

Abstract
The R47H variant in the microglial TREM2 receptor is a strong risk factor for Alzheimer’s disease (AD). Loss-of-function mutations in TREM2 or its adaptor TYROBP cause polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy, a systemic disease with early onset dementia. To characterize processes affected by the R47H we performed integrative network analysis of genes expressed in brains of AD patients with TREM2 R47H and sporadic AD without the TREM2 variant. In AD patients with TREM2 R47H we identified upregulation of interferon type I response and pro-inflammatory pathways, accompanied by induction of NKG2D stress ligands. In contrast, sporadic AD brains had few perturbed microglial and immune genes. In a human myeloid cell line, THP1, overexpression of normal TREM2 or its knockout revealed a profound effect of TREM2 dosage on gene networks. The effect of TREM2 R47H was complex, consistent with a partial loss of activity in conjunction with some dominant effect on pathways related to vasculature and angiogenesis. Changing the dosage of normal TREM2 in THP1 cells affected the IFN type 1 response signature, however, overexpression of TREM2 R47H was not sufficient to stimulate this pathway. We conclude that TREM2 is involved in control of the IFN type I response in myeloid cells and that its activation in TREM2 R47H AD brains is likely due to a reduced dosage of the normal TREM2 allele. Our findings indicate existence of a microglia-driven AD subtype caused by malfunction of TREM2 and possibly other genes of its network that is distinguished by gene expression phenotype.
Introduction
A proactive role of brain immune cells, microglia, in the pathogenesis of Alzheimer’s disease (AD) gained recognition after discovery of pathogenic variants in the Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) gene that is only expressed on immune cells of myeloid origin. Further support came from large-scale GWAS that identified multiple additional immune risk factors (OR 0.9-1.15, (1). Many of these immune molecules have either microglia-specific expression (e.g., CD33, the MS4As, INPP5D, SPI1) or are enriched in microglia (e.g., ABCA7, CR1, CLU). Of all microglial genetic AD risk factors, TREM2 variants confer the highest risk of disease development, especially the R47H variant that confers a risk similar to APOEε4 (OR 4.5-2.6, (2, 3)). Of note, pathogenic TREM2 variants are linked to other neurodegenerative diseases, suggesting involvement of TREM2 in shared pathogenic mechanisms. Loss-of-function mutations in the TREM2 receptor or its adaptor, TYROBP, cause polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL; OMIM # 221770), a recessive systemic disorder that presents with early onset dementia (4) or familial frontotemporal dementia (5).
TREM2 is involved in multiple aspects of myeloid cell function and interaction with other cell types. In microglia, it regulates cell chemotaxis (6), enhances phagocytosis (7, 8) and influences microglia survival, proliferation and differentiation (9). A complexity of TREM2 functioning is revealed in cell/tissue content dependent responses to stimuli of different nature, as well as their timing. For instance, whereas isolated TREM2-deficient cells increased production of pro-inflammatory molecules in vitro (7, 10), studies of intact tissues demonstrated that both pro- and anti-inflammatory roles of TREM2 in brain immunity are dependent on timing after stimulation (11), stage of pathology (12) and genetic background (13). In addition, a soluble form of TREM2, a product of cleavage or alternative splicing, may have a separate function as an extracellular signaling molecule that promotes cell survival (14, 15). Although bona fide TREM2 ligands are yet to be identified, in vitro it binds ApoE and ApoJ/CLU, known AD risk factors of lipoprotein nature (16-18). Also, TREM2 binds amyloid beta peptide activating microglia cytokine production and degradation of internalized peptide (8).
Recently, by exome sequencing of families affected with late onset AD we identified multiple carriers of the TREM2 R47H variant (19). AD patients with this variant (TREM2 R47H AD) demonstrated a shortened course of disease, pronounced synucleinopathy, changed microglial morphology and decreased level of the microglial marker Iba1, suggesting that compromised TREM2 signaling has a strong effect on microglia function in the disease. Herein, we performed RNA expression profiling of brain tissues from subjects with TREM2 R47H AD, sporadic AD (sAD) and normal aged-matched controls to assess the effect of R47H variant on brain immune gene networks. In parallel, we evaluated the effect of TREM2 knockout versus overexpression of either the wildtype protein or one with a pathogenic variant in a human myeloid cell line with active TREM2 signaling.
Results
RNA-seq reveals upregulated immune signatures in the brain transcriptome of TREM2 R47H carriers
We chose hippocampus for RNA expression analysis because this area had the most pronounced changes in microglial markers in our study of brains of R47H carriers (19). To identify gene expression changes associated with the R47H variant, we performed RNA-seq of total RNA isolated from flash frozen tissues of two TREM2 R47H carriers and three normal age-matched controls. To identify affected pathways and functions we used Gene Set Analysis (GSA) that detects coordinated changes in the expression of multiple functionally related genes (20). Table 1 shows the top 1% of differentially regulated gene sets in brains of TREM2 R47H carriers (23 of 2076 sets). Of the 16 most upregulated sets 10 belonged to immune process/function, such as cytokine metabolism (5 sets, 71 genes), antiviral response/TRAF6-mediated IRF7 activation (1 set, 30 genes) and immune response (4 sets, 61 genes). The second most upregulated group consisted of four signatures related to cell cycle/mitosis. Seven most down-regulated sets were related to ion and amino acid transport, endocytosis, glutamine metabolism and synaptic transmission (Table 1).
