

Abstract
Viperin is an interferon-inducible protein that is critical for eliciting an effective immune response against many diverse viral pathogens. As such, viperin has been implicated in interactions with many functionally unrelated host and viral proteins, making it increasingly difficult to determine a unifying mechanism of viperin’s antiviral activity. We report here that viperin acts synergistically to enhance the innate immune dsDNA signalling pathway to limit viral infection. Viperin co-localised with the key signalling molecules of the innate immune dsDNA sensing pathway, STING and TBK1, via direct binding to STING; inducing enhanced K63-linked ubiquitination of TBK1. Consistently, viperin’s interaction with these molecules resulted in an enhanced type-I interferon response to Hepatitis B virus and significantly lowered Hepatitis B viral levels in an in vitro transfection model. Subsequent analysis identified the necessity of N-terminal sequences and viperin’s radical SAM domain to enhance the type-I interferon response to aberrant dsDNA. Here we show that viperin facilitates the formation of a signalling enhansosome, to coordinate efficient signal transduction following activation of the dsDNA signalling pathway; which results in an enhanced antiviral state. This data further defines viperin’s role as a positive regulator of innate immune signalling, complementary to its role in TLR7/9 signalling, offering a mechanism of viperin’s broad antiviral capacity.
Introduction
Innate immunity constitutes the first line of host defence against viral invasion, acting to both prevent as well as clear infection [1]. A range of germ-line encoded pattern recognition receptors (PRRs) are responsible for detecting various viral structural motifs, termed pathogen associated molecular patterns (PAMPs), upon which a signalling cascade is initiated to illicit an innate immune response [2, 3]. This response is primarily associated with the production of cytokines and chemokines, such as interferon (IFN), which acts in both an autocrine and paracrine manner to induce hundreds of interferon stimulated genes (ISGs) [4]. The products of these ISGs act to both clear the viral infection within the infected cell while simultaneously resisting infection in neighbouring cells (reviewed in [5]).
The induction of ISGs creates a highly effective antiviral state within a cell, yet the specific antiviral activity has only been characterised for a handful of these genes. Viperin (also known as cig5 and RSAD2) is one of the most well described and potent ISGs, implicated in limiting many viruses from multiple viral families (reviewed in [6]). Viperin was first identified to have antiviral properties against human cytomegalovirus [7], and has since been shown to directly target multiple stages of the viral life cycle to inhibit infection of viruses including HCV, DENV, TBEV, HIV, BUNV and IAV (reviewed in [6]). Viperin has been shown to target the budding of IAV, HIV-1 and RABV, by directly disrupting cellular lipid rafts through interactions likely involving the host protein FPPS [8–10]. Additionally, the replication of viruses such as HCV and DENV is also a target for viperin’s antiviral activity, whereby viperin has been demonstrated to associate with the viral non-structural proteins NS5A and NS3 respectively at the replication complexes of each virus to inhibit infection [11, 12]. However, there is not one direct means of viperin inhibition described for these multiple viral families, which all employ different routes of infection and mechanisms of replication. (reviewed in [6, 13]). As such, viperin has been implicated in interactions with many functionally unrelated host and viral proteins, making it increasingly difficult to determine a unifying mechanism of viperin’s antiviral activity.
Viperin is a member of the radical SAM enzymatic family, and recent work has demonstrated for the first time that mammalian viperin’s substrate is cytidine triphosphate (CTP), and that it catalyses the formation of ddhCTP, a novel nucleotide [14]. Furthermore, ddhCTP was shown to act as a chain terminator for viral RNA-dependent RNA polymerases (RdRp) from multiple Flavivirus members [14], and this offers an explanation for viperin’s ability to broadly limit this viral genus (reviewed in [6]). Viperin’s generation of ddhCTP was significantly enhanced by the co-expression of the host protein CMPK2, which was found to ensure a sufficient supply of the substrate CTP by facilitating its conversion from CDP [14]. However, ddhCTP is unable to inhibit the polymerase activities of the Picornaviridae members HRV-C and poliovirus, despite viperin’s previously identified antiviral capacity against HRV [15]; which highlights the fact that this highly evolutionarily conserved anti-viral host protein, may still be involved in other as yet unidentified anti-viral mechanisms.
Viperin is one of a small group of ISGs that are capable of acting as both direct antiviral effectors and indirectly as enhancers of innate immune signalling, to inhibit viral infection (reviewed in [16]). Viperin can enhance the activation of key signalling molecules of both the TLR7 (ssRNA sensing) and TLR9 pathways (CpG DNA sensing), and is able to directly bind IRAK1 and TRAF6, to enhance the activation of IRAK1, via augmenting the molecule’s K63-linked ubiquitination [17]. This in turn resulted in a heightened production of interferon, and an enhanced anti-viral response. Multiple viruses have PAMPs that activate these pathways, including IAV and VSV [18, 19], which are known to be limited by viperin (reviewed in [6]), and this mechanism may help explain some of the broader anti-viral activities of viperin.
Here we show for the first time that viperin is also able to directly interact with the signalling adaptor molecule, stimulator of IFN genes (STING), and enhance production of anti-viral cytokines following activation of dsDNA signalling pathways. Viperin’s ability to augment both dsDNA signalling pathways, as well as TLR7 and 9 signalling, perhaps offers further explanation for this host protein’s ability to limit such a broad range of viral families.
