IgAhigh and IgAlow properties (Fig. 1g and Supplementary Table 3), which suggests that their distinct IgA-inducing function is shared amongst the species rather than representing an evolutionarily distinct group within the species.
To rule out a bias in our preliminary screen for IgAlow strains within the Bacteroides genus, we assayed whether additional strains could induce high fecal IgA (Fig. 1a). We found no strain-specific differences in fecal IgA induction when GF mice were monocolonized with three distinct strains of B. caccae, B. thetaiotaomicron and B. vulgatus (Supplementary Fig. 1h). The IgA-inducing function of additional common species from the order Bacteroidales, including Parabacteroides johnsonii, Bacteroides intestinalis and Bacteroides fragilis, were tested but also induced much less gut IgA than B. ovatus (Supplementary Fig. 1h). These results indicate that the high IgA-inducing ability of B. ovatus is unique to this gut bacterial species and only to a subset of strains.
To examine the influence of B. ovatus strain variation on host fecal IgA production in the context of more complex gut microbiotas, we colonized GF mice with one of the seven microbiota arrayed culture collections originally isolated from different human donors with each collection consisting of 15-20 unique species32. The arrayed culture collections were assembled to reconstitute a donor microbiota each containing a unique B. ovatus strain, which was already functionally tested by earlier monocolonization (Fig. 1f). We observed a significant positive correlation between the fecal IgA concentrations induced by an individual B. ovatus strain and the fecal IgA concentrations elicited by a culture collection representing the entire B. ovatus-containing microbiota from the same donor (Fig. 1h; R2 = 0.859, p = 0.0027). Again, these results suggest that the B. ovatus strain composition is a major contributor of gut IgA responses even when considered in the context of complex microbial communities.
Unlike inbred laboratory mice housed in a highly controlled environment, human beings, with different genetic background, are exposed to more complex continuum of factors including some that were demonstrated to affect fecal IgA production such as genetics and diet47,48. To determine whether B. ovatus could drive robust gut IgA responses also in humans, we measured the fecal concentration of IgA in multiple human donors and correlated this concentration with that of fecal IgA generated by GF mice monocolonized with a B. ovatus strain isolated from identical donors. Though no significant correlation was observed, there was a clear trend towards a positive correlation even in an uncontrolled condition (Fig. 1i; R2 = 0.2071, p = 0.0765).
In total these results demonstrate that a subset of B. ovatus strains induce high fecal IgA levels, which broadly influence the total fecal IgA output of the host even in the context of a diverse gut microbiota.
IgAhighB. ovatus strains induce more IgA production in the large intestine
To interrogate the mechanisms underpinning gut IgA induction by different B. ovatus strains, GF mice were colonized with a representative IgAhigh or IgAlow strain (B. ovatus strain E and Q, respectively). We quantified bacteria-bound IgA in the stool of mice. Monocolonization with the IgAhigh strain E not only induced more free fecal IgA but also more fecal bacteria-bound IgA than the IgAlow strain Q did (52.9% vs. 21.0% IgA-coated B. ovatus) (Fig. 2a). In contrast, no significant difference was observed in serum immunoglobulin isotypes (i.e. IgA, IgG1, IgG2a, IgG2b, IgG3, IgM and IgE) in monocolonized mice harboring either B. ovatus strain E or Q (Supplementary Fig. 1e, 2a).

(a) Representative flow cytometry plot and quantification of IgA-coated B. ovatus in feces of gnotobiotic mice harboring either B. ovatus strain E or Q. (b) Representative images of IgA+ cells in small intestine and the colon are shown. IgA+ cells were stained with anti-IgA (green); Nuclei were counter-stained with DAPI (4’,6-diamidino-2-phenylindole) (blue). n = 5~6. Scale bar = 50 μm. (c) Percentage of IgA+ B cells in small intestinal and colonic tissues is shown. LP lymphocytes were analyzed by flow cytometry. Number adjacent to gate represents percentage. (d) Free IgA concentration in luminal contents along the length of the intestine. S.I.: small intestine. Data shown are mean ± standard error of the mean. Each dot represents an individual mouse. p-values with statistical significance (assessed by two-tailed Student’s t test) are indicated: ***p < 0.001; ns, not significant.