Top gene sets in TREM2 R47H brains are enriched by immunity-related GO terms. Shown are the top 1% of sets up/down regulated in carriers of TREM2 R47H, GSA scores ≥1 or ≤ −1. (GSA of RNA-seq data, this study). Enrichment of the same gene sets in sporadic AD was assessed separately by performing GSA of the publicly available dataset of the three sAD cohorts (GSA Blalock et al, 2004).
To distinguish whether the upregulation of immune genes was a characteristic of AD pathology (effect of disease) or due to abnormal function of the TREM2 immune receptor (effect of mutation) we performed GSA analysis on sporadic AD (sAD) data from the Blalock study (GDS810, (21)). This dataset has been used in several meta-analyses of differential gene expression in AD (22-25). The study compared hippocampi of the non-demented control group (N=9) with three progressive stages of AD: “incipient” (N=7), “moderate” (N=8) and “severe” (N=7). The TREM2 R47H and sAD groups shared a number of downregulated sets (Table 1), and their enrichment scores increased with disease progression. Notably, immune sets upregulated in the TREM2 R47H group were not enriched in sAD. We assembled a custom 156 gene set (“all immune”) that included genes from the top up-regulated immune sets. GSEA, that uses different statistics, confirmed that the “all immune” signature retained high TREM2 mutation-specific enrichment scores and was not enriched in sAD (Figure 1).

Upregulation of immune gene signatures in brains of TREM2 R47H carriers. (A) Hierarchical clustering of enriched gene sets in R47H carriers and three groups of sAD. (B-E) GSEA enrichment plots for “ALL_IMMUNE” signature in R47H TREM2 (B), and three groups of sAD (C-E).
Nanostring profiling reveals perturbed immune, microglial and AD-related genes in TREM2 R47H AD and few alterations in sAD
We aimed to confirm the immune gene upregulation in brains of TREM2 R47H carriers by an independent method on a larger set of samples. To this end, we used Nanosting nCounter, a midthroughput nucleic acid hybridization-based platform especially suitable for RNA expression analysis of degraded RNA from FFPE material. We chose a predesigned PanCancer Immune panel (770 genes) that contained the main human immunity-related pathways. We complemented this panel with a custom set of 30 neurodegeneration and microglia-related genes. AD patients with R47H (TREM2 R47H AD, N=8) were compared with sAD (N=15) and normal aged controls (N=17, Table S1). As an AD-unrelated neurodegenerative condition, we included XPDS (N=2), a monogenic disorder, X-linked Parkinsonism with Spasticity, caused by pathogenic variants in the ATP6AP2 gene ((26), OMIM# 300911). Previous studies found high concordance of RNA expression in frozen and FFPE material obtained by Nanostring nCounter (27), and we confirmed it for our samples and experimental settings. RNA isolated from flash frozen or FFPE tissue of the same individual showed high Pearson correlation of gene expression (r2 = 0.84). Of 800 genes on the panel, 551 had a detectable expression in FFPE samples and 493 of these were included in our analysis (see Materials and Methods).
The TREM2 R47H AD group had the largest number of differentially expressed genes (104 vs 20 in sAD, FDR<0.1, Figure 2A, B). 75% of differentially expressed genes in sAD (15 of 20) were shared with TREM2 R47H AD (Figure 2D). No upregulation of immune genes was observed in XPDS brains. We previously reported that XPDS in the family included herein is caused by a splicing mutation in ATP6AP2 that results in skipping of its exon 4 (26). As expected, the XPDS group showed a highly upregulated e3-e5 ATP6AP2 splice-isoform (Figure 2D).