Results
Viperin enhances the STING-dependent type-I IFN response to dsDNA downstream of ligand detection
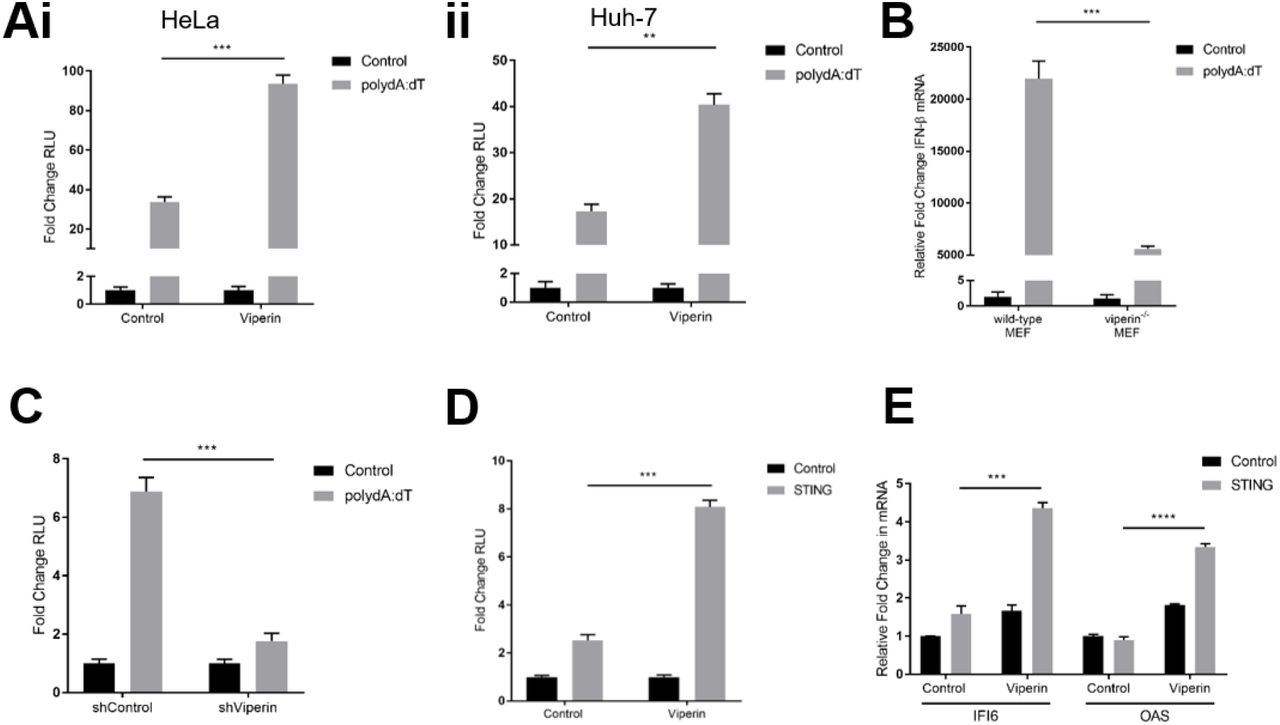
To investigate whether viperin plays a role in the enhancement of the IFN response to dsDNA we initially utilised in vitro cell culture based luciferase assays. Ectopic expression of viperin in both HeLa and Huh-7 cells was observed to enhance the activity of the type-I IFN - IFN-β promoter in dual luciferase reporter assays following stimulation of the DNA viral mimic poly dA:dT by approximately 2.5-fold and 2-fold respectively (Figure Ai & ii). These results were confirmed with the use of our previously developed primary viperin−/− MEFs [20] as well as a polyclonal Huh-7 cell line stably expressing shRNA targeting viperin mRNA [11]. As can be seen in Figure 1B, primary viperin−/− MEFs displayed an approximate 4-fold reduction in their expression of IFN-β relative to wild-type MEFs, and the activity of the IFN-β promoter was significantly reduced in the shViperin cells compared to the shControl cell line (Figure 1C). Together this data demonstrates the ability of viperin to enhance the type-I interferon response to exogenous dsDNA stimulus.

(Ai & ii) Luciferase production driven by the IFN-β promoter in HeLa (i) and Huh-7 (ii) cells transfected with either viperin or control constructs 24 hrs prior to stimulation with poly dA:dT for 8 hrs. (B) Expression of IFN-β mRNA in wild-type and viperin−/− primary MEFs following 8 hr stimulation with poly dA:dT. (C) Luciferase production driven by the IFN-β promoter in Huh-7 cells stably expressing shRNA targeting viperin mRNA and its control stimulated with poly dA:dT for 8 hrs. (D) Luciferase production driven by the IFN-β promoter in Huh-7 cells transfected with combinations of viperin, STING or control constructs for 24 hrs. (E) Expression of IFI6 and OAS mRNA in HeLa cells transfected with combinations of viperin, STING or control constructs for 24 hrs. Luciferase measurements were controlled by constitutive expression of renilla and presented as fold changes in relative luminometer units (RLU) from control unstimulated conditions. Data is presented as mean ± SEM from at least three independent experiments. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
The detection of exogenous dsDNA within the host cell relies on the activity of multiple DNA sensors, however upon recognition of their ligands these receptors predominantly converge on the adaptor molecule STING [21]. To identify whether viperin’s enhancement of the type-I IFN response to dsDNA involves an interaction with the downstream adaptor molecule STING, Huh-7 cells were co-transfected to ectopically express both viperin and STING in the absence of poly dA:dT stimulation. Cells expressing STING alone displayed a 3-fold increase in IFN-β promoter activity compared to cells transfected with a control plasmid (Figure 1D), indicating that the overexpression of this adaptor molecule is sufficient to auto-activate the pathway. Furthermore, in the absence of poly dA:dT stimulation, co-transfection of STING with viperin resulted in significantly higher IFN-β promoter activity compared to STING alone (Figure 1D), suggesting viperin’s enhancement of type-I IFN following dsDNA signalling occurs downstream of exogenous DNA recognition. Furthermore, viperin’s co-transfection with STING also significantly upregulated the production of key antiviral ISGs IFI6 and OAS compared to STING alone (Figure 1E), indicating that this positive augmentation of the type-I IFN pathway results in a functional upregulation of ISGs downstream of dsDNA ligand recognition.
