

Abstract
Inherited transposition events are important drivers of genome evolution but because transposable element (TE) mobilization is usually rare, its impact on the creation of genetic variation remains poorly characterized. Here, we used a population of A. thaliana epigenetic recombinant inbred lines (epiRILs) to characterize >8000 de novo insertions produced by several TEs families also active in nature. Integration was strongly biased towards genes, with evident deleterious effects. Biases were TE family-specific and associated with distinct chromatin features. Notably, we demonstrate that the histone variant H2A.Z guides the preferential integration of Ty1/Copia LTR-retrotransposons within environmentally responsive genes and that this guiding function is evolutionary conserved. Finally, we uncover an important role for epigenetic silencing in exacerbating or alleviating the effects of TE insertions on target genes. These findings establish chromatin as a major determinant of the spectrum and functional impact of TE-generated mutations, with important implications for adaptation and evolution.
INTRODUCTION
Transposable elements (TEs) are sequences that self-propagate and accumulate to various levels in the genome of eukaryotic species. TEs fall into two broad classes, depending on their mechanism of transposition: DNA transposons, which move through a “cut and paste” mechanism and Long Terminal Repeat (LTR) as well as non-LTR retrotransposons, which move through an RNA intermediate that is reverse transcribed. The content of the different classes and subclasses of TEs varies markedly across species. Thus, mammalian genomes are laden with non-LTR retrotransposons whereas plant genomes are populated by LTR retrotransposons and DNA transposons mainly (Lisch 2013; Chuong, Elde, and Feschotte 2016). However, most of these sequences are relics of once active TEs and although TEs are arguably among the main drivers of the evolution of genome size, organization and function (Chuong et al., 2016), we still know little about their contribution to the generation of somatic or heritable mutations in extant genomes (Huang, Burns, and Boeke 2012; Sultana et al. 2017). This situation stems in large part from the fact that transposition is typically rare in nature, notably because of the strong epigenetic silencing mechanisms, such as DNA methylation in plants and mammals, that target TEs (Slotkin and Martienssen 2007).
With the advent of population genomics, it is now possible to make use of natural variation in the number and distribution of recent TE insertions to gain information about the set of TEs likely mobile in extant genomes. However, because of the deleterious effects typically associated with TE mobilization, natural selection prevents the proper assessment of the complete range of the mutations generated by transposition (Huang, Burns, and Boeke 2012; Sultana et al. 2017; Quadrana et al. 2016).
Here, we have exploited a population of A. thaliana epigenetic Recombinant Inbred Lines (epiRILs; Johannes et al., 2009) to obtain a first comprehensive assessment of the rate, spectrum, properties and genome-wide distribution of heritable mutations generated by TEs. The epiRILs were derived from a cross between a wild type and a near isogenic parent in which transposition was kick-started for several TEs as a result of compromised DNA methylation (Johannes et al., 2009; Figure 1A). Based on whole genome sequencing for more than 100 epiRILs and the characterization of >8000 de novo heritable insertions, we first establish that the epirRILs are operationally similar to mutation accumulation (MA) lines (Denver et al. 2009; Keightley et al. 2009; Zhu et al. 2014). We then show that two DNA transposons and one LTR-retrotransposon that are among the most active in nature target non-overlapping sets of genes and in relation to distinct chromatin features. We further demonstrate that the preferential targeting of environmentally responsive genes by LTR-retrotransposons of the Ty1/Copia superfamily is guided by the histone variant H2A.Z. We also provide evidence that this guiding function is ancestral, as it is conserved from plants to yeast and propose a plausible mechanistic explanation for the invasive success of Ty1/Copia retrotransposons exclusively within plant genomes. Finally, we show that epigenetic silencing of new insertions soon after their occurrence can either alleviate or exacerbate their effects on gene transcription. Our findings illustrate the role of chromatin as a major determinant of the spectrum and impact of TE-generated mutations, with important implications for adaptation as well as the evolutionary success of TEs and the organisms in which they reside.

A. Crossing scheme used to generate the epiRIL population. B. Number and identity of insertions accumulated in wild-type, ddm1 and the epiRILs. Number of heterozygous insertions are indicated within brackets. C. Overlap between TE families transcriptionally reactivated in ddm1, those potentially mobile in Col-0 and those that transposed in at least one epiRIL. D. Relation between number of private TE insertions and average expression ratio (ddm1/wild-type) for the 11 TE families that transposed in the epiRILs. E. Proportion of reference TE sequences upregulated in ddm1 and either silenced or stably upregulated in the epiRILs.
RESULTS
TE insertions accumulate in the epiRILs
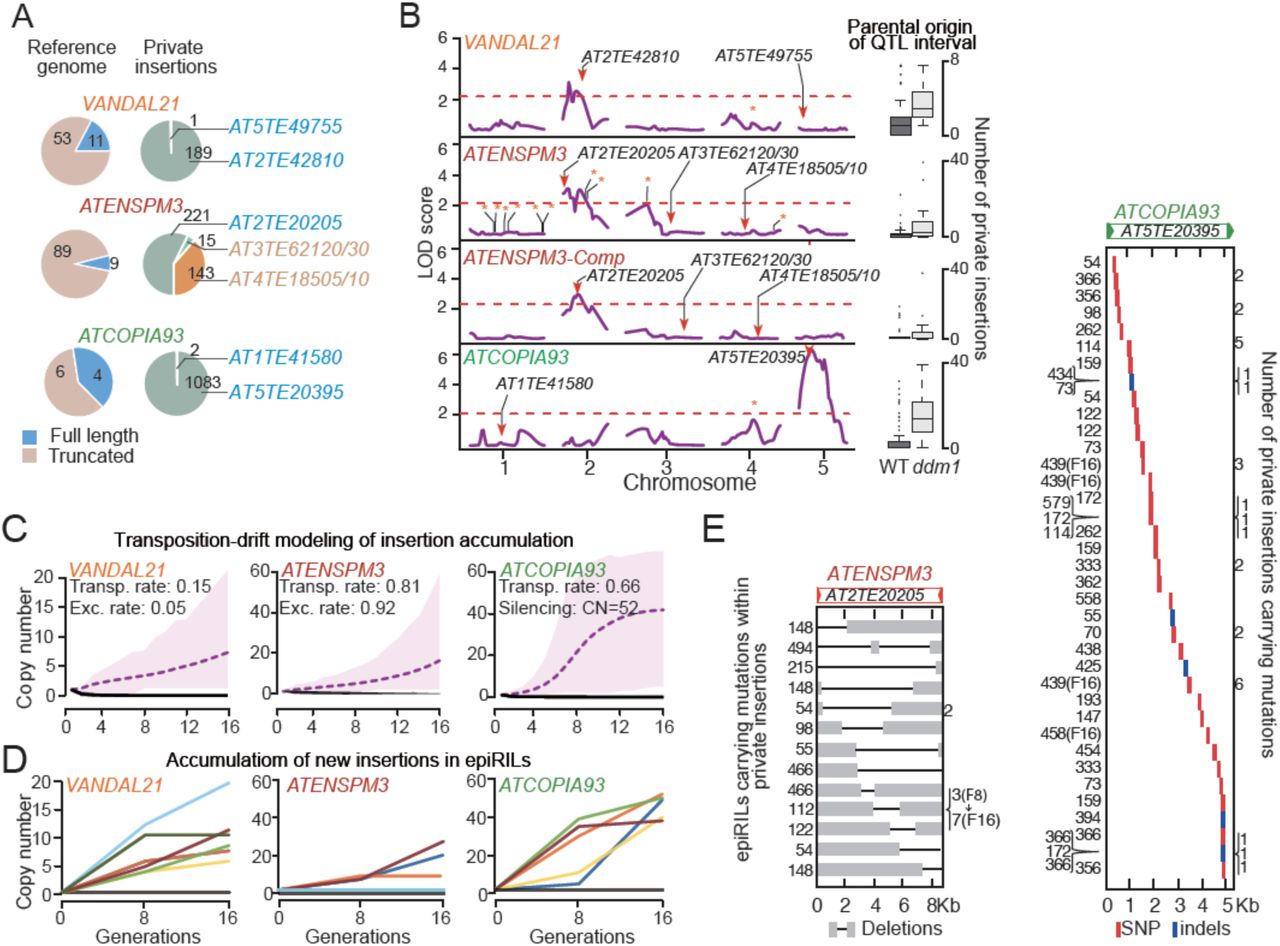
We produced high quality whole genome sequencing data for 107 epiRILs at generation F8 as well as of close relatives of the wild type and ddm1 parental lines (Figure 1A) using Illumina mate-pair libraries built from size-selected genomic fragments of ∼5.5 kb. Mate-pair reads were mapped to the reference genome sequence (accession Col-0) and non-reference (i.e. de novo) insertions were detected based on mapping discordance between mate-pair reads and using also split reads (Gilly et al., 2014; Quadrana et al., 2016; Figure S1A). Thanks to the large physical distance between mate-pair reads, the complete sequence of the de novo insertions was also determined, thus enabling the identification of the exact or most likely donor TE in each case. Consistent with the low frequency of TE mobilization in nature (Quadrana et al. 2016) there were no de novo TE insertions in the two wild type siblings sequenced nor in five A. thaliana MA lines (Ossowski et al. 2010). In contrast, two non-reference TE insertions were detected in the sequenced ddm1 individual and many more in the epiRILs. Specifically, 95% of the 107 epiRILs harbored between 1 and 97 heritable de novo insertions and only 8 had none (Figure S1B). Almost all (98.7%) of these insertions were private and therefore did not occur in the ddm1 parent but rather either in the inflorescences of the F1 individual used for the backcrossing or during the propagation of the epiRILs, starting from the F2 (Figure 1A). In fact, 1107 out of the 1670 private insertions detected in total were heterozygous (Figure 1B, S1B), which is strong evidence that transposition was ongoing in the epiRILs. Given that they were propagated by repeated selfing and single seed descent, the epiRILs could therefore be defined as TE accumulation (TEA) lines. In other words, notwithstanding the possibility of excision in the case of DNA transposons (see below), TE insertions that occurred during the propagation of the epiRILs and that were potentially heritable should be lost or fixed in a neutral fashion through random segregation, except for those disrupting essential genes.