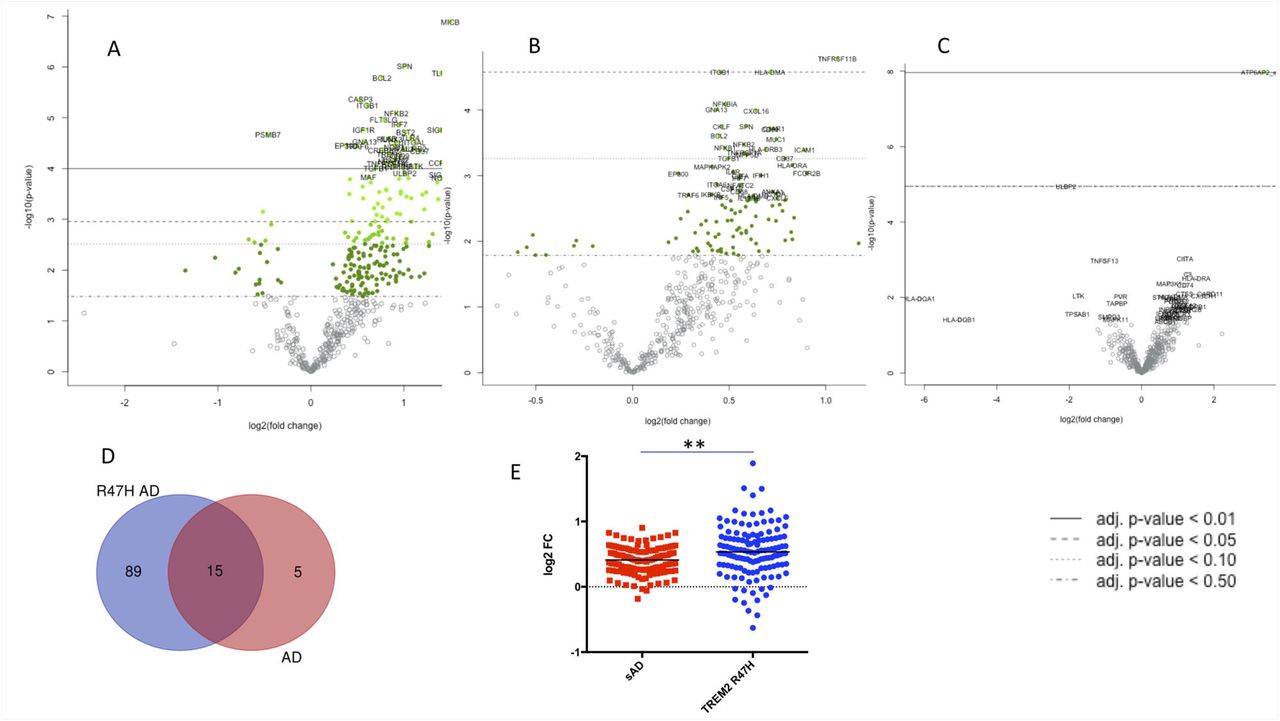
Differentially expressed (DE) genes in hippocampi of TREM2 R47H AD, sAD and XPDS patients. (A-D) Volcano plots displaying DE genes in TREM2 R47H AD (A), sAD (B), and XPDS (C) against a baseline of gene expression in controls. The y-axis corresponds to the log10(p-value), and the x-axis displays the log2(fold change) value. (D). Venn diagram showing shared DE genes (FDR < 0.1) between TREM2 R47H AD and sAD. (E) Microglia-specific genes (MG) perturbed in sAD and TREM2 R47H AD. Distribution of fold changes in MG (N=134); X-axis displays log2 of fold changes between gene expression in cases vs controls (**- p-value < 0.01; 1-way ANOVA, Tukey’s multiple comparison test).
In the brain, TREM2 expression is confined to microglia, so we assessed changes within the microglia-specific transcriptome. We took advantage of a recently identified human microglia signature, 880 genes expressed at least 10-fold higher in microglia than in other brain cell populations (28). Of 880 microglia-specific genes (MG), 156 were present and 134 had measurable expression on the Nanostring panel. MG expression was most perturbed in TREM2 R47H AD and much less in sAD (Figure 2E).
To assess the impact of TREM2-related microglial pathology on known AD genes and risk factors we compared their expression across the two AD groups. Six of 16 AD genes/factors were perturbed in TREM2 R47H AD compared to one in sAD (INPP5D, FDR < 0.1) (Figure 3). The gene encoding complement receptor 1 (CR1) was the most highly upregulated. TREM2 itself was up-regulated. Notably, PSEN2 and APP were downregulated in TREM2 R47H AD brain.

Perturbed expression of AD genes/risk factors in TREM2 R47H AD. AD-associated genes altered (FDR <0.1) in at least one condition are shown (* - adj p-value < 0.05; ** - adj p-value < 0.01); y-axis displays log2 (FC, fold changes) value. Shown is the average FC per group.
Pro-inũammatory immune networks and pathways are activated in TREM2 R4 7H AD brains
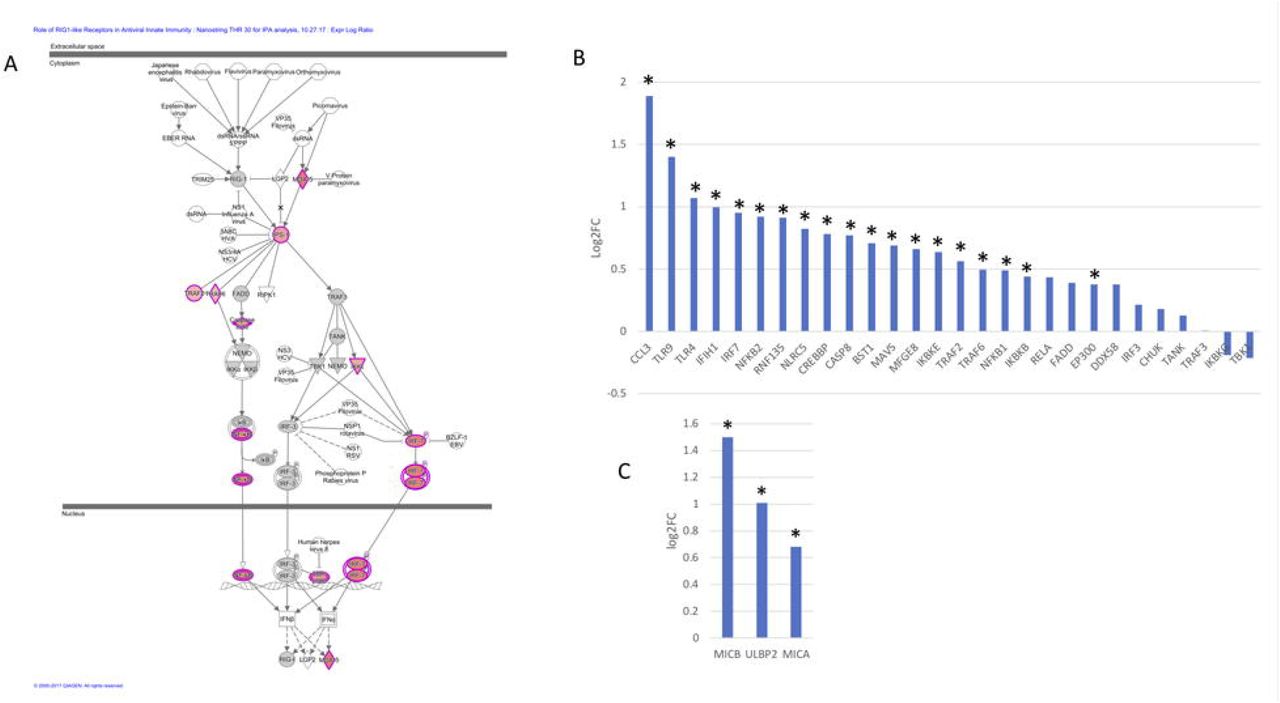
Ingenuity Pathway Analysis (IPA) of differentially expressed genes identified 14 significantly perturbed pathways in TREM2 R47H AD driven by 53 genes. The top TREM2 R47H AD specific pathways comprised “WNT/Ca++ Signaling”, “Role of RIG1-like Receptors in Antiviral Innate Immunity”, “Inflammasome Pathway” and “Neuroinflammation Signaling Pathway” (Table S2). Metascape analysis of protein-protein interaction (PPI) network revealed enriched GO terms corresponding to regulation of cytokine production and I-kappaB kinase/NF-kappaB signaling and identified three functional modules corresponding to type I interferon response, ligand-receptor interaction, and a module formed by PSEN2 and mediators of apoptosis (Figure S1). IPA of differentially expressed genes in sAD produced a heterogeneous group of overrepresented pathways driven almost exclusively by the TNF receptor superfamily members (TNFRSF11b and TNFRSF1A) and the NF-kB complex and its regulators (Table S2). These genes are known to have multifaceted roles in signal transduction in cells of immune and non-immune origin. Because they are also upregulated in TREM2 R47H AD they are likely to reflect perturbation common to the AD disease state.