Viperin relies on its N-terminus and binding to enzymatic 4Fe-4S cofactor to enhance type-I IFN response to dsDNA
Viperin relies on the action of specific functional domains to inhibit multiple families of viral pathogens [6], with its characteristic localisation to the lipid droplet (Figure 2A) being essential for its ability to enhance the type-I IFN response via TLR7/9 activation of plasmacytoid dendritic cells in the mouse [17]. To determine the potential role of certain viperin domains in its ability to enhance type-I IFN following poly dA:dT stimulation we utilised in vitro luciferase assays in combination with a panel of viperin mutants. As can be seen in Figure 2B, the 5’Δ33 viperin mutant, which redistributes the protein to appear more cytoplasmic in localisation due to its lack of the N-terminal amphipathic helix [11], and the SAM1 viperin mutant in which the Fe-binding cysteine residues of the protein’s radical SAM Motif 1 are mutated to alanine [11], both showed a significant decrease in IFN-β induction compared to those expressing viperin-wildtype following dsDNA stimulation, and resembled that of the cells entirely lacking viperin (control). Conversely, the 3’Δ17 viperin mutant which lacks 17 amino acids from the protein’s C-terminus [11], significantly enhances viperin’s ability to enhance the IFN-β promoter (Figure 2B). This would imply that viperin requires either its localisation to the lipid droplet or specific sequences within its N-terminus, in conjunction with its binding to the 4Fe-4S cluster, to enhance the type-I IFN response to dsDNA.

(A) Huh-7 cells were transfected with viperin-flag 24 hrs prior to immunofluorescence staining with a mouse monoclonal anti-flag antibody (Sigma), followed by an Alexa555-conjugated goat anti-mouse secondary (Invitrogen) as well as BODIPY and DAPI staining. Imaged on Ziess Confocal LSM 780 microscope. Scale bar represents 10 μm. Original magnification is X63. (B and C) Luciferase production driven by the IFN-β promoter in HeLa cells transfected with either (B) wild-type, 5’Δ33, 3’Δ17 and SAM1 viperin constructs or (C) wild-type and chimeric NS5A-viperin mutant constructs 24 hrs prior to stimulation with poly dA:dT for 8 hrs. Luciferase measurements were controlled by constitutive expression of renilla and presented as fold changes in relative luminometer units (RLU) from control unstimulated conditions. Data is presented as mean ± SEM from at least three independent experiments. **p<0.01, ****p<0.0001.
To further investigate the requirement of viperin’s N-terminus in its augmentation of the dsDNA signaling pathway, we utilised a chimeric viperin mutant (NS5A-TN50-Viperin) with its N-terminal amphipathic helix replaced with the alternate amphipathic helix of HCV NS5A (A kind gift from N.E. Marsh, University of Michigan [22]). This mutant was unable to enhance the induction of the IFN-β promoter to the same degree as wildtype viperin (p<0.01) (Figure 2C) following poly dA:dT stimulation, suggesting that localization to the lipid droplets is not of itself, sufficient for viperin’s enhancement of the type-I IFN response to dsDNA.
Viperin co-localises with TBK1 and STING, via a direct interaction with STING
The successful transduction of signalling events initiated by the dsDNA receptors relies on the activity of two major adaptor molecules, STING and TBK1 [23, 24]. The ER-resident protein STING assembles with TBK1 after dsDNA stimulation to facilitate the phosphorylation of IRF3, culminating in the induction of type-I IFN [25, 26]. As viperin has previously been shown to co-localise and interact with alternate signalling adaptor molecules to enhance the efficacy of the TLR7/9 innate immune signalling pathways [17], we investigated viperin’s ability to co-localise with STING and TBK1.
Utilising proximity localisation assays (PLA) in both Huh-7 and HeLa cells in conjunction with ectopically expressed viperin, we observed the co-localisation of viperin with endogenous STING and to a lesser degree endogenous TBK1 irrespective of poly dA:dT stimulation (Figure 3A). This co-localisation was only enhanced between viperin and TBK1 during poly dA:dT stimulation, while the viperin-STING co-localisation appeared to remain constant irrespective of poly dA:dT stimulation. To confirm these observations, we utilised confocal microscopy, where similar co-localisation was observed between viperin and either TBK1 or STING, however considerable co-localisation was only observed between viperin and TBK1 following dsDNA stimulation (Figure 3B and C). As can be seen in Figure 3B, the cytoplasmic localisation of TBK1 appeared to converge with viperin on lipid droplets 2 hrs following poly dA:dT stimulation. Unlike viperin’s localisation with TBK1, viperin appears to co-localise with STING with and without poly dA:dT stimulation (Figure 3C), localising to the lipid droplet with viperin, as well as in discrete punctate locations throughout the cytoplasm.

