

Abstract
Systematic variation in the methylation of cytosines at CpG sites plays a critical role in early development of humans and other mammals. Of particular interest are regions of differential methylation between parental alleles, as these often dictate monoallelic gene expression, resulting in parent of origin specific control of the embryonic transcriptome and subsequent development, in a phenomenon known as genomic imprinting.
Using long-read nanopore sequencing we show that, with an average genomic coverage of approximately ten, it is possible to determine both the level of methylation of CpG sites and the haplotype from which each read arises. The long-read property is exploited to characterise, using novel methods, both methylation and haplotype for reads that have reduced basecalling precision compared to Sanger sequencing. We validate the analysis both through comparison of nanopore-derived methylation patterns with those from Reduced Representation Bisulfite Sequencing data and through comparison with previously reported data.
Our analysis successfully identifies known imprinting control regions as well as some novel differentially methylated regions which, due to their proximity to hitherto unknown monoallelically expressed genes, may represent new imprinting control regions.
Introduction
Methylation of the 5th carbon of cytosines (5mC or simply mC) is an epigenetic modification essential for normal mammalian development. Methylation differences between alleles contribute to establishing allele-specific expression patterns. As obtaining genome-wide haplotyped methylomes with short reads remains challenging, we evaluated the ability of long read, nanopore-based sequencing to improve allele-specific methylation analyses.
We apply the technique to the study of genomic imprinting, where differential expression of the maternal and paternal alleles in the offspring is at least partially set by the differential methylation (1–5). Imprinting is proposed to arise from the diverging interests of the maternal and paternal genes (6). In accordance with its primordial role in allocation of resources from the mother to the offspring, the placenta, along with the brain, is the organ where parental conflict results in the most pronounced imprinted expression (7–9). We thus conduct a survey of differential methylation and expression in murine embryonic placenta.
Recent studies have increased the number of genes identified as subject to imprinting in mouse to about 200 (10–15). The cause of the differential expression between paternal and maternal alleles is only known for a subset of these genes; maternal histone marks can play a role (14), and in other cases it involves the differential methylation of adjacent regions (5). The differential methylation patterns may be established in the gametes and persist through the epigenetic reprogramming occurring after fertilisation (16). These differentially methylated regions (DMRs) are called primary DMRs, or imprinting control regions (ICRs). Alternatively, differential methylation may arise during development, perhaps as a downstream effect of differential expression, in which case the regions are called somatic or secondary DMRs (17).
Apart from the parent of origin of the allele, genetic differences can also be associated with differential methylation. In this case, F1 hybrids of distinct mouse strains will display DMRs between the alleles according to the strain of origin (18), and not the parent. Genetically determined DMRs can have profound effects on phenotype, for instance in humans by altering the expression of mismatch repair genes important in cancer (19). Therefore, we also investigate the link between DNA methylation and expression for strain-biased genes.
Reconstructing haplotyped methylomes necessitates the simultaneous measurement of DNA methylation and singlenucleotide polymorphisms (SNPs) differentiating the alleles. This can be achieved by deep sequencing of bisulfite-converted DNA on the Illumina platforms, although the short reads combined with the reduced complexity of the bisulfite-treated DNA make the process inefficient, meaning many regions with low SNP density remain unresolved. Long reads provided by third generation sequencing technologies can overcome the requirement of a high SNP density, while several methods allow the assessment of base modifications on native DNA (thus also avoiding the reduction in complexity associated with bisulfite conversion). These methods include: analysis of polymerase kinetics for PacBio SMRT sequencing (20), and detection of deviations in the electric signal for Oxford Nanopore sequencing, via Nanopolish (21), signalAlign (22), mCaller (23), Tombo (24), or DeepSignal (25). We note that, for the dominant eukaryotic genome base modification at 5mC, the PacBio technology requires very high coverage making it impractical for use in the analysis of mammalian genomes (26).
Here we use the MinION and PromethION long-read nanopore sequencers to generate whole-genome haplotyped methylomes from murine embryonic placenta. With a mean coverage of 10X we successfully identify known imprinting control regions as well as novel parent-of-origin DMRs near imprinted genes, as well as strain-specific DMRs close to both strain-biased genes and structural variants. We show the improved efficiency of this strategy over existing workflows to resolve allele-specific methylation, and highlight its utility in investigating the mechanisms of genomic imprinting.