Fecal IgA mostly derives from polymeric IgA released by IgA+ plasma cells residing in the intestinal LP and translocated to the gut lumen across epithelial cells via transcytosis49. This process is mediated by a basolateral IgA (and IgM) transporter termed polymeric immunoglobulin receptor (pIgR)49. Independent groups have reported that the expression of pIgR by gut epithelial cells is influenced by bacteria stimulation both in vivo and in vitro50,51. To determine if B. ovatus strain variation impacts fecal IgA level by modulating pIgR-mediated transcytosis, we imaged the expression of pIgR by immunofluorescence staining in the small intestine and the colon of mice colonized with either B. ovatus strain E (IgAhigh) or Q (IgAlow). However, no noticeable difference in pIgR expression was observed (Supplementary Fig. 2b). To further interrogate the mechanism underpinning the increased fecal IgA in B. ovatus strain E colonized mice, we then quantified, by histology and flow cytometry, IgA+ B cells in both small intestine and the colon. We found more IgA+ B cells in the colonic LP of mice harboring B. ovatus strain E compared to mice harboring strain Q, while no significant strain-specific difference was observed in the small intestine (Fig. 2b,c). Although PPs and MLNs usually serve as dominant IgA inductive sites52,53, we did not observe a significant difference in IgA+ B cells at these sites by strain E or strain Q (Supplementary Fig. 3).
Given the preferential expansion of IgA+ B cells in the colons of monocolonized mice harboring B. ovatus strain E, we then explored whether luminal IgA levels would vary between small and large intestinal regions. In the small intestine, we found that mice monocolonized with B. ovatus strain E or strain Q had comparable luminal IgA levels (Fig. 2d). In contrast, mice monocolonized with strain E had significantly more luminal IgA from cecum to distal colon than those colonized with strain Q (Fig. 2d). Similar results were also observed across all tested IgAhigh and IgAlow B. ovatus strains (Supplementary Fig. 4). Thus, the IgAhigh B. ovatus strains induce more colonic IgA-secreting cells compared to IgAlow B. ovatus strains, which results in the secretion of more IgA in the large intestinal lumen.
To determine if these observations were unique to GF C57BL/6 mice, we recapitulated our monocolonization strategy in GF Swiss Webster mice and found that fecal IgA was largely comparable in gnotobiotic C57BL/6 and Swiss Webster mice colonized with identical bacterial strains (Supplementary Fig. 5a-d; R2 = 0.601, p = 0.0011). Moreover, IgAhigh strain colonized gnotobiotic Swiss Webster mice also secreted more intraluminal IgA in the large intestine compared with IgAlow strain colonized mice (Supplementary Fig. 5e). Thus, bacteria-induced gut IgA production is similar across different host genetic backgrounds.
B. ovatus elicits gut IgA production via both TD and TI B cell-activation pathways
Gut IgA responses occur through TD or TI B cell-activation pathways52,54. To determine the influence of CD4+ T cells on the gut IgA production induced by B. ovatus, we depleted CD4+ T cells in mice by injecting with an anti-CD4 antibody five days prior to and for three weeks after monocolonization with B. ovatus strain E (Fig. 3a and Supplementary Fig. 6a-c). On day seven post-colonization, fecal IgA increased in both T cell-depleted and T cell-sufficient gnotobiotic mice, which suggests that CD4+ T cells are not a dominant factor in early stage IgA induction. By day 14 post-colonization, control mice receiving an isotype-matched irrelevant antibody generated significantly more fecal IgA than mice receiving anti-CD4 antibody (Fig. 3b). In addition to reduced free IgA, B. ovatus-bound IgA also decreased in the stool of CD4+ T cell-depleted mice (Fig. 3c). In both small intestine and the colon, the frequency of IgA+ B cells was reduced by approximately 1/3 compared to that of IgA+ B cells being detected in the control CD4+ T cell-sufficient mice (Fig. 3d and Supplementary Fig. 6d,e). In addition, these control mice showed more intraluminal IgA than CD4+ T cell-depleted mice across the whole intestinal tract (Fig. 3e).

(a) Schematic representation of CD4+ T cells depletion in GF B6 mice is illustrated. Red arrows represent i.p. injection of anti-CD4 antibody or isotype control. Black arrow indicates B. ovatus strain E inoculation and blue arrows represent time. (b) Dynamics of fecal IgA concentration in B. ovatus strain E inoculated gnotobiotic B6 mice treated with either anti-CD4 antibody or isotype control. (c) Representative flow cytometry plot and quantification of IgA-coated bacteria in feces of B. ovatus strain-E-colonized gnotobiotic mice treated with either anti-CD4 antibody or isotype control. (d) Representative flow cytometry plot and percentage of IgA+ B cells in small intestine and the colon are shown. Numbers adjacent to gates represent percentage. (e) Concentration of free IgA in the intestinal content collected from different regions of the whole intestinal tract is shown. S.I.: small intestine. Data shown are mean ± standard error of the mean. Each dot represents an individual mouse. p-values with statistical significance (assessed by two-tailed Student’s t test) are indicated: **p < 0.01, ***p < 0.001; ns, not significant.