(Ai and ii) (i) Huh-7 and (ii) HeLa cells were transfected with a viperin-flag construct 24 hrs prior to stimulation with poly dA:dT for 2 hrs and probing with mouse monoclonal anti-flag (Sigma) and rabbit monoclonal anti-STING (Millipore) or anti-TBK1 (Cell Signalling) antibodies, then subject to Duolink® In Situ Red Mouse/Rabbit PLA and DAPI staining. Imaged on Nikon Eclipse Ti-E fluorescence inverted microscope. Scale bar represents (i) 100 μm or (ii) 150 μm. Original magnification is X20. (B and C) Huh-7 cells were transfected with viperin-flag and either (B) TBK1-myc or (C) STING-myc constructs 24 hrs prior to stimulation with poly dA:dT for 2 hrs and immunofluorescence staining with rabbit monoclonal anti-flag (Sigma) and mouse monoclonal anti-myc (Millipore) antibodies followed by an Alexa555-conjugated goat anti-mouse (Invitrogen) and Alexa488-conjugated goat anti-rabbit (Invitrogen) secondary, as well as DAPI staining. Imaged on Ziess Confocal LSM 780 microscope. Scale bar represents 15 μm. Original magnification is X63. (D and E) Huh-7 cells were transfected with viperin-mCherry or control-mCherry and either (D) TBK1-myc or (E) STING-myc constructs 24 hrs prior to stimulation with poly dA:dT for 2 hrs, and cell extracts were immunoprecipitated with rabbit monoclonal anti-mCherry antibody (Biovision) and subject to immunoblot analysis with indicated antibodies. (D) Cell extract was subject to DSS crosslinking prior to lysis and immunoprecipitation.
To further investigate the ability of viperin to form a complex with either STING or TBK1, co-immunopreciptation assays were performed. Preliminary immunoblot analysis was unable to detect TBK1 with immunoprecipitated viperin (Supplementary Figure 1A). To elucidate whether this was the result of potentially weaker or transient binding interactions between the two proteins, a DSS cross-linker was utilised. Despite the addition of the cross-linker, TBK1 failed to be co-immunoprecipitated with viperin (Figure 3D). However, STING was successfully detected following co-immunoprecipitation assays with viperin, irrespective of poly dA:dT stimulation (Figure 3E). Further analysis identified the central domain of viperin to be responsible for this binding to STING (Supplementary Figure 1B). Together these findings highlight the strong interaction between viperin and STING, and imply the formation of a complex between these two proteins, while also indicating that the co-localisation between viperin and TBK1 does not involve direct binding.
Viperin enhances the ubiquitination-dependent activation of TBK1
The adaptor molecules involved in innate immune signalling events are commonly regulated by post-translational modifications such as ubiquitination [27]. The addition of both K27- and K63-linked ubiquitin chains to STING has been shown to facilitate optimal trafficking of the protein, enabling efficient activation of downstream signalling adaptors [28, 29]. To delineate viperin’s mechanism of enhanced IFN-β promoter activity in the presence of dsDNA, we first investigated STING activation in the presence of viperin. However, viperin expression was not found to impact either K27-or K63-linked ubiquitination of STING in HEK293T cells (Figure 4A and B). Moreover, the presence of dimerised and phosphorylated forms of STING, which are associated with the protein’s ligand binding affinity and recruitment of downstream adaptor molecules respectively [30, 31], were unaffected by the co-expression of viperin in HEK293T cells (Figure 4C).

(A and B) HEK293T cells were transfected with viperin-mCherry, STING-myc and either K63-Ub-HA, K27-Ub-HA or wt-Ub-HA constructs 24 hrs prior to (A) visualisation by fluorescence microscopy, after which (B) cell extracts were immunoprecipitated with mouse monoclonal anti-myc antibody (Millipore) and subject to immunoblot analysis with indicated antibodies. Imaged on Nikon Eclipse Ti-E fluorescence inverted microscope. Scale bar represents 200 μm. Original magnification is X20. (C) HEK293T cells were transfected with combinations of viperin-flag, STING-myc and TBK1-myc constructs 24 hrs prior to immunoblot analysis with indicated antibodies. (D and E) Huh-7 cells were transfected with viperin-GFP, TBK1-mCherry and K63-Ub-HA constructs 24 hrs prior to (D) visualisation by fluorescence microscopy, and (E) stimulation with poly dA:dT for 2 hrs, after which cell extracts were immunoprecipitated with rabbit monoclonal anti-mCherry antibody (Biovision) and subject to immunoblot analysis with indicated antibodies. Imaged on Nikon Eclipse Ti-E fluorescence inverted microscope. Scale bar represents 100 μm. Original magnification is X20. (F) Wild-type and viperin−/− MEFs were stimulated with poly dA:dT for 2 hrs, and cell extracts were immunoprecipitated with mouse monoclonal anti-K63-Ub antibody (Enzo) and subject to immunoblot with indicated antibodies.