We also identified 44 microglia-specific and microglia-enriched genes among 104 differentially expressed genes of TREM2 R47H AD (28, 29). Protein-protein association network analysis revealed a highly interconnected network of microglial genes involved in cytokine production, innate immune response, myeloid cell differentiation and regulation of locomotion (Figure S2).
TREM2 R47H AD brain manifests activation of interferon type I response and “induced-self” NKG2D ligands
The activated antiviral interferon type I (IFN I) response was one of the top immune signatures initially identified in the RNA-seq analysis of the TREM2 R47H brains and subsequently confirmed by Nanostring profiling of a larger set of samples (Table 1, Figure 4 A, B). We also identified additional upregulated genes known to act in the antiviral response (RNF135/Riplet, TLR9, TLR4, MFGE8 and BST2/tetherin) that were not part of the canonical IFN I response signature. It is well established that during IFN I response multiple downstream interferon stimulated genes (ISG) are upregulated. We therefore queried a database of ISG (INTERFEROME, (30) and found that differentially expressed genes in TREM2 R47H AD, were ISG enriched (hypergeometric test p-value is 0.03). Consequently, the activation of the IFN I response in the microglia expressing TREM2 R47H may in part explain a large number of upregulated genes seen in the TREM2 R47H AD brain. Of note, not all genes upregulated in the classical IFN I response to viral infection were perturbed in TREM2 R47H AD. For instance, expression of STAT1, STAT2, TAP1, CD38, ISG15, IFIT1, IFIT2, ISG20, and CD164 was not changed.

TREM2 R47H AD features upregulated Interferon type I response and MHC class I-like NKG2D ligands. (A) Role of RIG1-like receptors in antiviral innate immunity, IPA-generated diagram shows perturbed gene expression in TREM2 R47H AD brains. Red denote up-regulated genes, grey - not changed compared to control brains, white - not assessed. Expression of INF I type response/antiviral genes (B) and MHC class I-like NKG2D ligands (C) (* - DE genes in TREM2 R47H AD, FDR<0.1); y-axis - log2 (fold changes) value.
Prolonged stimulation of IFN I response causes cellular stress and cytotoxicity (31). In TREM2 R47H AD brains, we observed upregulation of several MHC class I-like NKG2D ligands (MICB, MICA and ULBP2, Figure 4C). These molecules are known as “induced self” because their expression on the cell surface is induced by diverse stress agents, such as viruses, heat shock or oxidative damage. In the brain, MICB is expressed primarily by the microglia (28), and it is the most significantly upregulated gene in TREM2 R47H AD (Figure 2A). Because multiple pro-inflammatory mediators are upregulated in TREM2 R47H AD brain, we tested whether pro-inflammatory stimulation in vitro would upregulate MICB expression in microglia-like myeloid cells. Pro-inflammatory simulation with a combination of LPS and IFNγ upregulated MICB mRNA expression in all tested cell lines, whereas stimulation with type 2 cytokines IL-4 or IL-10 had no effect (Figure S3).