Multiplex cocktail of B. ovatus strains robustly modify gut IgA production
Given the potential of gut microbiota manipulation as a therapeutic, we next determined whether the high-IgA phenotype could be transferred to mice harboring microbiotas that induce a low level of fecal IgA. For this purpose, we recolonized GF C57BL/6 mice with either B. ovatus strain E (IgAhigh) or Q (IgAlow) for three weeks, followed by cohousing these mice for an additional three weeks (Fig. 4a). After cohousing, mice monocolonized with B. ovatus strain Q showed no significant change in fecal IgA. In contrast, mice colonized initially with B. ovatus strain E had reduced fecal IgA, which raised the possibility that the low-IgA phenotype behaves as a dominant character in the context of this simple bacterial community (Fig. 4b). Interestingly, the IgAlow B. ovatus strain Q also dominated the relative abundance of the microbiota, as it represented ~95% of the microbiota compared with ~5% of B. ovatus strain E (Fig. 4c). In an attempt to overcome this resistance to transfer of the high-IgA phenotype to mice with low-IgA phenotype, we performed a similar experiment but added three more IgAhigh B. ovatus strains. Under these conditions, the high-IgA phenotype was transfered to the cohoused mice initially monocolonized with the IgAlow strain (Supplementary Fig. 7a). However, B. ovatus strain Q still represented a substantial proportion (32.5 ~ 53.8%) of the relative abundance in this bacterial community (Supplementary Fig. 7b). Thus, a multiplex cocktail of bacterial effector strains that each individually can induce a specific phenotype provides a more robust strategy for transferring a high-IgA phenotype.
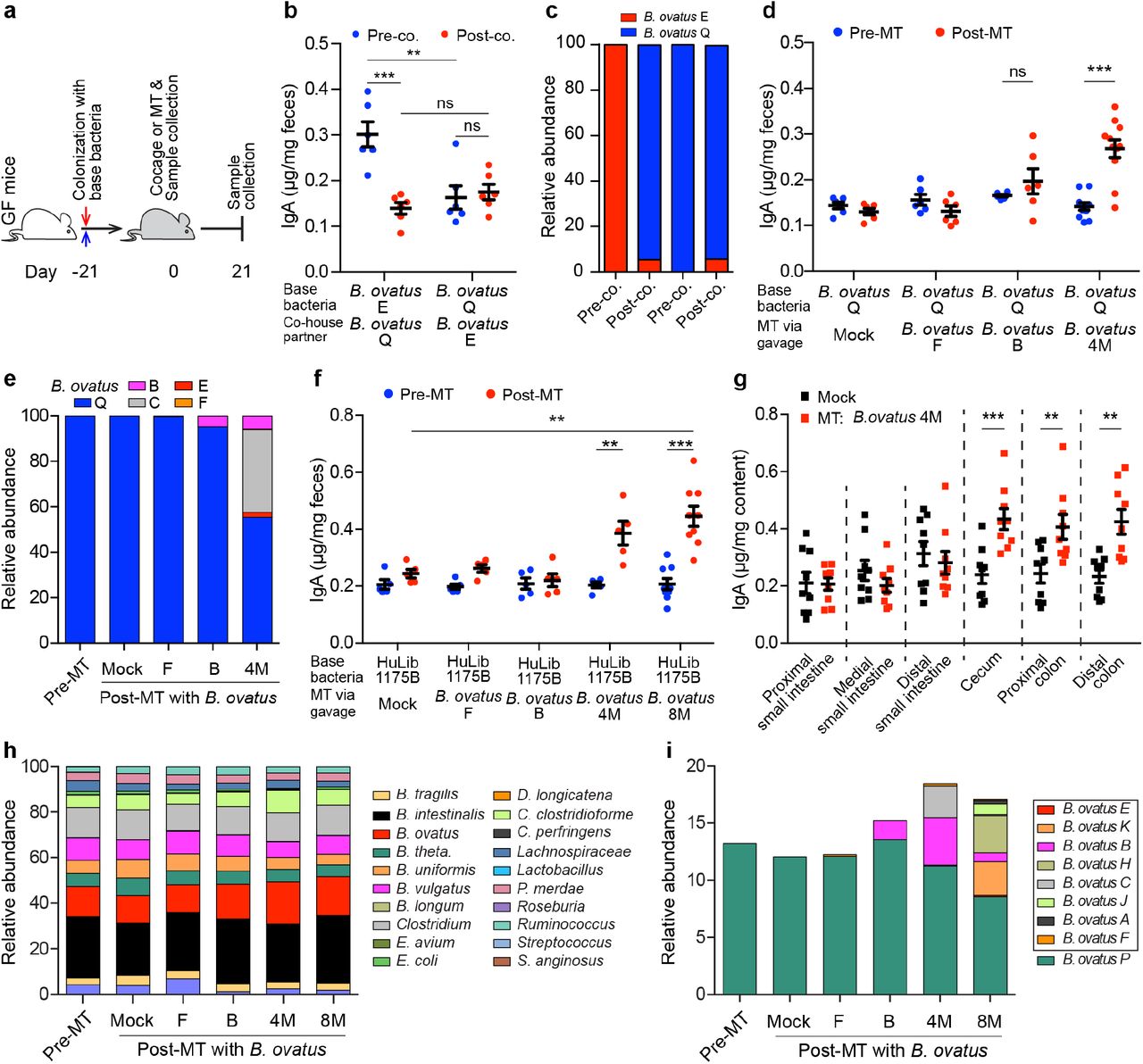
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(a) Schematic representation of cohousing and microbial therapeutic strategies. (b and c) Fecal IgA concentration (b) and relative abundance of each B. ovatus strain (c) in pre- and post-cohoused gnotobiotic mice, which were pre-colonized with either B. ovatus strain E or Q. (d and e) Fecal IgA concentration (d) and relative abundance of each B. ovatus strain (e) in mice pre- and post-microbiota-based therapeutic (MT). Mice were first colonized with B. ovatus strain Q for three weeks and subsequently administered a microbial therapeutic comprised of either an individual IgAhigh B. ovatus strain or a cocktail of IgAhigh B. ovatus strains. (f) Fecal IgA concentration in mice pre- and post-MT, which were pre-colonized with human microbiota arrayed culture collection (e.g. HuLib1175B) for three weeks. The therapeutic was either an individual IgAhigh B. ovatus strain or a cocktail of IgAhigh B. ovatus strains. B. ovatus 4M: a cocktail of 4 different IgAhigh B. ovatus strains; B. ovatus 