The activation of TBK1 is regulated through the conjugation of K63-linked ubiquitin chains to lysine residues 30 and 401 along the protein [32, 33]. To determine the impact of viperin on this ubiquitination event, ectopically expressed viperin was visualised in Huh-7 cells (Figure 4D), prior to the determination of the ubiquitination status of TBK1 through immunoprecipitation coupled with immunoblot analysis. There was substantial K63-linked ubiquitination of TBK1 observed in cells containing viperin following a 2 hr poly dA:dT stimulation (Figure 4E), in contrast to the control. Furthermore, in primary wild-type MEFs, K63-linked ubiquitination of endogenous TBK1 was observed to be markedly increased following poly dA:dT stimulation, compared to viperin−/− MEFs (Figure 4F). Collectively this data demonstrates that viperin enhances the K63-linked ubiquitination of TBK1.
Viperin interacts with STING to enhance the type-I IFN response to limit HBV
To investigate whether viperin’s ability to enhance a type-I IFN response to dsDNA would functionally affect the outcome of a DNA viral infection, we utilised a well-characterised HBV in vitro viral system [34]. A 1.3 mer HBV plasmid transfection model for two prevalent HBV genotypes (HBV-D & HBV-A) was utilised in HepG2 cells ectopically expressing a combination of viperin and the central dsDNA signalling adaptor STING.
To determine the involvement of viperin in eliciting a type-I IFN response, the above-mentioned HBV infection model was utilised in conjunction with a dual luciferase reporter assay. At 48 hrs post transfection with either HBV-D or HBV-A, the cells expressing both viperin and STING showed an approximate 20-fold and 60-fold increase respectively in the induction of IFN-β compared to those only expressing STING (Figure 5A); suggesting that the interaction between viperin and STING drives an enhanced type-I interferon response to HBV infection.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A, B and C) HepG2 cells were transfected ectopically with viperin, STING and/or control plasmids in conjunction with either HBV-D or HBV-A 1.3 mer constructs 48 or 96 hrs prior to lysis and; (A) IFN-β promoter driven luciferase production was detected, while (B and C) supernatants were collected for detection of HBsAg and HBeAg by Roche Cobas Elecsys quantitative serology for both HBV-D and HBV-A genotypes. Luciferase measurements were controlled by constitutive expression of renilla and presented as fold changes in relative luminometer units (RLU) from control unstimulated conditions. Data is presented as mean ± SEM from at least three independent experiments. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
To evaluate the effect of the enhanced type-I IFN response to HBV infection in the presence of viperin, cell supernatants were collected 48 and 96 hrs post HBV transfection and analysed by quantitative serology for the presence of the HBV surface antigen (HBsAg) and HBVe antigen (HBeAg). At 96 hrs post HBV-A transfection, the ectopic expression of viperin in combination with STING significantly reduced the presence of both HBsAg and HBeAg circulating in cell supernatants, compared to cells either expressing STING or viperin alone (Figure 5B). Similarly, a significant reduction in HBsAg and HBeAg was also observed in supernatants derived from cells expressing both viperin and STING compared to those solely expressing STING or viperin when transfected with the genotype-D HBV for 96 hrs (Figure 5C). Collectively, these results demonstrate that the enhanced type-I IFN response elicited by the interaction between viperin and the dsDNA signalling adaptor STING, enhances the ability of the cellular immune response to limit HBV infection.
Discussion
Viperin is a potent antiviral host protein, associated with the inhibition of a broad range of viral infections (reviewed in [6]), however the scope of viperin’s antiviral capacity makes it increasingly difficult to discern the protein’s mechanism of viral inhibition. To date viperin has been demonstrated to target multiple stages of the viral lifecycle through interactions with many, often functionally unrelated, host and viral proteins (reviewed in [6, 13]). The recent discovery of the novel molecule ddhCTP, a by-product of viperins enzymatic function offers an explanation for its ability to inhibit Flavivirus infection and unequivocally instigates the protein’s enzymatic function in its antiviral activity [14]. However, these described mechanisms of viperin’s antiviral activity fail to account for each instance of the protein’s ability to limit viral infection. Recent literature has suggested that viperin’s ability to positively regulate innate immune responses may elucidate a unifying antiviral mechanism for this potent antiviral protein [16, 17]. Here we show for the first time that viperin is able to enhance the innate immune response to dsDNA viral mimics and to DNA viral infection, which adds to the limited knowledge of viperin’s alternate innate immune regulatory capacity.
Viperins enhancement of the dsDNA signalling pathway has many commonalities with its ability to positively augment signalling activation following ssRNA and CpG DNA detection in a host cell. Here we show that viperin interacts with signalling adaptor proteins, STING and TBK1, which are central to the dsDNA signalling pathways, to enhance the activation of TBK1. Likewise viperin was also shown to interact with the signalling adaptor protein IRAK1 which is central to the TLR7 & 9 signalling pathway [17]; However, in this instance viperin was also demonstrated to interact with the E3 ligase TRAF6 which was responsible for the ubiquitination of IRAK1. While it is evident viperin directly binds to the adaptor protein STING (Figure 3E), which is the upstream adaptor protein of TBK1, the E3 ligase responsible for ligating the K63-linked ubiquitin chains to TBK1 remains unknown. Microscopy based analysis confirmed that viperin co-localises with the E3 ligases TRAF6 following dsDNA stimulation (Supplementary Figure 2A and B), in a similar manner to that seen following activation of TLR7 and TLR9 [17]. We were also able to show that this is the case for viperin and TRAF3 following dsDNA stimulation (Supplementary Figure 2A and C). Both TRAF3 and TRAF6 have been previously implicated in the STING-TBK1 signalling axis [24, 35], however, further analysis is required to determine the functional relevance of this co-localisation to viperin’s enhancement of TBK1 activation.