Overall, we detected private insertions for two DNA transposon and eight retrotransposon families as well as for three composite DNA transposons (Figure 1B). These composite elements were uncovered thanks to our mate-pair sequencing approach and are made up for two of them of a protein-coding gene of unknown function flanked by two distinct truncated copies of ATENSPM3. The third composite DNA transposon is more complex and contains several truncated TEs, the largest of which is related to the ATMU5 family (Figure S1C). The number of transposition events varied considerably among the eleven TE families, with the two DNA transposons VANDAL21 and ATENSPM3 (together with the two composite ATENSPM3 elements) and the LTR-containing retrotransposon ATCOPIA93 contributing 11.2%, 22.5% and 64.4% of the de novo insertions, respectively (Figure 1B).
We showed previously that 73 of the 326 TE families annotated in the A. thaliana genome transposed in the recent past in the reference accession Col-0 (Quadrana et al. 2016), which was used to derive the epiRIL population. Consistent with this finding the 10 annotated TE families with private insertions in the epiRILs are also mobile in nature and belong to the Col-0 mobilome. Nonetheless, the epiRIL mobilome is only a small subset of that of Col-0, which indicates that the widespread loss of DNA methylation induced by ddm1 does not translate in an equally widespread remobilization of TE families. To determine if differences in the level of ddm1-induced transcriptional reactivation between TE families could explain at least in part the limited mobilome of the epiRILs, we performed RNA-seq on whole seedlings for five epiRILs as well as one sibling of the two parents. Between 731 and 1056 (considering unique or multiple mapping reads, respectively) TEs, mostly full-length, were significantly overexpressed at least 2-fold (P<0.05) in ddm1 compared to wild type, consistent with previous RNA seq data sets (Figure S2). Moreover, >75% of the 73 TE families that compose the Col-0 mobilome were upregulated in ddm1, including eight of the 11 TE families that are part of the epiRIL mobilome (Figure 1C). However, levels of upregulation in ddm1 did not correlate with the number of private insertions in the epiRILs (Figure 1D). Finally, 72% of the ddm1-upregulated TEs for which expression could be ascertained unambiguously in the epiRILs remained transcriptionally active when derived from the ddm1 parent (Figure 1E). Thus, levels of transcription in seedlings did not reflect transposition activity. Whether this also holds true in the cells, starting from the zygote, that will ultimately pass on their DNA to the next generation remains to be determined, as these cells are where transposition must occur for insertions to be inherited.
Invasion dynamics differ between TE families
The large number of private insertions for VANDAL21, ATENSPM3 and ATCOPIA93 enabled us to investigate in depth for each of these three TE families their mode and tempo of mobilization across generations. Mate-pair reads were first used to determine the sequence of the private insertions. In the case of VANDAL21 and ATCOPIA93, almost all (99%) were identical to a single one of the 11 and four full-length copies present in the reference genome, respectively. This single copy is the same that was reported previously to be mobile in ddm1 or another DNA methylation mutant background (Fu et al. 2013; Mirouze et al. 2009). In contrast, the identity of the ATENSPM3 private insertions was more diverse, with 58% matching a single one of the nine full-length copies and the remaining private insertions matching either one of the two composite copies also present in the reference genome (Figure 2A). Each of these two composite ATENSPM3 copies contains a single gene of unknown function with no similarity to any known transposase gene, indicating that they were likely mobilized in trans, presumably using the transposase encoded by the mobile full-length ATENSPM3 reference copy. Thus, for each of these three TE families, a single ddm1-derived reference element is at the origin of most transposition events that accumulated in the epiRILs.

A. Identification of donor copies for each transposition event. The number of truncated and full-length copies annotated in the A. thaliana reference genome is also shown. B. QTL mapping results for the mobilization of VANDAL21, ATENSPM3, the two composite ATENSPM3 and ATCOPIA93. TEs annotated in the reference A. thaliana genome as well as donor and shared insertions are indicated by bars, arrowheads and stars, respectively. Box-plots on the right indicate for each TE family the numbers of insertions in the epiRILs in relation to the parental origin of the relevant QTL interval. C. Predicted dynamics of insertion accumulation for the three TE families based on the best fitted transposition-drift model. Estimates of the transposition and excision rates as well as of the number of TE insertions required for triggering concerted epigenetic silencing are indicated. D. Number of VANDAL21, ATENSPM3, and ATCOPIA93 insertions observed at generation F8 and F16 in ten epiRILs. E. Mutations detected within private insertions of ATENSPM3 and ATCOPIA93. Carrier epiRILs as well as the number of private insertions with the mutation are indicated in each case on the left and right side of each panel, respectively.
To confirm this conclusion and explore in more detail the genetic architecture underlying the large variation in the number of private insertions accumulated in the epiRILs, we performed a quantitative trait locus (QTL) analysis for each of the three TE families. We took advantage of the fact that the epiRILs are virtually isogenic but segregate hundreds of parental differentially methylated regions (DMRs) that can be used as bona fide genetic markers (Colomé-Taché et al, 2012; Cortijo et al., 2014) to identify loci whose epigenetic state is associated with transposition activity. Using the number of private insertions as the quantitative trait, this approach revealed a single QTL interval for each TE family, which included the full-length reference copy identified in the previous step (Figure 2B). Furthermore, the same QTL interval was obtained whether all ATENSPM3 private insertions were considered or only those that match the two composite ATENSPM3 present in the reference genome, thus demonstrating that these were mobilized in trans by the single full-length, ddm1-derived reference copy of ATENSPM3.