8M: a cocktail of 8 different IgAhigh B. ovatus strains. (g) Free IgA concentration along the intestinal tract of mice after gavage with Mock or B. ovatus 4M. (h) Relative abundance of bacterial species in mice pre- and post-MT. (i) Relative abundance of different B. ovatus strains in mice pre- and post-MT. Data shown are mean ± standard error of the mean. Sequencing plots display the average abundance from five mice. Each dot represents a biological replicate. Detailed strain information is listed in Supplementary Tables 2 and 6. p-values with statistical significance (assessed by two-tailed Student’s t test) are indicated: **p < 0.01, ***p < 0.001; ns, not significant.
Beyond cohousing, we further validated the above findings by transferring IgAhigh strains therapeutically by oral gavage. Consistent with the cohousing results, mice first colonized with an IgAlow B. ovatus strain and then orally gavaged with an additional IgAhigh strain did not alter gut IgA secretion. In contrast, mice receiving a cocktail of four IgAhigh B. ovatus strains (B. ovatus 4M) produced significantly more fecal IgA (Fig. 4d and Supplementary Table 4). Metagenomic sequencing results demonstrated that multiple B. ovatus strains colonized the recipient mice (Fig. 4e). Of note, IgAlow B. ovatus strain Q still dominated the relative abundance of the gut microbiota in individual strain transfers (Fig. 4e). IgAhigh strains accounted for 44% of the gut microbiota in the B. ovatus 4M transfer with each individual IgAhigh strain having a distinct relative abundance (Fig. 4e). Finally, we replicated these results in mice pre-colonized with another IgAlow B. ovatus strain R (Supplementary Fig. 7c,d).
To validate these results in the setting of more complex gut microbiotas, we performed similar experiments using either gnotobiotic mice colonized by a synthetic cocktail of diverse bacterial species that included B. ovatus IgAlow strain Q (Supplementary Table 5) or gnotobiotic mice colonized with arrayed culture collections established from donors harboring a functionally validated IgAlow B. ovatus strain (Fig. 1h and Supplementary Table 6). As with simpler communities, transfer of the high-IgA phenotype was robustly achieved with B. ovatus 4M or a multiplex cocktail of eight IgAhigh B. ovatus strains (B. ovatus 8M) (Supplementary Table 4) but not by individual IgAhigh B. ovatus strains (Fig. 4f and Supplementary Fig. 7e). Consistent with our previous findings, IgA was elevated only in the large intestine (Fig. 4g and Supplementary Fig. 7f). The relative proportions of each IgAhigh strain and total relative abundance of all IgAhigh strains in the stool of multiplex bacterial cocktail recipient mice varied across recipient microbiota communities (Fig. 4h,i and Supplementary Fig. 7g,h).