Results
Nanopore methylation calls are concordant with other technologies
We sequenced the embryonic portion of placenta derived from a male embryonic day 14.5 (E14.5) conceptus from a C57BL/6 (Black6, or B6) × Castaneus (Cast) F1 on the MinION platform to a depth of about 8X (Fig. S1) and called methylation using Nanopolish (21). The genome-wide methylation data successfully recapitulated known patterns: CpG islands (CGIs), as defined by Irizarry et al. (27), separated into two groups of high and low methylation (Fig. 1A); the methylation level dipped at transcriptional start sites (TSSs) (Fig. 1B), and the average genome-wide methylation level was around 50%, as previously reported for placental tissue (28).

A. Metaplot of nanopore methylation calls across CpG islands, clustered in two groups of high and low methylation. B. Metaplot of nanopore methylation calls across the aggregated gene bodies of all protein-coding genes recapitulated known methylation structures. C. Density of methylation calls (β, the average methylation based on all reads covering that position) for sites covered by both nanopore and RRBS. D. Joint density of nanopore and RRBS methylation calls for the same sites as in C. Darker regions indicate regions of higher density, while lighter regions indicate regions of lower density. The density plot is split into four quadrants according to a RRBS threshold of 0.5 and a nanopore threshold of 0.36, and the percentage of sites in each quadrant is displayed.
To further validate the accuracy of the nanopore methylation calls, we compared them to Reduced Representation Bisulfite Sequencing (RRBS) data on the same sample at sites covered by both methods. Nanopore methylation calls showed an overall similar distribution to RRBS methylation calls, albeit with a bias toward intermediate values of methylation (Fig. 1C). Per-site methylation also correlated well between the two methods, with a linear model giving an R-squared of 0.56 (Fig. 1D).
Sequencing of E14.5 embryonic placenta from the reciprocal cross (Cast × B6) on the PromethION platform at 12X coverage (Fig. S1) generated comparable results.
Increased haplotyping efficiency with nanopore reads
We next used high-confidence SNPs between the Cast and B6 strains to haplotype RRBS and nanopore reads. In order to mitigate the high sequencing error rates of nanopore sequencing, we used two methods of haplotype assignment, denoted the ‘basecall method’, based on FASTQ data, and the ‘signal method’, based on the phase-reads module from nanopolish, a HMM which uses the raw nanopore signal to predict genotype (29). Additionally, we only assigned a haplotype to those nanopore reads with at least five high-confidence SNPs (Fig. S3). All RRBS reads overlapping at least one SNP were assigned a haplotype (30). Only 24% of the mapped RRBS reads could be assigned to one haplotype, whereas 74% of mapped nanopore reads were haplotyped in the expected proportions (Fig. 2A and B): roughly half of the haplotyped reads were assigned to the maternal haplotype, and half to the paternal haplotype, albeit showing a slight bias towards the paternal haplotype (due to an increased number of split reads in regions where sections of the Cast genome has a deletion with respect to the B6 genome). The pattern of haplotype assignment was consistent across the autosomal chromosomes, while, as expected for a male sample, almost all (91%) of the reads aligned to the X chromosome were assigned to the maternal haplotype (Fig. 2B). Haplotyping of the Cast × B6 cross gave similar results (Fig. S2A). The lack of maternal bias in read haplotype indicates minimal maternal contamination, which is also reflected in consistent RNA-seq library sizes (Fig. S4).

A. Percentages of mapped reads from RRBS and nanopore sequencing that were assigned to the B6 genome (maternal), Cast genome (paternal), or that could not be haplotyped (filtered) for the B6 × Cast F1 sample. B. Percentages of mapped reads from nanopore sequencing that were assigned to each haplotype on each chromosome. C. Scatter plot of haplotype scores for nanopore reads according to signal (x-axis) and basecall (y-axis) methods. Only 10,000 randomly selected reads are shown for ease of visualisation. D. Signal and basecall haplotype scores for reads from the sequencing of the pure parental Cast strain.
We further evaluated the accuracy of the haplotyping of the nanopore reads by sequencing the same tissue from the parents (B6 only, and Cast only). Following the same haplotyping procedure, 85.7% of the reads were correctly assigned to the relevant genotype, and 1.5% were misassigned (Fig. S2B and C).