To determine if these observations were compatible with a transposition-drift scenario, we modeled TE mobilization for each of the three TE families based on three key parameters. These are the rate of transposition as well as of excision in the case of the DNA transposons, as this is an integral part of their mechanism of transposition, and copy-number dependent inhibition of transposition, as ATCOPIA93 was shown to be epigenetically silenced when reaching around 40 copies (Marí-Ordóñez et al. 2013). For each TE family we selected the model that produced the best fit to the observed number and ratio of homozygous vs heterozygous private insertions at generation F8 and ran each model for another eight generations (F16; Figure S3A). The selected models predicted that in addition to the original reference TE donor, all new insertions are equally able to transpose, at a rate of 0.15, 0.81 and 0.66 new copy/per donor/per generation for VANDAL21, ATENSPM3 and ATCOPIA93, respectively (Figure 2C and S3B). The best-fit models also predicted low and high rates of excision for VANDAL21 and ATENSPM3, as was observed in maize for TEs belonging to the same two superfamilies Mu and En/Spm, respectively (Doseff, Martienssen, and Sundaresan 1990; Masson et al. 1987). In agreement with these predictions, the mobile reference copy of VANDAL21 was almost always retained in the epiRILs, unlike that of ATENSPM3, which was systematically missing from the corresponding ddm1-derived chromosome interval (Figure S3C). Furthermore, indels compatible with ATENSPM3 excision footprints were consistently detected across the genome and their number correlated positively with that of ATENSPM3 private insertions (Figure S3D). Finally, our modeling predicted that transposition of ATCOPIA93 should stop when reaching 52 copies (Figure 2C and S3B). To evaluate further the predictive power of our models, we sequenced ten epiRILs that were propagated for another 8 generations (F16). Consistent with numerical simulations (Figure 2C), VANDAL21 and ATENSPM3 continued to transpose in most of the lines with initial transposition activity, with no evidence of copy-number-dependent transposition inhibition. In contrast, although ATCOPIA93 continued to transpose beyond F8, none of the F16 lines had accumulated more than 50 copies (Figure 2D). Furthermore, almost all copies were in the homozygous state at F16, consistent with the concerted epigenetic silencing expected at this copy number (Marí-Ordóñez et al. 2013).
Last, we took advantage of the fact that mobilized TEs tend to mutate during the transposition process to obtain direct evidence that in addition to the initial ddm1-derived donor copy, new insertions were also mobilized in the epiRILs. While all new insertions were identical to the mobile reference element for VANDAL21, approximately 5% of ATENSMP3 and ATCOPIA93 new insertions identified at F8 and/or F16 contained large internal deletions and point mutations or small indels, respectively (Figure 2E). Some mutations were carried by more than one new insertion within an epiRIL, but rarely between epiRILs. Notably, for one of the 10 epiRILs also sequenced at F16, we observed an increase in the number of ATENSPM3 insertions containing the same mutation at that later generation (Figure 2E). These findings provided therefore further evidence that newly inserted copies were also mobilized in the epiRILs.
TEs exhibit strong and diverse chromatin-associated insertion biases towards genes
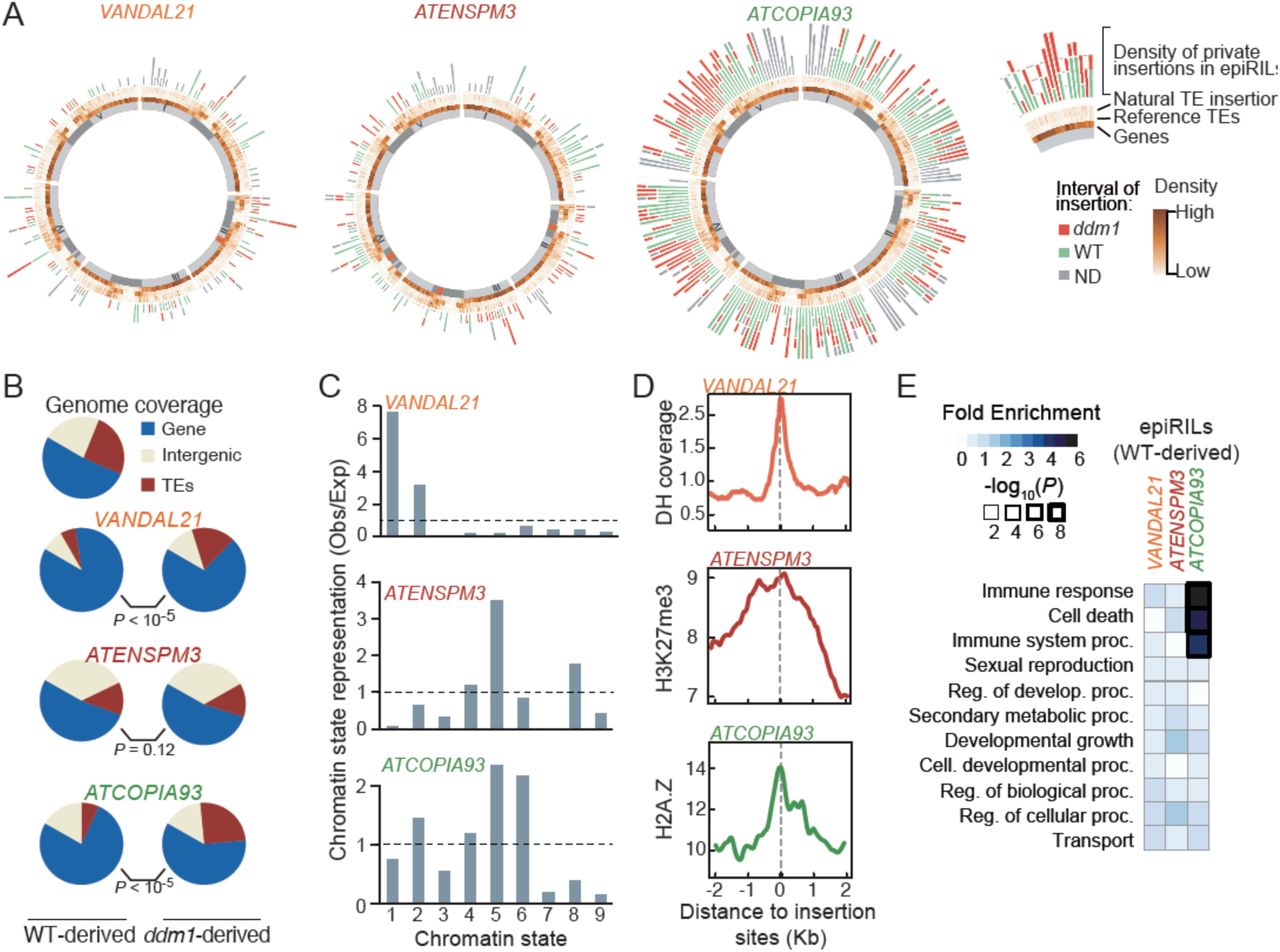
As in nature (Quadrana et al. 2016), private insertions for VANDAL21, ATENSPM3 and COPIA93 were distributed evenly across the five chromosomes in the epiRILs, overall (Figure 3A). However, because the genomes of the epiRILs are epigenetic mosaics, each being composed on average of 25% ddm1- and 75% wild-type-derived segments (Colomé-Tatché et al, 2012), we searched for potential differences in insertion frequency associated with parental origin. For all three TE families, the percentage of insertions in ddm1-derived intervals was slightly higher than expected by chance at the whole genome level but much higher (35-55%) when considering the pericentromeric regions only (Figure 3A and S4A), which lose their heterochromatic features in ddm1 as well as in subsequent generations (Soppe et al. 2002; Lippman et al. 2004; Colome-Tatche et al. 2012). These findings reinforced the notion that euchromatin is the preferred substrate for the integration of VANDAL21, ATENSPM3 and COPIA93.