To further validate the IgA-inducing properties of our multiplex IgAhigh B. ovatus cocktails, we tested these cocktails in two additional gnotobiotic mouse models colonized by human microbiota arrayed culture collections with low-IgA potential (Supplementary Table 6). Again, we found that the multiplex IgAhigh B. ovatus cocktails robustly increased fecal IgA (Supplementary Fig. 8a-f). Across all of the tested B. ovatus 4M and B. ovatus 8M recipients, we did not find a correlation between the total relative abundance of IgAhigh strains and the fecal IgA levels, which indicates that maximizing the total abundance of IgAhigh B. ovatus strains does not necessarily increase gut IgA production (Supplementary Fig 8g-i). In summary, our results demonstrate that transfer of multiplex IgAhigh B. ovatus strain cocktails, but not that of individual IgAhigh strains, consistently and robustly modulates the immune system (e.g. IgA phenotype) across several complex pre-existing gut microbiota.
Discussion
Functional differences of pathogenic bacteria at the strain level have been intensively studied in the past decades and are a fundamental component of infectious disease clinical practice. More recently the functional impact of bacterial strain variation is becoming apparent in the context of the protective or disease-enhancing properties of the commensal microbiota11,12,32,55,56. Here, we identified that approximately half of the isolated strains from B. ovatus species, which is one of the most common species of our gut commensal microbiota, drive increased IgA production in the distal intestinal tract. Interestingly, we did not find that the variation in fecal IgA induced by different B. ovatus strains was related to unique genetic lineages amongst strains or the density of the bacteria in the feces. Through manipulation of the pre-existing gut microbiota composition, we discovered that cocktails of IgAhigh B. ovatus strains were more efficient than individual IgAhigh B. ovatus strains in converting mice with low gut IgA production into mice producing large amounts of gut IgA.
IgAhigh B. ovatus strains increased IgA production in distal but not proximal intestinal segments by enhancing the ratio of IgA-secreting B cells. Remarkably, this induction was not dominated by the migration of IgA+ B cells from canonical IgA inductive sites, as gnotobiotic mice colonized with either IgAhigh or IgAlow B. ovatus strains showed comparable IgA+ B cells in PPs and MLNs. One possibility is that IgAhigh B. ovatus strains locally elicit IgA production in the large intestine including cecal patches, ILFs and LP2,54,57. Interestingly, mice harboring specific B. ovatus strains showed no significant differences in the intestinal abundance of B. ovatus, which further highlights the unique IgA-inducing properties of individual strains.
After IgAhigh B. ovatus strain colonization, CD4+ T cell-depleted mice showed a reduced ratio of IgA+ B cells in the gut, in turn leading to decreased luminal IgA along the entire intestinal tract. Of note, both CD4+ T cell-sufficient and T cell-depleted mice produced comparable level of gut IgA at the beginning post colonization, which suggests CD4+ T cells play less of a role during the very early stage of IgA induction likely due to the dominance of the TI B cell-activation pathway. Interestingly, a protein from the gut commensal Lactobacillus rhamnosus was recently shown to locally elicit IgA production via gut epithelial cells58. Thus, further studies will be needed to delineate the precise mechanisms whereby IgAhigh B. ovatus strain colonized mice generate gut IgA. Nevertheless, our study highlights the important contribution of T cells in bacteria-mediated IgA production, especially in the large intestine59,60.