The ability of viperin to enhance the dsDNA signaling pathway presents a novel mechanism for the protein’s antiviral capacity against DNA viruses. To investigate this in the absence of potential direct antiviral activity, we utilised a plasmid based induction model of HBV replication in a hepatocyte cell line. In this study, overexpression of viperin resulted in enhanced activity of the IFN-β promoter in HepG2 cells transfected with both HBV genotypes D and A (Figure 5A), which correlated with a reduction in HBsAg and HBeAg present in the culture media (Figure B and C). Through viperin’s direct interaction with STING and enhancement of TBK1 activation, we postulate that viperin enhances the type-I interferon response to HBV, enabling a more effective clearance of the viral infection in vitro. Additionally, viperin knockdown in HeLa cells was able to significantly enhance the viral titre of the DNA virus HSV-1 (Supplementary Figure 3). This is in contrast to a previously described lack of HSV-1 inhibition attributed to the ectopic expression of viperin in HEK293T cells [36]. However STING and cGAS protein is undetectable in HEK293T cells [37], which would undermine viperin’s ability to limit HSV-1. Together this data further highlights the importance of viperin’s interaction with adaptor molecules of the DNA signalling pathway to inhibit the infection of DNA viruses.
Viperin’s ability to generate ddhCTP to inhibit the replication of members of the Flavivirus genus via inhibition of polymerase function, relies on its enzymatic cleavage of SAM [14]; however ddhCTP was found to not inhibit the polymerases of members of the Picornaviridae family. Additionally, the ability of ddhCTP to inhibit the HCV RdRp was considerably lower than either of the Flaviviruses, DENV or WNV RdRps in an ex vivo assay [14], and other more diverse viruses such as HIV [8] and BUNV [38] are also inhibited by the functions of viperin’s enzymatic radical SAM domain in an as yet unspecified manner. Moreover, previous studies have demonstrated that the deletion of viperin’s N-terminus significantly abrogates its inhibition of not only HCV [11], but also CHIKV [39], and WNV [40]; a domain absent in the recombinant Rattus norvegicus viperin utilised to generate ddhCTP in the Gizzi et al. study [14]. This evidence is suggestive of a potentially alternate antiviral role to viperin’s generation of ddhCTP but reliant on its radical SAM domain, to achieve the observed levels of viperin mediated inhibition of multiple viruses.
The radical SAM domain of viperin may confer multiple functions to the protein and be regulated by the varied mechanisms of viperin Fe/S-dependent maturation. Here we demonstrate viperin’s requirement for the insertion of the 4Fe-4S cluster, a co-factor necessary for viperin’s enzymatic activity, within its radical SAM domain to enhance the type-I IFN response to dsDNA (Figure 2B). The insertion alone of this 4Fe-4S cluster has previously been shown to stabilise viperin [41], and is primarily inserted by the cytosolic iron-sulphur protein assembly (CIA) targeting complex CIA1-CIA2B-MMS19, via localisation sequences in the C-terminus of viperin [42]. However the significance of this interaction appears limited as the deletion of viperin’s C-terminus, and hence the insertion of the enzymatically-required 4Fe-4S cluster, significantly increased the protein’s ability to enhance dsDNA signalling (Figure 2B). However, viperin has been shown to also independently interact with the alternate CIA targeting factor CIA2A at its N-terminus [42]. The two isoforms of CIA2 (CIA2A and CIA2B) represent distinct branches of the CIA pathway and their combined interaction with a single protein is unprecedented [43]. Binding of CIA2A to viperin may offer altered conformational stability of viperin independent of viperin’s utilisation of the 4Fe-4S cluster, as is the case for iron regulatory protein 2 (IRP2) [43], one of the few other binding partners of CIA2A – which intrinsically lacks 4Fe-4S binding capacity (as reviewed in [44]).
The interaction between viperin and CIA2A may underpin viperin’s role in augmenting innate signalling events to limit viral infection. Following depletion of any component of the alternate CIA1-CIA2B-MMS19 targeting complex, the interaction between viperin and CIA2A is significantly improved [42]. Here we show that a viperin mutant (viperin 3’Δ17) which would be incapable of interacting with CIA1-CIA2B-MMS19, but retain association with CIA2A [42], significantly enhances viperin’s ability to enhance the IFN-β promoter following dsDNA stimulation (Figure 2B). Conversely, the viperin-5’Δ33 mutant which lacks its N-terminus – the region necessary for association with CIA2A – is unable to enhance the type-I IFN response to the same degree as wild-type viperin following dsDNA stimulation (Figure 2B). Additionally, the chimeric NS5A mutant which also lacks its N-terminus but retains the N-terminal attributed ER/lipid droplet localisation of viperin, displays similar abrogation in its ability to enhance the IFN-β promoter compared to the viperin-5’Δ33 mutant (Figure 2C). Together this data suggests the presence of an N-terminal sequence which is linked to the function of viperin’s radical SAM domain and is required for the protein’s enhancement of the dsDNA signalling pathway. We hypothesis that this sequence confers its interaction with CIA2A, and that viperin’s independent interaction with both the CIA2A and the CIA1-CIA2B-MMS19 complexes at opposing termini may represent a novel regulatory mechanism of viperin activity; switching between the innate immune regulation and the enzymatic generation of ddhCTP respectively.