A. Circos representation of private TE insertions detected for VANDAL21, ATENSPM3 and ATCOPIA93. The exterior circle represents the density of private insertions within wild-type- and ddm1-derived intervals. Density of non-reference (natural) TE insertions, reference TEs as well as genes are represented inwards in this order. B. Fraction of TE insertions in wild-type- and ddm1-derived intervals in genes, TEs and intergenic regions. Statistical significance for each comparison was obtained using the Chi-square test. C. Observed/expected ratio (Obs/Exp) of insertion sites within wild-type-derived regions in relation to the nine chromatin states defined in A. thaliana. Error bars represent the 95% confidence interval obtained by 1000 boots-traps. D. Coverage of DNAse hypersensitivity (DH; top panel), H3K7me3 (middle panel) and H2A.Z (bottom panel) around private insertion located within wild-type-derived regions for VANDAL21, ATENSPM3 and ATCOPIA93, respectively. E. GO term analysis of genes with private TE insertions in the epiRILs.
Given that the methylome as well as the transcriptome of wild-type-derived intervals in the epiRILs are with few exceptions identical to their wild type parental counterparts (Colome-Tatche et al., 2012; Ito et al., 2015; Figure S2), we then used the private insertions located within these intervals to obtain information on integration preferences. Insertions were strongly overrepresented within or adjacent to genes for the three TE families (Figure 3B). However, while the fraction of essential genes (Lloyd et al. 2015) that were targeted was that expected by chance for VANDAL21, it was much lower for ATENSPM3 and ATCOPIA93, suggesting that their integration, unlike that of VANDAL21, strongly affects the expression of target genes. Consistent with this interpretation, the fraction of essential genes targeted by ATCOPIA93 was further reduced when only considering insertions in the homozygous state (Figure S4B). Unexpectedly though, we found an opposite pattern for ATENSPM3 insertions within essential genes, which were less frequent in the heterozygous than the homozygous state. Given the high rate of excision associated with ATENSPM3 mobilization, this last result suggested stronger deleterious effects after excision of ATENSPM3. Consistently, none of the 98 homozygous excision footprints detected in the epiRILs were located within essential genes (Figure S4B).
Using the nine main chromatin states defined in A. thaliana based on epigenomic maps obtained from diverse organs and tissues (Sequeira-Mendes et al. 2014), we found that VANDAL21 mainly targeted promoters and 5’ UTRs of genes characterized as active (chromatin states 1&2; Figure 3C). In contrast, ATENSPM3 and ATCOPIA93 preferentially inserted within or close to genes that are typically marked by H3K27me3 and slightly enriched for the variant histone H2A.Z (chromatin state 5) and, in the case of ATCOPIA93, also in or adjacent to genes that were only enriched for that histone variant (chromatin state 6; Figure 3C).
Meta-analysis of insertion sites confirmed these findings and revealed that VANDAL21 integration within wild-type-derived intervals tended to coincide with peaks of DNase I hypersensitivity (Figure 3D) at the transcriptional start site (TSS) of genes (Figure S4C). Furthermore, insertions were typically in the same orientation as the target genes (Figure S4C). These results support previous observations reported for VANDAL21 in ddm1 mutant plants (Fu et al.,2013) as well as in nature (Quadrana et al. 2016). In contrast, insertion sites were enriched for H3K27me3 in the case of ATENSPM3 and for nucleosomal DNA as well as H2A.Z in the case of ATCOPIA93 (Figure S4D). However, ATCOPIA93 did not insert preferentially within well-positioned nucleosomes (Figure S4E; Lyons and Zilberman, 2017) nor within so-called “+1 nucleosomes”, which tend to incorporate H2A.Z (Henikoff and Smith 2015), but rather within genes (Figure S4F) that are enriched in H2A.Z throughout their body (Figure S4G). Such pattern of H2A.Z deposition is specific to plants and was shown previously to concern mainly responsive genes (Coleman-Derr and Zilberman 2012; Zahraeifard et al. 2018). In agreement, this class of genes was strongly overrepresented among ATCOPIA93 targets (Figure 3E).
H2A.Z directs the integration of ATCOPIA93
The Ty1/copia superfamily of LTR-retrotransposons is one of the main contributors of genome size inflation seen in many plant species (Huang, Burns, and Boeke 2012). We therefore explored further the function of chromatin in the integration of ATCOPIA93 and first noted that the proportion of insertions within TE sequences was much higher in ddm1-derived than in wild-type-derived intervals (Figure 3B). This finding is entirely consistent with the fact that TE sequences tend to acquire H2A.Z when hypomethylated (Zilberman et al. 2008). Indeed, H2A.Z enrichment levels measured in the hypomethylated background met1 that was used for this previous analysis were on average much higher, in ddm1-derived intervals, for TE sequences targeted by ATCOPIA93 than for those that were not (Figure S4H).
To demonstrate the involvement of H2A.Z in the integration of ATCOPIA93, we crossed one epiRIL (epiRIL54, F8) containing 23 active (mainly heterozygous) ATCOPIA93 copies to a wild type or an hta9 hta11 double mutant parent, which lacks most H2A.Z (March-Díaz et al. 2008). Two F1 individuals were selfed in each case and two homozygous wild type as well as two homozygous double mutant F2 lines were selected (Figure 4A) to produce F3 progeny for TE-sequence capture (Quadrana et al. 2016). DNA was extracted from approximately 1000 F3 seedlings from each of the four F2 lines. Over 2000 new ATCOPIA93 insertions were detected in each of the two wild type F3 progenies and less than twice that number in the two hta9 hta11 F3 progenies (Figure 4B). In addition, while the strong ATCOPIA93 integration preferences observed in the epiRILs was confirmed in wild type F3 seedlings, they were totally abolished in the hta9 hta11 lines (Figure 4C, 4D, 4E). Moreover, the proportion of essential genes targeted by ATCOPIA93 almost tripled in the double mutant (Figure 4F). These results demonstrated that H2A.Z acts at two levels, to promote ATCOPIA93 retrotransposition and to guide integration within environmentally responsive genes. To investigate at the nucleosome-scale the guiding function of H2A.Z, we crossed the list of the ATCOPIA93 insertion sites detected in the wild type F3 progeny with the list of well-positioned nucleosomes previously produced for A. thaliana (Lyons and Zilberman 2017). A major peak of integration was observed ∼55 bp away from the nucleosome dyad (Figure 4G). This position is a main point of contact between DNA and H2A or H2A.Z and it is also where these two histones differ by several amino acids (Suto et al. 2000; Zlatanova and Thakar 2008). These findings further support a direct role of H2A.Z in guiding ATCOPIA93 integration.

A. Experimental strategy for determining the role of H2A.Z in the integration of ATCOPIA93. epiRIL54 was crossed with wild type or the double mutant hta9 hat11. Two wild type (HTA9 HTA11 L1 and L2) and two double mutant (hta9 hta11 L1 and L2) F2 plants were selected and 1000 F3 seedlings were collected in each case to perform TE-sequence capture. B. Number of new insertions detected in 1000 HTA9 HTA11 and hta9 hta11 F3 seedlings. C. Meta analysis of levels of H2A.Z around ATCOPIA93 insertion sites. D. Observed/expected ratio (O/E) of insertion sites in relation to the nine chromatin states defined in A. thaliana. Error bars represent the 95% confidence interval obtained by 1000 boots-traps. E. GO term analysis of genes containing new ATCOPIA93 insertions in HTA9 HTA11 or hta9 hta11. F. Fraction of essential genes among those targeted by ATCOPIA93 in HTA9 HTA11 or hta9 hta11 F3 seedlings. Statistically significant differences were calculated using the chi-square test. G. Density of ATCOPIA93 insertions over the surface of mouse H2A.Z containing nucleosomes (PDB 1F66). Only 73bp of DNA and associated proteins are viewed down the superhelical dyad and each number (0-7) represents one DNA double helix turn, starting from the central base pair. The exterior circle shows the density of new ATCOPIA93 insertions per base pair.