FMT-based manipulation of the gut microbiota has a high success rate in the treatment of recurrent C. difficile infection61. However, its success in other indications, such as ulcerative colitis, is more limited62,63. Although improving bacteria engraftment remains a key therapeutic goal of microbiota manipulation62-64, identifying new strategies that optimize the transfer of a specific immune phenotype constitutes a therapeutic goal with a potentially larger range of applications. Using IgA induction as an example of immunomodulatory phenotype transfer, our data showed that multiplex bacterial cocktails of IgAhigh B. ovatus strains elicited a more robust phenotype transfer than any individual strain, even in mice with complex gut ecosystem. This multiplex effector strain cocktail strategy was robust across multiple recipients, who had low-IgA phenotype and pre-colonized with different microbiotas, and could represent an effective approach to therapeutically modify gut immune parameters in addition to IgA. Of note, across the tested inductions of IgA via gut microbiota manipulation with IgAhigh B. ovatus strains, we found that no single strain consistently dominated over the others. Thus, multiplex bacterial cocktails do not appear to have “super strains” with dominant IgA-inducing function. Rather, the combination of multiple IgAhigh effector strains in these cocktails has an IgA-inducing potential superior to that of any individual strain. Intriguingly, the relative abundance of total B. ovatus species remained largely stable even after the introduction of one to eight new strains suggesting that these new strains largely share the same ecological niches as that occupied by the pre-existing B. ovatus.
In summary, our results highlight the importance of bacterial strain variation on the IgA-inducing potential of the gut microbiota. In addition, we also identify a new strategy (e.g. multiplex bacterial strain cocktail) for the exploitation of strain variation in the development of robust microbiota-based immunomodulation therapeutic strategies.
Methods
Mice
Germ-free C57BL/6 and Swiss Webster mice were bred and maintained in flexible film gnotobiotic isolators (Class Biologically Clean, Ltd.). All mice were group housed with a 12-hour light/dark cycle and allowed ad libitum access to diet and water. All animal studies were carried out in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) in Icahn School of Medicine at Mount Sinai.
Colonization of germ-free mice with cultured bacteria
Germ-free mice (~8 weeks old) were colonized 200-μl aliquot of bacteria suspension via oral gavage. Colonized mice were housed in flexible film vinyl isolators or in filter top cages using previously described techniques43.
Growth and isolation of bacterial strains
All bacterial strains were obtained from previously banked stool, public culture repositories or human gut microbiota arrayed culture collections27. All bacterial strains isolated for this study were isolated from deidentified stool samples from individuals under a Mount Sinai IRB approved protocol (IRB-16-00008). All bacteria apart from E. coli were grown under anaerobic condition at 37°C in Brain Heart Infusion (BHI) medium supplemented with 0.5% yeast extract (Difco Laboratories), 0.4% monosaccharide mixture, 0.3% disaccharide mixture, L-cysteine (0.5 mg/ml; Sigma-Aldrich), malic acid (1 mg/ml; Sigma-Aldrich) and 5 μg/ml hemin. E. coli was cultured in LB Broth Miller (EMD Chemicals, Inc.) under aerobic condition at 37°C.
Quantification of immunoglobulins by ELISA
Total fecal and serum IgA were measured by sandwich ELISA. For total IgA detection, ELISA plates (Corning 3690) were coated with 1 μg/ml goat anti-mouse IgA (SouthernBiotech, AL) capture antibody overnight at 4°C. Plates were washed and blocked with 1% BSA in PBS for 2 h at room temperature. Diluted samples and standards were added and incubated overnight at 4°C. Captured IgA was detected by horseradish peroxidase (HRP)-conjugated goat anti-mouse IgA antibody (Sigma-Aldrich). ELISA plates were developed by TMB microwell peroxidase substrate (KPL, Inc.) and quenched by 1 M H2SO4. Colorimetric reaction was measured at OD = 450 nm by a Synergy™ HTX Multi-Mode Microplate Reader (BioTek Instruments, Inc.). Other serum immunoglobulins (IgG1, IgG2a, IgG2b, IgG3, IgM and IgE) were also detected using sandwich ELISA with the following capture and detection antibody pairs (all the following antibodies were purchased from SouthernBiotech, AL): goat anti-mouse IgG1, goat anti-mouse IgG2a, goat anti-mouse IgG2b, goat anti-mouse IgG3, rat anti-mouse IgE, rat anti-mouse IgM, goat anti-mouse IgG-HRP, goat anti-mouse IgE-HRP and goat anti-mouse IgM-HRP. Corresponding mouse immunoglobulin isotypes were used as standards.
Depletion of CD4+ T Cells in germ-free mice
In vivo depletion of CD4+ T cells was performed as described65. Briefly, gnotobiotic mice (8 weeks old) were first injected intraperitoneally (i.p.) with anti-mouse CD4 monoclonal antibody (Bio X Cell, clone GK1.5) or matched isotype control (Bio X Cell, clone LTF-2) at 0.5 mg/day/mouse for 3 consecutive days. Then the injection was performed every 3 days for a period of 3 weeks. Five days after the first antibody injection, mice were inoculated via oral gavage with B. ovatus strain E. Efficacy of T cell depletion was evaluated by flow cytometry.