The effect of TREM2 R47H on THP1 transcriptome reveals partial inactivity of the variant
To evaluate the effect of R47H TREM2 expression on gene networks of a human myeloid cell we used the THP1 line. THP1 cells share many properties of cultured primary human microglia (32), and they express TREM2-TYROBP receptor at a high level. We generated derivatives with doxycycline-inducible WT TREM2, R47H TREM2 or GFP, as well as CRISPR/Cas9 TREM2 knockouts and analyzed them by mRNA-Seq. The knockout produced the largest number of perturbed genes (1,799) pointing to a profound effect of abrogated TREM2 signaling on gene networks in THP1 (Figure 5A). In overexpression experiments (OE), doxycycline induction increased TREM2 level by 17-fold for WT and 13-fold for R47H TREM2 (not shown). Surprisingly, OE WT altered 675 genes while OE R47H altered only 136 genes. The 5-fold difference in a number of perturbed genes is most consistent with a partial loss of TREM2 R47H activity. Accordingly, pathway analysis of DE genes identified a cluster of eight GO groups enriched in OE WT and TREM2 KO, but unaffected in OE R47H (Figure 5B). GO:0043062 (“extracellular structure organization”) and GO:0072358 (“cardiovascular system development”) were the most enriched GO groups in OE R47H. Of them, GO:0043062 was also among the top enriched groups in OE WT, while GO:0072358 was highly enriched in TREM2 KO. The latter suggests that in addition to partial loss of activity, R47H may acquire some negative function that is related to vasculature and angiogenesis. Interestingly, APOE, a proposed TREM2 ligand, was upregulated upon all TREM2 perturbations (Figure S4).

Effects of knockout and overexpression of TREM2 variants on THP1 transcriptome. (A) Venn diagram showing shared DE genes (FDR < 0.05) between TREM2 knockout (KO), overexpression of TREM2 wild-type (OE WT) and overexpression of TREM2 R47H in THP1 cells. (B) Clustering of enriched gene pathways (Metascape, GO:Biological_Process) formed by DE genes in TREM2 KO, OE WT and OE R47H in THP1 cells (see also Table S5).
We also queried RNA-Seq data for expression changes in the set of 28 biomarkers of IFN type I antiviral response upon knockout or overexpression of TREM2 in THP1. TREM2 KO demonstrated the largest number of up-regulated genes (N=13) followed by the OE WT (N=8), surprisingly, OE R47H did not result in the upregulation of the IFN gene signature (Table S3).
Discussion
Pathogenic TREM2 variants of different effect are found in several neurodegenerative diseases. In PLOSL, homozygous inactivating mutations include premature stop codons (33, 34), splice site mutations (35, 36) and missense mutations D134G, K186N (4), Y38C, T66M, and V126G (37),(38),(5). Heterozygous missense variants of dominant effect associated with AD include R47H and R62H. Whereas missense mutations in PLOSL affect protein folding and stability decreasing TREM2 surface expression, AD-linked mutations alter the ligand-binding interface of TREM2 (39). Analysis of TREM2 R47H function in microglial cells showed weakened phagocytosis and changes in binding affinity to phospholipids (40) consistent with altered function not compensated by the normal allele. A recent morphometric analysis of the mouse model of AD found that animals completely or partially deficient for TREM2 alleles had microglia with reduced ability to envelop amyloid deposits and an increased axonal dystrophy (41). A similar phenotype was also observed in TREM2 R47H AD patients (41).
Studies of the R47H mutation in the murine Trem2 found phenotypes suggesting complete loss of function (42, 43). The variant activated a cryptic splice site in the mouse gene that reduced the levels of the correct transcript and TREM2 protein. However, the effect of R47H on TREM2 splicing does not occur in humans (44), so phenotypes reported in Trem2 R47H knock-in mice may not be relevant to the human pathology. The results of the most recent mouse study with the humanized TREM2 are consistent with partial loss of function (45). It was found that R47H impaired amyloid-β-induced microgliosis and microglial activation and abrogated soluble TREM2 binding to neurons and plaques, but did not alter receptor shedding. Indeed, if R47H merely inactivates the TREM2 allele, then heterozygous carriers of loss-of-function TREM2 variants in PLOSL families should have increased AD susceptibility. To date, pedigrees with nine distinct TREM2-inactivating variants have been described across the world (46), and a study of AD burden in heterozygous mutation carriers in these PLOSL families would be required test this hypothesis. Of several homozygous R47H individuals reported in Iceland none manifested a PLOSL-like early onset form of dementia, indicating that the variant protein has retained essential function (2). In addition, neuropathological examination of R47H carriers did not reveal any of PLOSL-like features (19).