H2A.Z-directed integration is evolutionarily conserved
Unlike Ty3/Gypsy LTR-retrotransposons, those of the Ty1/Copia superfamily tend to insert within euchromatin (Sultana et al. 2017). To determine if the guiding function of histone H2A.Z is evolutionary conserved, we first focused on ATCOPIA78, which is distantly related to ATCOPIA93 (Figure 5A). There were no private ATCOPIA78 insertions in the epiRILs, but previous work showed that ATCOPIA78 can be transcriptionally reactivated by heat stress and mobilized if stressed plants are defective in RNA-directed DNA methylation, such as in the nrpd1 mutant background (Ito et al., 2011). We therefore assessed the mobilization of ATCOPIA78 in pools of nrpd1 seedlings that were subjected or not to heat stress and subsequently grown under normal conditions to produce seeds. One thousand F1 seedlings from each pool were grown under normal conditions and used to perform TE-sequence capture (Figure 5B). A total of 279 ATCOPIA78 insertions were recovered in the progeny of heat-stressed plants, compared to only two in the progeny of non-stressed plants (Figure 5C). Insertion preferences were similar to those observed with ATCOPIA93 (Figure 5D, E, F).

A. Phylogenetic analysis of Ty1/Copia LTR-retrotransposons from A. thaliana as well as Tos17 and Ty1 from rice and budding yeast, respectively. B. Experimental strategy for studying transposition landscape in A. thaliana of the heat-responsive ATCOPIA78 LTR-retrotransposon. 1000 nrpd1 F1 seedlings derived from plants grown under control conditions (Ctl F0) or subjected to heat-stress (HS F0) were subjected to TE-sequence capture. C. Number of new insertions detected in Ctl F0 or HS F0. D. Metagene analysis showing the distribution of new ATCOPIA78 insertions detected in heat-stressed nrpd1 mutant plants (HS F0) or in natural population of A. thaliana (Natural insertions). UTR, untranslated transcribed region. E. GO term analysis of genes containing new ATCOPIA78 insertions in heat-stressed nrpd1 mutant plants (HS F0) or in natural population of A. thaliana (Natural insertions). F. Metanalysis of A. thaliana, rice (O. sativa) and yeast (S. cerevisiae) H2A.Z levels around ATCOPIA78, Tos17 and Ty1 insertion sites, respectively. For A. thaliana, experimental and natural insertions are depicted in green and red, respectively.
Unlike for ATCOPIA93, numerous recent ATCOPIA78 insertions were found in nature (Quadrana et al. 2016). Although patterns of integration were similar in the experimental and natural settings (Figure 5D, E and F), the purifying effect of selection was already evident in nature. Specifically, the fraction of natural private insertions within exons was much reduced compared to that found experimentally (Figure 5D). Similarly, none of the 147 recent natural insertions examined were within essential genes, compared to 2.8% of the ATCOPIA78 insertions detected experimentally (Figure S5). These findings establish a key role for H2A.Z in directing the integration of COPIA retrotransposons in nature and further highlight the strong deleterious effects they can cause.
Last, we investigated the integration patterns of Ty1/Copia retrotransposons in relation to H2A.Z in a distant plant (rice; Miyao et al., 2003) and in widely divergent species (S. cerevisiae; Mularoni et al., 2012). In both cases, experimentally induced insertions were located at sites enriched for H2A.Z (Figure 5F), thus indicating that the guiding role of H2A.Z has been evolutionarily conserved since the last common ancestor of plants and fungi.
Intronic ATCOPIA insertions create epigenetically-inducible alternative transcripts
To assess the functional impact of the strong, chromatin-based insertion biases towards genes observed in the epiRILs, we measured gene expression in six lines by RNA-seq. None of the homozygous VANDAL21 and ATENSPM3 insertions had detectable effects on the expression of target genes (Figure 6A), consistent with findings in natural accessions (Quadrana et al. 2016). In marked contrast, six of the 16 distinct homozygous ATCOPIA93 insertions located within genes affected negatively their expression (Figure 6A). We also performed RNA-seq on one of the six epiRILs taken at a later generation (F16, epiRIL394). At this generation, there were approximately 40 ATCOPIA93 insertions (counting homozygous insertions as two) per epiRIL and all were silenced and presumably methylated (Figure 6B).

A. Expression ratios between epiRILs and wild type plants for genes harboring homozygous VANDAL21, ATENSPM3 and ATCOPIA93 insertions. Genes with exonic, intronic or nearby insertions are indicated by circles, boxes or diamonds, respectively. Expected expression ratios and 95% confidence intervals were obtained by sampling 1000 random set of 55 genes and calculating their expression ratio. B. q-PCR analyses of DNA methylation, copy number and expression of ATCOPIA93 in wild type, ddm1 and epiRIL394, taken at F8 and more advanced generations. Data are mean ± s.d. (n = 2 independent biological experiments). C. Genome browser view of RNA-seq coverage over selected genes containing new ATCOPIA93 insertions. Exon-exon junctions detected by split-reads are represented by arcs that connect exons. Samples containing or lacking TE insertions are highlighted in red and blue, respectively. Expression levels for the gene ADR1 and PR-1 are shown on the upper right corner. D. RT–PCR analysis of genes containing new ATCOPIA93 insertions in epiRIL394. Primers are indicated by arrow-heads in B. E. Genome browser view of RNA seq data for a gene containing an ATCOPIA78 insertion in Col-0 in plants grown under control conditions (Ctl) or subjected to heat stress (HS). Percentage of splicing for the TE-containing intron (indicated by dashed lines) is shown on the right panel. Data are mean (n = 2 independent biological experiments). F. qRT-PCR analyses of AT1G11270 expression in response to heat stress (HS) in accessions containing (Col-0) or lacking (Bl-1) the intronic ATCOPIA78 insertion. Primers are indicated in E by arrow-heads. Data are mean ± s.d. (n = 2 independent biological experiments).
One ATCOPIA93 insertion present at F8 and F16 was located in the 5’UTR of gene AT1G33560, which encodes an NBS-LRR disease resistance protein, and had a much stronger dampening effect on gene expression at F16 than at F8 (Figure 6C). This result resembles that obtained for another gene in another epiRIL (Marí-Ordóñez et al. 2013). Moreover, reduction in AT1G33560 expression correlated with down-regulation of PR-1 (Figure 6C), a gene that is implicated in the response of plants to pathogens and whose expression depends on that of AT1G33560 (Collier et al., 2011). These findings indicate that epigenetic silencing of newly inserted ATCOPIA93 insertions can reinforce gene repression with potential phenotypic consequences.
Two ATCOPIA93-containing genes, AT4G17190 and AT5G20280, which are implicated respectively in defense against aphids and in nectar secretion (Lin et al. 2014; Bhatia et al. 2015), also showed reduced expression, but only at generation F8. In both cases, ATCOPIA93 was located within an intron and either in the same or the opposite orientation relative to gene transcription and was associated with transcript truncation at F8, but not at F16 (Figure 6C), when the insertions are epigenetically silent. Examination of DNA methylation, copy number and expression of ATCOPIA93 as well as expression of the two genes across several generations between F8 and F16 revealed that DNA methylation was established at F12 and coincided with the silencing of all ATCOPIA93 copies (Figure 6B) as well as the proper splicing of the insertion-containing intron (Figure 6D). These results demonstrate that the deleterious effects of intronic ATCOPIA93 insertions can be alleviated once they become epigenetically silent.
We last investigated a natural intronic ATCOPIA78 insertion that is present in the reference Col-0 genome but absent from other accessions (Figure S6). Analysis of publicly available RNA-seq data (Pietzenuk et al. 2016) indicated that the insertion is epigenetically reactivated by heat stress and that reactivation is associated with the production of a truncated transcript from the target gene (Figure 6E). To demonstrate causality, we compared the splicing level of the second intron of the target gene (AT1G11270) in Col-0 plants subjected or not to heat stress as well as in plants of the Bl-1 accession, which does not contain any ATCOPIA78 insertion within this gene. While the second intron was spliced in heat-stressed and control Bl-1 plants, this was not the case for Col-0 plants, which showed splicing only under non-stress conditions (Figure 6F). Given that ATCOPIA78 is among the most active TE families in nature and that its activity correlates with several geo-climatic variables (Quadrana et al. 2016), it is likely that numerous natural ATCOPIA78-containing alleles of genes are similarly endowed with the possibility to modulate their expression in response to environmental stress. Whether such alleles confer selective advantages remains to be determined.