Lymphocyte isolation from tissues
To isolate mononuclear cells from Peyer’s patches (PPs), PPs were excised from mouse small intestines and incubated in dissociation buffer, containing Hank’s Balanced Salt Solution (HBSS) without Ca2+ and Mg2+ (GIBCO), 10% fetal bovine serum (FBS), 5 mM EDTA and 15 mM HEPES, at 37°C for 30 min. Later, tissues were mechanically separated by pushing them through a 70 μm strainer into Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 2% FBS. Filtered cells were spun down, washed and resuspended in IMDM/2%FBS. Lamina propria lymphocytes were isolated as described27. Briefly, small intestines and colons were excised, followed by removing visceral fat and intestinal contents. Tissues were opened longitudinally, washed twice in HBSS and incubated in dissociation buffer for 30 min at 37°C with mild agitation to remove epithelium and intraepithelial lymphocytes. Tissues were then washed three times in ice cold HBSS, cut into ~2 cm pieces and digested with collagenase (Sigma-Aldrich), DNase I (Sigma-Aldrich) and dispase I (Corning). Cell suspensions were filtered through 70 μm cell strainers, washed three times, and resuspended in IMDM/2%FBS. Mesenteric lymph nodes were separated from mesenteric fat and dissociated in IMDM/2%FBS by physically pressing the tissues between the frosted portions of two glass microscope slides. The cell suspension was filtered through a 70 μm cell strainer, washed three times and resuspended in IMDM/2%FBS
Detection of IgA-coated bacteria in feces
IgA-coated fecal bacteria were measured by flow cytometry as previously described12,13. Briefly, mouse fecal pellets, stored at −80°C freezer after collection, were dissolved in PBS to a final concentration of 100 milligram per milliliter PBS by weight, thawed at room temperature, homogenized in vortex mixer and centrifuged at 4°C to remove large particles. The supernatant was passed through a 40 μm sterile nylon filter and 50 μl aliquot of the bacteria suspension was collected for staining. Bacteria were pelleted by centrifugation and washed in 1ml of PBS/1%BSA/2mM EDTA for 3 times. Non-specific binding sites were first blocked with 50 μl PBS/1%BSA/20% rat serum for 20 min at 4°C. Bacteria were then stained with 50 μl of PBS/1%BSA/2mM EDTA buffer containing 1:100 dilution of monoclonal rat anti-mouse IgA antibody (eBioscience, clone mA-6E1) for 30 min at 4°C. After washing 3 times, bacterial pellets were resuspended in PBS containing SYBR Green I (1:100,000 dilution; Invitrogen). Samples were run through a BD LSR Fortessa™ cell analyzer and further analyzed by FlowJo software (Tree Star, Inc.). Only SYBR positive events were regarded as bacteria and gated for further quantification of IgA-coated bacteria (Supplementary Fig. 9).
Flow cytometry analysis and antibodies
Isolated mononuclear cells were washed in PBS and incubated with Zombie Aqua™ dye (BioLegend) to distinguish live and dead cells. Before surface staining, non-specific binding of immunoglobulin to Fc receptors was blocked by anti-mouse CD16/32 antibody (BD Biosciences). Cells were stained in FACS buffer (PBS without Ca2+/Mg2+ supplemented with 2% FBS and 2 mM EDTA) containing a mix of antibodies for 30 min at 4°C. The following antibodies were purchased from BioLegend if not indicated otherwise: anti-mouse CD45 (clone 30-F11), anti-mouse/human CD45R/B220 (clone RA3-6B2), anti-mouse GL7 (clone GL7), anti-mouse CD4 (clone GK1.5), anti-mouse IgA (eBioscience, clone mA-6E1). For the staining of IgA+ cells, both surface and intracellular staining were performed. Multi-parameter analysis was conducted with BD™ LSR II flow cytometry and analyzed with FlowJo software (Tree Star, Inc.). Only live cells and singlets were used in all analyses (Supplementary Fig. 9).