Our findings that numerous immune genes/pathways are altered in TREM2 R47H AD brains may be explained by partial loss of TREM2 activity, as well as by gain of pathologic function that occurs in conjunction with decreased dosage of the normal TREM2 allele. To test whether TREM2 R47H has a dominant stimulatory effect on immune pathways we overexpressed it in THP1, a myeloid cell line with a high level of endogenous TREM2. Inflammatory response and myeloid cell signaling pathways affected by an increased level of normal TREM2 highlights active TREM2 signaling in these cells (Figure 5). Comparison of gene networks affected by the gene knockout or overexpression of TREM2 variants reveal a complex effect of R47H on the function of the receptor. Eight of 20 top enriched pathways for OE WT were unaffected by OE R47H indicating functional deficiency of the mutant. At the same time, OE R47H shared a characteristic pathway only with the KO, suggesting some dominant negative effect related to vascular integrity/angiogenesis. Recent co-expression network analysis in the TREM2 knockout mouse uncovered an unexpected role of TREM2 in homeostasis of brain endothelium (47). Analysis of the IFN type 1 response signature in THP1 confirmed the effect of changed TREM2 dosage on this pathway (Table S6). Surprisingly, TREM2 R47H expression per se was not sufficient to induce the IFN type 1 response. Hence, haploinsufficiency for the normal allele in TREM2 R47H heterozygotes is a plausible explanation for the observed activation of IFN type I response in their brains. We cannot rule out the possibility that the effect of R47H on IFN type I response is amplified via microglia interaction with other cell types.
The IFN I response is an important line of defense against viruses. It senses presence of viral nucleic acids inside the cell and activates a cascade of immune responses that restrict viral propagation and eliminate infected cells. In the absence of viral infection, unchecked IFN I response is harmful, being a cause of Aicardi-Goutières syndrome (AGS), a group of monogenic interferonopathies that affect the CNS (48). Mutations causing AGS occur in sensors of viral nucleic acids and in certain DNA/RNA metabolic enzymes. A common RNA expression phenotype, “IFN I response signature”, features upregulation of interferon-stimulated genes in AGS patients. This IFN I response signature is also a characteristic of brain aging, expressed by microglia of choroid plexus in mice and humans (49). In the aged mouse, a chronic IFN I response negatively affects brain function, and it is ameliorated by drugs that block IFN I signaling. We speculate that a chronically activated IFN I response in R47H carriers triggers premature aging of microglia and contributes to its transition from the homeostatic to disease-associated state.
In our study, expression of several AD-associated genes was altered in TREM2 R47H AD indicating a profound effect of TREM2 dysfunction on the AD genes of microglia-specific (TREM2, INPP5D) and non-MG origin (CR1, CLU/APOJ, PSEN2). CR1 encodes a complement receptor with functions in phagocytosis, clearance of immune complexes and inhibition of complement (50). CLU/APOJ encodes an apolipoprotein that is predominantly expressed by astrocytes. Besides its roles in protein folding and lipid metabolism, it may function as an inhibitor of the terminal complement complex (51, 52). CLU is a potential TREM2 ligand (16), and in brains of AD patients there is regional increase in its level that correlates positively with amyloid plaque burden and APOEε4 genotype (53). INPP5D/SHIP1 encodes an inositol polyphosphate-5-phosphatase that regulates immune signaling in lymphocytes and myeloid cells, including microglia and osteoclasts, and controls cell proliferation, survival and chemotaxis (54). INPP5D negatively regulates PI3K-AKT signaling by at least two mechanisms: 1) it dephosphorylates phosphatidylinositol-3,4,5-triphosphate (PIP3), a substrate phosphorylated by PI3K, thus antagonizing PI3K action; 2) INPP5D binds directly to the phosphorylated ITAM motif of activated TYROBP, working as a switch that prevents PI3K interaction with the TREM2-TYROBP receptor (55). INPP5D was identified as an AD risk factor in GWAS screens (56). The AD-associated allele increases INPP5D gene expression in whole blood (57) indicating that its level may be a biomarker for AD susceptibility. In brain, INPP5D expression is microglia-specific (28) and its level is increased in AD patients (58). INPP5D also forms a complex with CD2AP, another AD risk factor (59).
PSEN2 was part of a perturbed module that also included CASP1 and CASP3 and several other apoptotic molecules (the MCODE_3”, Figure S1). In humans, PSEN2 is predominantly expressed by neurons, astrocytes and endothelium and less by microglia (60), making it unlikely that the PSEN2 perturbation observed in TREM2 R47H AD brain is of microglial origin. At present, it is unclear how microglia-expressed R47H TREM2 may cause PSEN2 downregulation in neurons. A recent study of soluble forms of human TREM2 receptor in the mouse AD model offers a clue about the possible mechanism of such an interaction. While normal soluble TREM2 readily bound neurons, the mutant R47H was unable to bind them (45). It will be of interest to establish whether soluble TREM2 R47H itself or other molecules secreted by TREM2 R47H microglia exert deleterious effects on neurons.
In summary, our findings support a hypothesis that carriers of R47H TREM2, and likely of other AD-associated missense TREM2 variants, comprise a distinct subtype of AD, in which an activated immune response exacerbates the neurodegenerative process. Screening of individual AD transcriptomes for presence of immune/microglial signatures may delineate the contribution of known immune risk factors and facilitate discovery of novel AD biomarkers. Because brain microglia share expression of multiple immune molecules with osteoclasts and macrophages of the periphery, these accessible cell types can be interrogated Alternatively, microglia-like cells can be obtained by differentiating induced pluripotent stem cells generated from patients. Also, expression of numerous soluble cytokines and chemokines characteristic of TREM2 R47H AD can be measured in the CSF of patients and used for development of diagnostics of microglia-driven neuropathologies.