Discussion
TEA lines: when mobile TEs are caught in the act
Whole genome sequencing of MA lines has been extremely valuable in determining the rate, spectrum and genome-wide distribution of spontaneous point and other small-size mutations in several model organisms (Ossowski et al. 2010; Zhu et al. 2014; Denver et al. 2009; Keightley et al. 2009). Results of these experiments indicated that small-size mutations occur almost randomly throughout the genome, although local modulations were observed, notably in relation to certain DNA sequences, recombination, transcription activity, and chromatin marks, including DNA methylation (Makova and Hardison 2015). However, MA lines did not provide much information about the heritable mutational landscape generated by TE mobilization, mainly because transposition is typically rare and also because non-reference TE insertions are difficult to detect using standard short-read sequencing strategies.
Based on a unique experimental system of A. thaliana epiRILs, in which transposition activity was kick-started but that otherwise resembled MA lines, we documented >8000 new TE insertions. These were produced mainly by three TEs, which belong to two DNA transposon (VANDAL21 and ATENSPM3) and one LTR-retrotransposon (ATCOPIA93) families that are among the most active in nature (Quadrana et al. 2016). Furthermore, our modeling indicated that the insertion mutations produced by each of these three TEs in the epiRILs likely accumulated following a transposition-genetic drift scenario. Thus, the epiRILs can be defined operationally as TEA lines and as such they provide a first comprehensive assessment of the actual rate and genome-wide distribution of heritable mutations generated by TE mobilization in any species.
Using the epiRILs and additional experimental populations, we showed that VANDAL21, ATENSPM3, ATCOPIA93 as well as another member (ATCOPIA78) of the Ty1/Copia superfamily of LTR-retrotransposons preferentially integrate within or close to three distinct sets of genes, each characterized by specific chromatin features. Moreover, we obtained strong evidence that ATENSPM3 and the two ATCOPIA families generate highly deleterious mutations that are immediately purged by natural selection.
Collectively, our findings provide a first comprehensive experimental demonstration that mutations generated by TEs have radically distinct properties than the spontaneous small size mutations documented in MA lines. First, because of strong TE-specific integration preferences linked to chromatin, TE-caused mutations are distributed non-uniformly across the genome, with the implication also that their repertoire may vary substantially for any given TE as a result of changes in chromatin states, such as the loss of heterochromatin. Second, because many TEs target chromatin states that are associated with genes, their insertion, as well as their excision in the case of DNA transposons, tend to have drastic effects on gene expression. This is however not an obligate outcome, as exemplified by the lack of any discernible consequences of VANDAL21 integration on gene transcription. Third, the functional impact of TE insertions can vary in a reversible manner over very short times, through their epigenetic silencing, which can be influenced by the environment, as in the case for ATCOPIA78. Last, TE insertions accumulate discontinuously, as a result of episodic, TE-specific reactivation, and at rates that may differ widely between TEs and environments.
Retrotransposition-driven allelic heterogeneity associated with adaptive traits
Although WGS has revealed that TEs are powerful engine of genome as well as organism evolution (Chuong, Elde, and Feschotte 2016), we still lack a clear understanding of the impact of their mobilization within any given species. Our finding that LTR-retrotransposons belonging to the Ty1/Copia superfamily preferentially integrate within environmentally responsive genes provide valuable information in this respect. As we have also shown previously that mobilization of diverse ATCOPIA elements is associated with climate (Quadrana et al. 2016), it is tempting to speculate that this superfamily of TEs facilitates adaptation to changing or local environments. Consistent with this hypothesis, disease resistance genes are characterized by a high load of COPIA insertions and extensive allelic heterogeneity in nature (Quadrana et al, 2016; Kawakatsu et al., 2016). Thus, recurrent retrotransposition within these genes may play a critical role in the rapid evolution and expansion of the innate immune system in plants. We have demonstrated however that most ATCOPIA insertions have severe deleterious effects, athough some of these effects may be mitigated or fully erased once the insertions are epigenetically silenced. This mitigation can be environmentally dependent, thus endowing TE-containing alleles with unique properties in fluctuating environments (Figure 7A).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
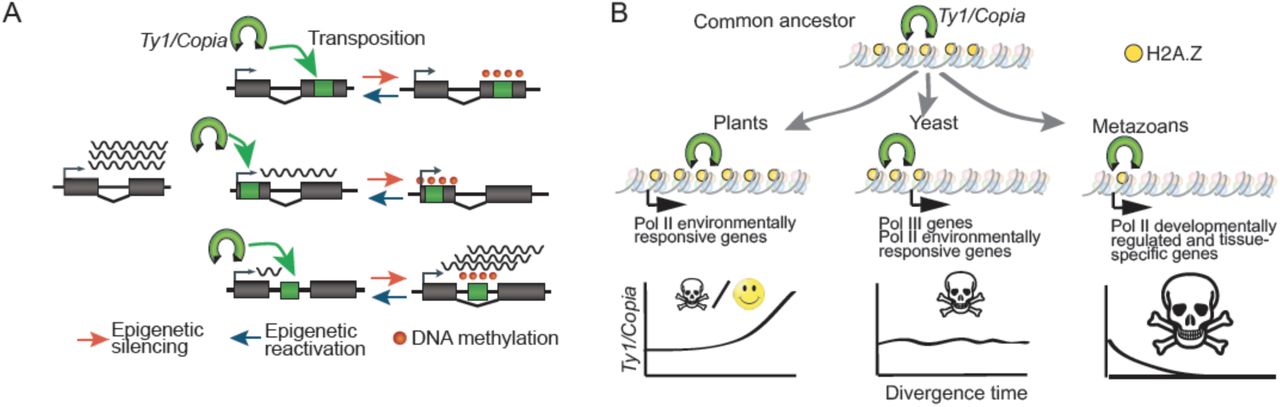
A. De novo retrotransposon insertions within genes impact their expression in multiple ways. While retrotransposition within internal exons systematically disrupt gene expression, insertions in 5’ regions or introns have an immediate dampening effect, which can be either aggravated or mitigated following epigenetic silencing of the inserted TE, respectively. Note that DNA methylation is depicted as spreading from the COPIA insertion into adjacent sequences, which remains to be determined experimentally. B. H2A.Z-guided integration of Ty1/Copia is ancestral. Functional diversification of H2A.Z between kingdoms determined the preferential integration of Ty1/Copia towards fast evolving genes in plants and yeast and towards developmentally regulated genes in animals. This differentiation may explain the high and low invasions success of Ty1/Copia retrotransposons in plants and animals, respectively.
H2A.Z and the different fate of Ty1/Copia retrotransposons in plants, yeast and animals
TEs need to keep moving in order to prevent their demise by the accumulation of inactivating mutations. However, because uncontrolled mobilization compromises host survival and that of TEs, different mechanisms have evolved that limit TE activity, notably epigenetic silencing, or that target integration to reduce the mutational load they generate (Sultana et al. 2017). For example, the LTR-retrotransposon Ty5 from yeast interacts with the heterochromatic factor silent information regulator 4 (Sir4), which directs Ty5 integration within gene-poor regions (Sultana et al. 2017). Here, we have uncovered another targeting mechanism based on the histone variant H2A.Z, which is among the most conserved histone H2As (Henikoff and Smith 2015). H2A.Z guides the integration of Ty1/Copia elements not only in A. thaliana but presumably also in rice and yeast. Furthermore, as in plants, the S. cerevisiae Ty1 retrotransposon integrates preferentially within the arrays of H2A.Z-containing nucleosomes that are located upstream of RNA polymerase III (Pol III)-transcribed genes (Figure 5F; Albert et al., 2007; Baller et al., 2012), aided by the Pol III subunit AC40 (Bridier-Nahmias et al. 2015). Disruption of the AC40-integrase interaction leads to a redistribution of Ty1 insertions towards subtelomeres (Bridier-Nahmias et al. 2015), which are enriched in environmentally responsive genes (Brown, Murray, and Verstrepen 2010). As in plants, these genes are enriched in H2A.Z-containing nucleosomes (Meneghini, Wu, and Madhani 2003; Sadeghi et al. 2011; Albert et al. 2007). These findings suggest therefore that AC40 serves to further restrict the integration of Ty1 to a subclass of genes containing arrays of H2A.Z, which otherwise would integrate preferentially within or around responsive genes.