The evolution of viperin predicates the protein’s role in innate immune regulation. Viperin is highly conserved, showing high amino acid identity across not only vertebrates including mammals, fish (reviewed in [6]) and reptiles [45], but also invertebrates such as oysters [46]. A recent study of the type-I ‘interferome’ identified viperin as a core IFN-induced antiviral factor across numerous vertebrate species [47], highlighting the protein’s ancestral role in antiviral innate immunity. Interestingly, a separate study of transcriptional divergence of the innate immune response between species revealed the high conservation of genes encoding proteins involved in immune response regulation as opposed to those with more direct acting effects on viral invasion [48]. Together this data provides evidence for viperin’s ancestral role as a regulator of the innate immune response to viral infection, and here we describe another instance of viperin’s enhancement of innate immune signalling events, complementary to its role in positively regulating TLR7/9 signalling [17].
The continued understanding of viperin’s antiviral activity offers valuable insight into the development of novel antiviral therapeutics. The study of viperin’s enzymatic function has already lead to the synthetic generation of the metabolic precursor of ddhCTP – ddhC – which shows promise as an antiviral therapeutic for its ability to enter human cells and inhibit the replication of ZIKV in vitro [14]. A clearer understanding of viperin’s role in augmenting a multitude of diverse innate signalling events may additionally highlight complementary approaches to a broad range of small-molecule agonist therapies to combat anti-viral therapeutics.
Materials and Methods
Cells and Culture Conditions
All mammalian cell lines were maintained at 37°C in a 5% CO2 air atmosphere. Huh-7 human hepatoma cells, HeLa human epithelial cells, HEK293T human embryonic kidney cells, as well as primary murine embryonic fibroblast (MEF) cells were all maintained in DMEM (Gibco) containing 10% (v/v) FCS. HepG2 human hepatoma cells were maintained in MEM (Gibco) containing 10% (v/v) FCS. The viperin−/− MEFs were generated and prepared as previously described [20]. The polyclonal Huh-7 cell line stably expressing shRNA targeting viperin mRNA was as previously described [11].
Real Time PCR
All experiments involving real-time PCR were performed in 12-well plates with cells seeded at 7 × 104/well, 24 hrs prior to transfection, and performed at least in triplicate. Total RNA was extracted from cells using TriSure reagent (Bioline), with first strand cDNA being synthesized from total RNA and reverse transcribed using a Tetro cDNA synthesis kit (Bioline). Quantitative real-time PCR was performed in a CFX Connect Real-Time Detection System (BioRad) to quantitate the relative levels of IFN and ISG mRNA in comparison to the house keeping gene RPLPO. Primers sequences were as follows: RPLOPO-FP 5’-AGA TGC AGC AGA TCC GCA T-3’, RPLPO-RP 5’-GGA TGG CCT TGC GCA-3’, IFI6-FP 5’-CCT GCT GCT CTT CAC TTG CA-3’, IFI6-RP 5’-CCG ACG GCC ATG AAG T-3’, OAS-FP 5’-TCC ACC TGC TTC ACA GAA CTA CA-3’, OAS-RP 5’-GGC GGA TGA GGC TCT TGA G-3’, mIFN-β-FP 5’-AGA AAG GAC GAA CAT TGG GAA A-3’, mIFN-β-RP 5’-TAG CAG AGC CCT TTT TGA TAA TGT AA-3’.
Immunoprecipitation Analysis
Where stated, prior to immunoprecipitation, cells were incubated with No-Weigh™ Format DSS crosslinker (Thermo-Fisher Scientific) for 30 mins at RT in ice-cold PBS (1.35mM DSS, pH 8.0), and then in quench solution (15 mM Tris, pH 7.5) for 15 mins at RT. Cell extracts were prepared with 0.5% (w/v) CHAPS lysis buffer supplemented with protease inhibition cocktail (Sigma). Lysates were pre-cleared with protein A/G PLUS-agarose beads (Santa Cruz Biotechnology) washed with 0.5% (w/v) CHAPS, immunoprecipitated with 2 μg/sample of indicated antibodies overnight at 4 °C before addition of washed protein A/G PLUS-agarose beads (Santa Cruz Biotechnology) for 1 hr with rotation at 4 °C. After extensive washes with the same lysis buffer, the immunoprecipitates were subject to immunoblot analysis.
Immunoblot Analysis
Lysates were subjected to SDS-PAGE. Proteins were transferred to 0.2 μm nitrocellulose membranes (Bio-Strategy) and probed with indicated primary antibodies. The protein bands were visualized using a SuperSignal West Femto Maximum Sensitivity Substrate (Thermo-Fisher Scientific) for horseradish peroxidase (HRP) conjugated secondary antibodies. The probing with the monoclonal mouse anti-β-actin antibody (Sigma) was used as a loading control. Membranes were scanned using an Amersham 600 chemiluminescent imager.