Extraction of bacterial DNA from feces
Each murine fecal pellet was collected into a 2 ml screw cap tube (Axygen Scientific, SCT200SSC) and stored at −80°C freezer until processing. Each sample was mixed with 1.3 ml of buffer, composed of 282 μl of DNA buffer A (20 mM Tris pH 8.0, 2 mM EDTA and 200 mM NaCl), 200 μl of 20% SDS (v/w), 550 μl of Phenol:Chloroform:IAA (25:24:1) (Ambion, AM9732) and 268 μl of Buffer PM (Qiagen, 19083), and 400 μl of 0.1 mm diameter zirconia/silica beads (BioSpec, 11079101z). Next, the sample was mechanically lysed with a Mini-Beadbeater-96 (BioSpec, 1001) for 5 min at room temperature. After centrifuging for 5 min at 4000 rpm (Eppendorf Centrifuge 5810 R), all aqueous phase was collected, mixed with 650 μl of Buffer PM thoroughly before running through a Qiagen spin column. The column was washed twice with Buffer PE (Qiagen, 19065). Attached DNA was eluted with 100 μl of Buffer EB (Qiagen, 19086) and quantified with Qubit™ dsDNA Assay Kit (Thermo Fisher Scientific, Q32853/Q32854). Bacteria density was calculated by the following equation: Bacteria Density = DNA yield per sample (ug) / weight of sample (mg)66.
Bacterial genome and metagenomic sequencing
Purified bacterial template DNA (~250 ng) was sonicated and prepared using the NEBNext® Ultra™ II DNA Library Prep kit. Samples were pooled and sequenced with an Illumina HiSeq 4000 with pair-end 150nt reads. Metagenomic sequencing reads were mapped back to the reference genomes for each experiment to determine the relative abundance of each strain. To uniquely distinguish each strain, 100K sequencing reads for each sample were mapped to the unique regions of each genome and final abundances were scaled by the unique genome size of each strain (i.e. genome equivalents), as previously described67.
Immunofluorescence staining
Immunofluorescence staining was performed as described previously25,60. Briefly, intestinal tissues were fixed in 10% neutral formalin overnight at 4°C, dehydrated in 15% and 30% sucrose buffer sequentially and mounted in O.C.T Embedding Compound (Electron Microscopy Sciences). Cryostat sections (~8 μm) were prepared, blocked with anti-CD16/32 antibody in 10% (v/v) rat serum/0.1% Triton-X100 in PBS for 30 min at room temperature and incubated with the indicated primary antibodies at 4°C overnight. The following primary antibodies were used: rat anti-mouse IgA-FITC (1/300 dilution; eBioscience, clone mA-6E1), goat anti-mouse pIgR (1/500 dilution; R&D Systems, cat #: AF2800). Slides were washed in PBS for three times, incubated with Alexa Fluor®-conjugated species-specific secondary antibody (1/400 dilution; Invitrogen) for 1 h at room temperature if needed and finally mounted with ProLong® Gold Anti-fade Reagent with DAPI (Invitrogen). Fluorescence images of sections were acquired with a LSM780 confocal laser-scanning microscope (Carl Zeiss) and further processed in ImageJ if necessary.
Statistical analysis
Data are shown as mean ± SEM. Statistical significance between two groups was assessed by an unpaired, two-tailed Student’s t test. Comparisons among three or more groups were performed using One-way ANOVA. Bimodality distribution of IgA levels induced by different B. ovatus strains was performed in R (R package ‘diptest’). For correlation test, Pearson correlation coefficient was employed. Data plotting, interpolation and statistical analysis were performed using GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA) or R statistical software (version 3.2.2). A p-value less than 0.05 is considered statistically significant.
Data availability
Bacterial genomes and metagenomic sequencing reads for this study are available via NCBI BioProject accession number PRJNA518912.
Author contributions
C.Y. and J.J.F conceived the study and designed the experiments; C.Y., I.M., E.J.C., J.B., V.A., E.G., D.H., M.D. and J.J.F. collected samples and conducted the experiments; I.M. and Z.L. provided bacterial isolates; C.Y., S.M., A.C. and J.J.F. analyzed data; C.Y. and J.J.F. prepared the manuscript. All authors read and approved the final manuscript.
Competing interests
J.J.F. serves as a consultant for Janssen Research & Development LLC. The other authors declare no conflict of interests.
Acknowledgments
We are grateful to C. Fermin, E. Vazquez, and G. Escano for the husbandry of gnotobiotic mice; Drs. Anuk Das, Dirk Gevers, C. Cunningham-Rundles, B. Brown and T. Moran for helpful discussions and comments. This work was supported in part by the staff and resources of the Gnotobiotic Mouse Core Facility, the Microbiome Translational Center, the Flow Cytometry Core Facility and the Scientific Computing Division in Icahn School of Medicine at Mount Sinai. This work was supported by National Institutes of Health Grants (NIGMS GM108505 and NCCIH AT008661) and Janssen Human Microbiome Institute (to J.J.F.) and NIH DK112679 (to E.J.C.).
References