In animals, H2A.Z is essential and mainly found at developmentally regulated genes, where it occupies nucleosomes that flank the promoter (Henikoff and Smith 2015). Given the conserved role of H2A.Z in guiding the integration of Ty1/Copia retrotransposons, their mobilization should therefore have catastrophic effects in animals, which may explain why this superfamily of retrotransposons is virtually absent from animal genomes (Huang, Burns, and Boeke 2012). In other words, functional diversification of H2A.Z between plants and animals may have directed the opposite fate of Ty1/Copia retrotransposons in these two kingdoms (Figure 7B). This in turn opens up the possibility that the evolutionary fate of other TEs could also be explained by similar chromatin-directed integration biases.
Author contributions
VC, PW and JMA conceived the project, with substantial additional input from LQ. ME and EC performed DNA and RNA extraction; JG, SE and JMA produced the WGS data; AG, ME, JMA and LQ performed the detection of TE insertions in the epiRILs; VB identified excision footprints in the epiRILs. LQ performed all of the other bioinformatic analyses, as well as the TE sequence capture and RT-QPCR experiments. LQ and AS performed the heat-stress experiments. LQ and VC interpreted the results. LQ and VC wrote the manuscript.
Declaration of interests
The authors declare no competing financial interest.
MATERIALS AND METHODS
Experimental model and subject details
The following A. thaliana plants were used: wild type Col-0 and Bl-1 accessions. The Col-0 ddm1-2 mutant and the epiRILs population (Johannes et al. 2009), the Col-0 nrpd1-3 mutant (H. Ito et al. 2011) and the Col-0 hta9-1 hta11-2 double mutant (March-Díaz et al. 2008). Unless stated otherwise, all plants were grown in long-days (16h:8h light:dark) at 23°C.
Genomic DNA sequencing using mate-pairs
Genomic sequencing was performed as described before (Gilly et al, 2014). Briefly, DNA was extracted from 10-20 seedlings grown under long day conditions, using DNeasy Qiagen kits. About 10 μg of genomic DNA was sonicated to a 4-6 kb size range using the E210 Covaris instrument (Covaris, Inc., USA). Libraries were prepared following Illumina’s protocol (Illumina Mate Pair library kit), starting with size-selected (approximately 5kb) fragments, which were end-repaired, biotin labeled and circularized. Linear DNA was eliminated by digestion and circularized DNA was fragmented to 300-700 bp using the E210 Covaris. Biotinylated DNA junctions were purified using streptavidin, end-repaired and 3’-adenylated in order to ligate Illumina adapters. Junction fragments were PCR-amplified using Illumina adapter-specific primers and amplified fragments within the 350-650 bp size range were selected for sequencing. Each library was sequenced using 100 base-length read chemistry in a paired-end flow cell on the Illumina GAIIx (2 lanes) or HiSeq2000 (1 lane) (Illumina, USA).
Mapping and detection of TE insertions using WGS
Reads were mapped with BWA v.0.6.1 using the parameters -R 10000 -l 35 -O 11, and the parameters n 10000 N 10000 -s for sampe, onto the TAIR10 reference sequence. Reads hanging over chromosome ends were removed using picard CleanSam and duplicate pairs were removed using picard MarkDuplicates. TE insertions were detected by implementing TE-Tracker software (available at http://www.genoscope.cns.fr/TE-Tracker) exactly as described before (Gilly et al., 2014). WGS did not produce sufficient coverage (<10X) for 16 of the 123 epiRILs analyzed at generation F8, and these 16 epiRILs were not considered further (Table S1). TE-Tracker is a computational method that we have previously developed for accurately detecting both the identity and destination of newly mobilized TEs in genomes re-sequenced using mate-pair libraries (Gilly et al, 2014). Importantly, TE-Tracker does not rely on prior annotation, yet is able to integrate it, making the results easily interpretable. Briefly, TE-Tracker uses paired reads mapping information to identify discordant reads that mapped partially over TE-sequences to detect the position of TE insertions. Insertion site positions were refined at the single nucleotide resolution by exploiting sequence information contained in split-reads. To this end, we implemented the software SPLITREADER (available at https://aithub.com/LeanQ/SPLITREADER: Quadrana et al., 2016). Homozygous and heterozygous insertions were defined based on the normalized number of reads supporting each insertion event (Table S2). In addition, this approach enabled us also to identify insertions that were likely present in only one of the 10-20 seedlings used to extract DNA and which reflect transposition during the reproductive phase of the parent. These insertions were also called heterozygous, as this was likely the case and to reflect their very recent ancestry. Conversely, our approach was designed to exclude poorly supported insertions, which could reflect either mapping artifacts or rare somatic events. Finally, visual inspection was carried out for a random sample of over 200 insertion events and their homozygous or heterozygous status was confirmed in each case.
TE-sequence capture
TE sequence capture was performed on exactly 1000 seedlings in all cases except for the F3 progeny of hta9 hta11 line 1, where only 477 seedlings were recovered (see main text and Figure 4A and 5B for details of the plant materials used). Seedlings were grown under control (long-day) conditions and genomic DNA was extracted using the CTAB method (Murray and Thompson 1980). In order to assess the sensitivity of TE-sequence capture, we added 1ng of genomic DNA extracted from epiRILs 394 (generation F16) to 1ug of genomic DNA extracted from the 477 F3 seedling of hta9 hta11 line 1 prior to library construction (i.e. 1:1000 dilution of the spiked-in genomic DNA). Libraries were prepared using 1μg of DNA and TruSeq paired-end kit (Illumina) following manufacturer instructions. Libraries were then amplified through 7 cycles of ligation-mediated PCR using the KAPA HiFi Hot Start Ready Mix and primers AATGATACGGCGACCACCGAGA and CAAGCAGAAGACGGCATACGAG at a final concentration of 2μM. 1μg of multiplexed libraries were then subjected to TE-sequence capture exactly as previously reported (Quadrana et al. 2016). Enrichment for captured TE sequences was confirmed by qPCR and estimated to be higher than 1000 fold. Pair-end sequencing was performed using one lane of Illumina NextSeq500 and 75bp reads. About 42 million pairs were sequenced per library and mapped to the TAIR10 reference genome using Bowtie2 v2.3.2 (Langmead and Salzberg 2012) with the arguments --mp 13 --rdg 8,5 --rfg 8,5 --very-sensitive. An improved version of SPLITREADER (available at https://github.com/LeanQ/SPLITREADER) was used to detect new TE insertions. Briefly, split-reads as well as discordant reads mapping partially on the reference sequence of ATCOPIA93 and ATCOPIA78 (obtained from RepBase update) were identified, soft clipped and remapped to the TAIR10 reference genome using Bowtie2 (Langmead and Salzberg 2012). Putative insertions supported by at least one split- and/or discordant-reads at each side of the insertion sites were retained. Insertions spanning centromeric repeats or coordinates spanning the corresponding donor TE sequence were excluded. In addition, putative TE insertions detected in more than one library were excluded to retain only sample-specific TE insertions (Table S3). More than 80% of new TE insertions present in epRIL394 F16 were detected, confirming that the sensitivity of our TE-sequence capture and computational approach is higher than 1:1000. In addition, no non-reference insertions were detected in 1000 F1 seedlings of wild type Col-0, highlighting the specificity of our approach.