Materials and Methods
Subjects
Familial AD and frontotemporal dementia (FTD) patients with R47H
Through the University of Washington (UW) Alzheimer’s Disease Research Center, we acquired brain autopsy material of nine TREM2 R47H carriers from four unrelated families affected with AD or FTD (Table S1). Flash-frozen brain tissues from two R47H carriers were used for RNA-seq analyses. One subject had an AD diagnosis confirmed by neuropathology and another had no AD pathology. RNA isolated from formalin-fixed, paraffin-embedded (FFPE) sections of eight R47H carriers with AD were used for gene expression by Nanostring nCounter.
Sporadic AD and controls
Frozen and fixed autopsy tissues from neuropathologically confirmed AD cases and aged non-demented controls were obtained from the UW Neuropathology Core Brain Bank (Table 1S). Under protocols approved by the Institutional Review Boards of the UW all subjects had previously given informed consent to share and study autopsy material. The average age of subjects in case/control groups was 75.8 years and the average postmortem interval was 10.9 □ hr.
Cell culture and cytokine stimulation
A human microglia cell line, HMC3, and human myeloid cell lines THP1, U937 and MOLM13 were obtained from the ATCC. Cells were cultured in DMEM (HMC3) or RPMI (the others) supplemented with 10% fetal bovine serum. Pro-inflammatory stimulation with LPS (Sigma, #055:B5, 0.5 μg/ml) and IFNγ (PeproTech, #300-02 150U/ml) was performed for 24 hrs. Stimulations with IL-4 (PeproTech, #200-04, 20 ng/ml) or IL-10 (PeproTech, #200-10, 20 ng/ml) were similarly performed for 24 hrs. Total RNA from harvested cells was isolated with RNAeasy kit (Qiagen), and cDNA was synthesized with SuperScript III kit (Invitrogen, #18080051). The following TaqMan Gene Expression Assays (ThermoFisher, #4331182) were used for qRT-PCR: MICB: Hs00792952_m1, and controls: GUSB: Hs00939627_m1 and TBP: Hs00427620-m1. Assays were performed on StepOnePlus real time PCR machine and analyzed with StepOne software (Applied Biosystems).
Antibodies and immunostaining
Live cells were immunostained with fluorescent polyclonal antibodies to TREM2 (R&D Systems #MAB17291) and analyzed by FACS. For western blotting, 50 μg of whole cell lysates were resolved on polyacrylamide gel, transferred to PVDF membrane and stained with polyclonal TREM2 antibodies (Cell Signaling Technology, #91068) at 1:1000 dilution.
TREM2 overexpression
Human TREM2 (NM_018965.3), with R47 (wt) or R47H, were sub-cloned in doxycycline-inducible lentiviral pCW57-MCS1-2A-MCS2 vector (Addgene, #71782). THP1 cells were infected with lentiviral particles expressing either of wild-type TREM2, R47H TREM2 or GFP proteins (Addgene; pCW57-GFP-2A-MCS, #71783). After 2-weeks of puromycin selection, resistant cells were induced with doxycycline (500 ng/ml) and tested for TREM2 and GFP expression by qRT-PCR and by FACS analyses.
Genome editing of TREM2 with CRISPR/Cas9
A THP1 derivative that stably expresses Cas9 (THP1-CAS9) was generated using a lentiviral construct (Addgene, lentiCas9-Blast, #52962) after blasticidine selection. sgRNA for targeting TREM2 were selected using the online CRISPR design tool (crispr.mit.edu) (61). THP1-CAS9 cells were nucleofected with sgRNA using a protocol recommended for THP1 (Lonza, Amaxa 4D Nucleofector, Protocol #292). Genomic cleavage detection nuclease assay was performed to determine cutting efficiency for each guide (ThermoFisher, GeneArt Genomic Cleavage Detection Kit, #A24372). Based on % indel efficiency, sgRNA within exon 2 (sense: ACTGGTAGAGACCCGCATCA) was chosen for subsequent experiments. Gene targeting followed by immunostaining and enrichment of TREM2-dim cells on the cell sorter was performed twice. Colonies grown from single sorted cells were sequenced; clones with frame-shifting indel mutations inactivating all four TREM2 alleles were tested for absence of protein by Western blotting.
Gene expression analyses
Whole transcriptome RNA-seq of brain tissues
Total RNA was isolated from flash-frozen brain tissues, and ribosomal RNA was depleted using the Ribo Zero Gold Magnetic system (Epicentre/Illumina, San Diego, CA). RNA-seq libraries were prepared with the ScriptSeq v2 kit (Epicentre) and subjected to paired-end sequencing (2 x 100 bp) on an Illumina HiSeq 2500 instrument. A mean read number of 6×107 was generated per sample. The following data analysis tools were used: FastQC 0.9.6 for visualizing read QC https://www.bioinformatics.babraham.ac.uk/projects/fastqc; Bowtie 2.2.1 https://sourceforge.net/projects/bowtie-bio/files/bowtie2/2.2.1/ and TopHat 2.0.11 https://ccb.jhu.edu/software/tophat/index.shtml for read alignment and splice site discovery; Cufflinks 2.1.1 http://cole-trapnell-lab.github.io/cufflinks/releases/v2.1.1/ for transcript assembly and quantification; MISO 0.5.2 https://sbgrid.org/software/titles/miso for quantitating isoform expression levels; cummeRbund 2.0.0 http://compbio.mit.edu/cummeRbund and IGV 2.3.14 http://software.broadinstitute.org/software/igv/download for data visualization.