Dual Luciferase Reporter Assay
Luciferase experiments were performed essentially as previously described [49]. Cells were seeded at 4 x 104 per well in 24-well plates, 24 hrs prior to transient transfection using Viafect (Promega) with 250 ng of a specified target construct as well as 250 ng pIFN-β-Firefly luciferase in combination with 2.5 ng of the constitutively expressing Renilla luciferase plasmid, pRL-TK. Following a further 24 hrs, cells were stimulated with synthetic viral mimics for specified time periods. Cells were lysed with 1 x PLB (Promega) and the luciferase outputs were measured with a dual-luciferase reporter assay system (Promega) on a CLARIOstar (BMG LABTECH) microplate reader. All conditions were performed in at least triplicate.
Viral Infection and Viral Mimics
Transfection of HBV was performed as previously described [34], in HepG2 cells using a recombinant 1.3-mer transient transfection model system for HBV genotypes A and D. Cells were seeded at 1 x 105 per well in 24-well plates, 24 hrs prior to transient transfection using Viafect (Promega) with a combined total of 500 ng per well of the specified HBV 1.3 mer plasmid as well as target and luciferase plasmids. Cell supernatants were harvested at specified time points for quantitative serology as previously described [34], and cell lysates were harvested for dual luciferase reporter assays.
Herpes simplex virus type I (HSV-1) strain KOS was used to infect HeLa cells washed with phosphate-buffered saline (PBS) at the indicated multiplicity of infection (MOI) in DMEM without FCS. After 1 hr of incubation, the infection medium was removed and replaced with DMEM containing 2.5% (v/v) FCS.
The viral mimics, poly dA:dT (Invivogen) was transfected into cells using DMRIE-C reagent (Life Technologies) as per manufacturer’s instructions at a concentration of 1 μg/ml.
Immunofluorescence Microscopy
All immunofluorescence staining was performed as previously described [50], and was visualised using either a Nikon Eclipse Ti-E fluorescence inverted microscope or a Ziess Confocal LSM 780 microscope, and images were captured using NIS Elements software or ZEN microscopy software respectively.
Proximity Ligation Assay
Cells were seeded at 7 x 104 per well in 12-well plates, 24 hrs prior to transient transfection using Viafect (Promega) with the specified viperin-flag constructs. Following a further 24 hrs, cells were trypsinised and seeded at 3 x 103 per well in a 96-well plate, allowed to recover and then stimulated with viral mimics. Cells were fixed with 4% (w/v) paraformaldehyde and the proximity ligation assay (PLA) was conducted using the Duolink® In Situ Kit (Merck) as per the manufacturer’s instructions. Positive interactions were visualized using a Nikon Eclipse Ti-E fluorescence inverted microscope and images were captured using NIS Elements software.
Plasmid Constructs and Transfections
All plasmid constructs were transiently transfected into the indicated cells using Viafect Transfection Reagent (Promgea) as per manufacturer’s instructions. Viperin-GFP is within the pEGFP-C1 (Clontech) backbone where GFP is expressed as an N-terminal fusion protein. The corresponding control was empty pEGFP-C1. Viperin-flag consists of the pFLAG-CMV™ Gateway® backbone (Invitrogen), where a CMV promoter drives expression of the viperin with a flag tag at the N-terminus. The viperin mutant plasmids viperin-5’Δ33-flag, viperin-3’Δ17-flag and Viperin-SAM1-flag, were created as previously described [11]. Viperin-mCherry and mCherry plasmids were created as previously described [12]. The reporter plasmid IFN-β-Luc (IFN-Beta_pGL3) was a gift from Nicolas Manel (Addgene plasmid # 102597) [51]. pRL-TK (Promega) consists of a TK promoter which drives the constitutive expresses of Renilla luciferase. The, pCMV-STING-3Xmyc and pCMV-TBK1-myc plasmids were a gift from Russell Diefenbach (Westmead Millennium Institute, Sydney). These are vectors expressing an N-terminally myc-tagged STING or TBK1 proteins in transfected mammalian cells and are expressed constitutively by a CMV promoter. TBK1-mCherry was within pLenti-V5-D-TOPO-mCherry backbone where a CMV promoter drives its constitutive expression. The NS5A-TN50-viperin (NS5A-viperin) plasmid, which replaces the amphipathic helix of viperin with that of HCV NS5A was a kind gift from Professor Neil Marsh, University of Michigan [22]. The ubiquitin plasmids pRK5-Ubiqutin-K48-HA and pRK5-Ubiquitin-K63-HA (Addgene) are constructed with a pRK5-HA backbone where the HA tag is at the N-terminus of ubiquitin. Hepatitis B virus 1.3 mer plasmid constructs for both genotype A and D are as previously described [34].
The Nucleofector transfection kit (Lonza) was used to transfect siRNA into cells, according to the manufacturer’s guide. In brief, 1 x 106 of HeLa cells were transfected with 50 nM of siRNA universal control (Sigma Aldrich) or siRNA specific for viperin (SASI_Hs02_00362416; Sigma Aldrich) by a Nucleofector 2b Device (Lonza). Post 24 hr transfection, cells were used for infection or collected for immunoblot analysis. The sequence of the siRNA specific for viperin was 5’-AGA GCG GAA AGT GGA ACG AGA-3’.
Statistical Analysis
Results are expressed as mean ± S.E.M. Student’s t test was used for statistical analysis, with p < 0.05 considered to be significant. All statistical analysis was performed using Prism 6 (GraphPad Software). All experiments were performed in at least triplicate.
Competing Interests
The authors declare no competing interests exist.
Acknowledgements
The authors would like to acknowledge the La Trobe University Microscopy Platform for their assistance in the microscopy aspects of this work.
References