Detection of Tos17 non reference insertions in rice genomes
A total of 16,784 Tos17 non-reference flanking sequences (Miyao et al. 2003) were retrieved from GeneBank and mapped to the reference rice genome using Minimap2 v2.11-r797 (H. Li 2018), which enabled us to identify 14,258 insertion points with high confidence (Table S4).
Detection of mutations within transposed copies
Discordant mate-pair reads mapping within a 6kb interval either upstream or downstream of each insertion site were extracted and re-mapped using Bowtie2 (Langmead and Salzberg 2012) over a library constructed with the specific donor TE sequence only. Sequence variants were detected using samtools mpileup V1.2.1 and only variants with a quality of at least 30 were kept. Long deletions were detected as regions without coverage and breakpoints were reconstructed by local assembly using Velvet V1.2.09 (Zerbino and Birney 2008).
Analysis of global and local enrichment of new TE insertions
To assess if new TE insertions are enriched in pericentromeric regions, their number within these regions was compared with that expected from a random distribution. The expected distribution was calculated by randomizing 104 times the position of new TE insertions across the chromosomes (genomic regions showing coverage deviation, the inner pericentromeres, or coordinates spanning the corresponding donor TE sequence were excluded). This set of random positions was used as a control for all subsequent analyses. Insertion distribution over wild-type- and ddm1-derived regions was obtained by counting the number of new TE insertions within intervals delimited by at least two consecutive stable hypermethylated or hypomethylated regions, respectively (Cortijo et al. 2014). Overrepresentation over genes and neighboring sequences was performed using a meta-gene analysis. Briefly, protein coding gene features were extracted from the TAIR10 annotation and coordinates of non-reference TE insertions with TSDs were crossed with the set of genic features according to the following stepwise hierarchy: 5’ UTR > 3’ UTR> exon > intron > intergenic regions. For insertions that do not overlap protein-coding genes, the distance to the closest gene was calculated and reported as negative or positive distance according to the gene orientation. Overrepresentation over chromatin states was performed by comparing the number of new TE insertions and randomly generated TE insertions located within each chromatin domain (Sequeira-Mendes et al. 2014). Density of ATCOPIA93 insertions were obtained by calculating the distance between insertion sites and the middle point of the nearest well positioned nucleosome mapped in Col-0 (Table S5; Lyons and Zilberman, 2017). Gene ontology (GO) analyses were performed using AGRIGO (http://bioinfo.cau.edu.cn/agriGO/) and as input the ID of genes that contain a TE insertion within the limits of their annotation.
Analysis of chromatin features at insertion sites
4kb regions centered around insertion sites were defined and used to extract normalized coverage of DnaseI hypersensitivity (Sullivan et al. 2014), Mnase accessibility (G. Li et al. 2014), H3K27me3 (C. Li et al. 2015), H2A.Z enrichment level (Coleman-Derr and Zilberman 2012) and well-positioned nucleosomes (Lyons and Zilberman 2017). The same approach was used for the analysis of H2A.Z enrichment in rice (Zahraeifard et al. 2018) and of htz1 from yeast (Gu et al. 2015). Average normalized coverage was then calculated for each bp and plotted using the smooth.spline function in R.
epiQTL mapping of transposition activity
Using TE copy number as phenotype and a total of 126 parental differentially methylated regions (DMRs) that segregate in a Mendelian fashion in the epiRILs (i.e. stable DMRs) as physical markers (Cortijo et al. 2014), we implemented the multiple QTL model (mqmsacn) from the R/qtl package. Genome-wide significance was determined empirically for each trait using 1000 permutations of the data. LOD significance thresholds were chosen to correspond to a genome-wide false positive rate of 5%.
Transposition-drift modeling of insertion accumulation
In the absence of selection, TE invasion is mostly determined by the rate of transposition and rate of fixation of insertions by random segregation (i.e. drift). Thus, we constructed individual-based transposition-drift models, all starting with an initial number of copies all equally active (with a rate K of new copies per generation) and with each new copy being also equally active. New copies arise in the heterozygous state and can be inherited following a Poisson distribution according to Mendelian segregation. The model also considers TE elimination by excision, which occurs at rate E per transposition event. Additionally, concerted silencing of all active copies may occur when copy number reach the threshold l. Simulations were run 1000 times using a wide space of parameter values (K={0,0.1,…,1}, E={0,0.2,…,1} and I={20,21,…,60}. Distribution of homozygous and heterozygous insertions between simulated and observed data at F8 was evaluated using a two-dimensional goodness-of-fit test.
Transcriptome analysis
RNA from wild type, ddm1, epiRIL55 (F8), epiRIL95 (F8), epiRIL260 (F8), epiRIL439 (F8), epiRIL454 (F8) epiRIL394 (F8) and epiRIL394 (F16) was isolated using Rneasy Plant Minikit (Qiagen) according to the supplier’s instructions. Contaminating DNA was removed using RQ1 DNase (Promega). One μg of total RNA was processed using TruSeq Stranded Total RNA kit (Illumina) according to the supplier’s instructions. About 20M 76nt-long single-end reads were obtained per sample on the Illumina HiSeq2000. Expression level was calculated by mapping reads using STAR v2.5.3a (Dobin et al. 2013) on the A. thaliana reference genome (TAIR10) with the following arguments --outFilterMultimapNmax 50 --outFilterMatchNmin 30 -- alignSJoverhangMin 3 --alignIntronMax 10000. Duplicated pairs were removed using picard MarkDuplicates. Counts were normalized and annotations were declared differentially expressed between samples using DESeq2 (Love, Huber, and Anders 2014). When specified, uniquely mapped reads (mapping quality > 10) were selected with samtools (H. Li et al. 2009). RNAseq data from heat-stressed plants were obtained from (Pietzenuk et al. 2016) and analyzed as described above. Splicing of intronic TE insertions was calculated as described previously (Teixeira et al. 2017). Briefly, the number of split-reads (SR) and non-split-reads (NSR, which should be fully and uniquely contained within the interval surrounding the same exon–intron junction) mapping to an exon-intron junction was extracted and the ratio SR/(SR+NSR)*100 was then calculated.
Quantification of expression level, copy number and DNA methylation
RNA was extracted using the RNeasy plant mini kit (Qiagen) from Col-0 or Bl-1 plants grown under normal conditions (10 days old seedlings grown in liquid medium) or subjected to heat shock treatment (H. Ito et al. 2011). RT-qPCR was performed as described previously (Quadrana et al. 2016). Primer sequences are provided in Table S6. RT-qPCR results (two biological replicates) are indicated relative to those obtained for a gene (AT5G13440) that shows invariant expression under multiple conditions. Copy number and DNA methylation of ATCOPIA93 was performed as described before (Marí-Ordóñez et al. 2013).
Data and software availability
Sequencing data has been deposited in the European Nucleotide Archive (ENA) under project XXXXX.
Acknowledgments
We thank members of the Colot group and especially Pierre Baduel for discussions and critical reading of the manuscript and S. Khouider for help with plant crosses. We thank the Genomics and Informatics facilities at IBENS for their help. Support was from the Agence National de la Recherche (ANR-09-BLAN-0237 to V.C. and P.W.), the Investissements d’Avenir ANR-10-LABX-54 MEMO LIFE, ANR-11-IDEX-0001-02 PSL* Research University (to V.C.), the European Union Seventh Framework Programme Network of Excellence EpiGeneSys (Award 257082, to V.C.) and the Centre National de la Recherche Scientifique (MOMENTUM program, to L.Q.). M.E. was supported by a PhD studentship from the French Ministry of Research and by a postdoctoral fellowship from MEMOLIFE. Initial support for L.Q. was provided by MEMOLIFE and EpiGeneSys. V.B. was supported by a PhD studentship from PSL Research University and from MEMOLIFE.
REFERENCES