mRNA-seq of modified THP1 cells
The RNeasy Mini Kit (Qiagen, #74104) was used to isolate total RNA from the following cell lines: THP1-CAS9, THP1 TREM2 KO line 1, THP1 TREM2 KO line2, induced THP1-GFP, induced THP1-TREM2 WT, induced THP1-TREM2 R47H, using 3 biological replicates per each cell line. mRNA-seq analysis was performed by Novogene (250-300 bp insert cDNA libraries were generated from 2 μg of RNA using the NEBNext Ultra Directional RNA library prep kit for Illumina, paired-end 150 bp sequencing was performed using Illumina HiSeq platform. A mean read number of 5.7×107 was generated per sample. Filtered reads were aligned to the most recent version of human genome using STAR software (62), differential expression analysis was performed using the DESeq2 R package with the threshold of padj<0.05.
Gene expression analysis by nCounter system (NanoString Technologies)
Total RNA was isolated from FFPE samples. For each sample, four 10 μm thick FFPE sections were processed using the Recover All RNA isolation kit (Ambion, Termo Fisher) following the manufacturer’s protocol. RNA yield and fragment-length distribution were measured by Qubit Fluorimeter (Molecular Probes, Termo Fisher) and 2100 Bioanalyzer (Agilent Technologies). Typical yield was 0.8-1μg per 10 μm section. As expected, RNAs from archived FFPE samples were highly degraded (RIN 2-3), however more than 80% of RNA species were over 80 bp allowing analysis by Nanostring technology (hybridization with the 70 nucleotides gene-specific probe). Total RNA (1 μg) was used for hybridization on a NanoString nCounter platform according to the manufacturer’s instruction. Samples in which >50% genes had raw counts below the cutoff of 20 (average of 8 negative controls plus 2 standard deviations, 95% confidence interval) were excluded from analysis. 80% of initially taken FFPE samples were suitable for gene expression analysis.
For gene expression analysis we used the PanCancer Immune panel, which contains 730 immunity-related and cancer genes together with 40 validated housekeeping genes, and a custom CodeSet containing 30 genes designed at Nanostring Technologies (NanoString Technologies). All probe sets were produced in a single batch. Data were normalized and log2 transformed using the nSolver 3.0 Analysis Software (NanoString Technologies). In brief, normalization was performed in two steps, 1) technical normalization using positive control spikes on each panel, and 2) input amount normalization using the mean expression of 30 housekeeping genes selected by a geNorm algorithm on the basis of stability of their expression through all experimental groups. All transcripts with raw counts below the threshold 20 (average of 8 negative controls plus 2 SD (~ 95% confidence interval) were labeled as undetected. Only genes detected at a level of 30 or more row counts in at least 50% of our samples were included in the analysis (N=493).
Gene set and pathway analyses
Gene set enrichment analysis (GSEA)(63) and its modification, Gene Set Analysis (GSA) (20), were used to detect significant enrichment of known transcriptional signatures in gene expression data. To compute an enrichment score for each pathway GSA uses a maxmean statistic while GSEA uses a weighted Kolmogorov-Smirnov test. We used 10,295 gene sets from the Molecular Signature Database version 4.0 (64). False discovery rate and P-value estimates were computed for each gene set based on 1000 separate permutation distributions.
To identify biologically relevant pathways, we used Ingenuity Pathway Analysis (IPA, Ingenuity Systems, Qiagen) and Metascape (metascape.org). IPA queries a proprietary knowledge database and calculates significance of genes/pathways enrichment using Fisher’s exact test. Metascape queries public databases (Reactome, MSigDB, GO, KEGG Pathway, CORUM), and enrichment is calculated by hypergeometric test. Additionally, Metascape performs analysis of protein-protein interactions (PPI, data from BioGrid database) in which subnetworks with high local network connectivity are found using the Molecular Complex Detection (MCODE) algorithm (65). Visualization of protein-protein association networks that include known protein interaction and functional connection was done using STRING database (http:/string-db.org) (66).
Funding
This work was supported by National Institute of Health grants [P30AG013280 to O.K., P41GM109824 and P50 GM076547 to A.R., 2R01 NS069719 to W.H.R.] and funds from the Department of Veterans Affairs (to T.B.D.).
Author contributions
Conceptualization O.K., K.K., W.H.R., Methodology A.R., K.K., M.O.D., T.K.B., Investigation O.K., K.K., A.R., M.M., W-M.C. Resources C.D.K., T.D.B, Writing – original draft O.K., K.K., Writing – review and editing T.B.D., W.H.R., Visualization O.K., A.R., K.K., Supervision O.K., W.H.R.
Competing interests
The authors declare no competing interests.
Acknowledgements
We are grateful to Drs. Zoran Brkanac and John Neumaier for critical comments.
Abbreviations
- AD
- Alzheimer’s disease
- sAD
- sporadic AD
- GWAS
- genome-wide association study
- PLOSL
- polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy
- XPDS
- X-linked Parkinsonism with Spasticity
- DE
- differentially expressed (genes)
- MG
- microglia-specific genes
- ISG
- interferon stimulated genes
- NK
- natural killers





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}