The large majority of nanopore reads showed strong agreement between the two haplotyping methods, though a small number showed confident assignment from the basecall method where the signal method gave an ambiguous assignment (Fig. 2C). This discrepancy was even more obvious in the haplotyping of the parental strains (Fig. 2D). This may be attributed to recent advances in nanopore basecalling beyond the Hidden Markov Model used in nanopolish (21). However, when examining the overall predictive performance of the two methods with AUROC (Area Under Receiver Operating Characteristic Curve) on the single-strain experiments, the signal method (0.988) outperformed the basecall method (0.976). We also found that the combination of the two methods further enhanced performance, with a logistic regression model (31) trained on the two scores achieving an AUROC of 0.992, with a maximum accuracy of 97% excluding filtered reads. The manual combination of methods (see Methods) further enhanced the total accuracy on classified reads to 98% and allowed classification of 30,000 additional reads over the logistic regression, which required both scores to be defined. In both nanopore and RRBS data, the main cause of haplotyping failure is the lack of SNPs in the region covered by the read (Fig. S3). While the proportion of successfully haplotyped nanopore reads could increase with optimisation for longer reads and anticipated improved sequencing accuracy, RRBS haplotyping efficiency is limited by the short read length.
Parent-of-origin and strain-biased gene expression
To investigate the correlation between differential methylation and differential gene expression, we performed RNA-seq on the same F1 placental tissue from reciprocal crosses of B6 and Cast, in quadruplicates. Maternal tissue contamination was unlikely as for each embryo maternal and paternal counts were similar (Fig. S4). We found 135 genes with a parental bias in expression (imprinted genes, 10% FDR, Fig. 3A): 88 with higher expression from the maternal allele and 47 with higher expression of the paternal allele. Among the 135 genes, 53 corresponded to well-characterised imprinted genes in classic databases (32–35). A further 17 of these genes, including Fkbp6, Smoc1/2, Gzmc/d/e/f/g, Zdhhc14 and Arid1b have been identified as imprinted in one or several recent studies (12–15). The remaining 65 genes constitute novel candidate genes with parent-biased expression in mouse placenta. The complete annotated list is reported in Additional File 1.
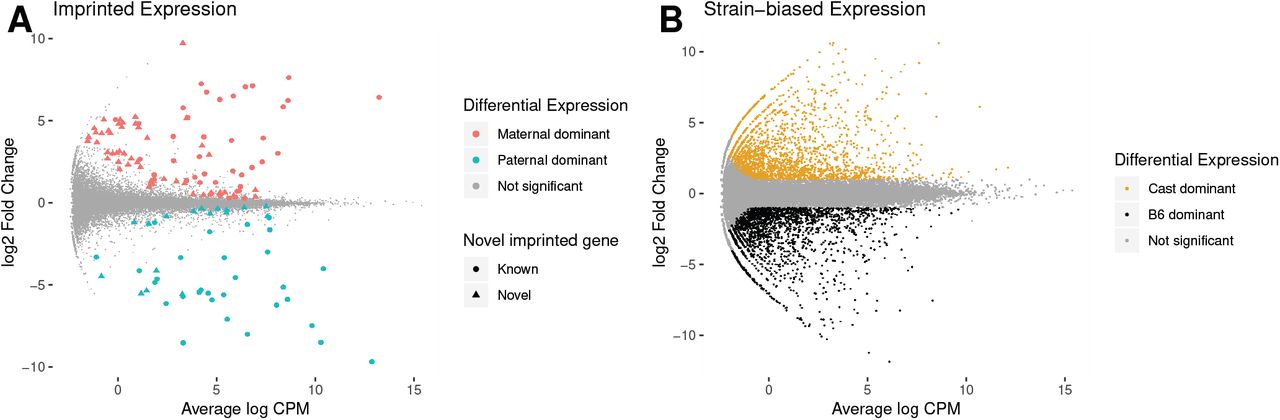
A. Differential expression between the maternal and paternal alleles. Genes with adjusted p-value < 0.1 are coloured in red when maternal expression dominates (positive log-fold change) and blue when paternal expression is greater (negative log-fold change). The shape of the point indicates whether the differentially expressed gene has previously been reported as imprinted. B. Differential expression between B6 and Cast alleles. Genes with adjusted p-value < 0.05 and log2 fold-change > 1 are coloured in black when B6 expression is higher and orange when Cast expression is higher. Interactive plots are available at http://bioinf.wehi.edu.au/haplotyped_methylome.
We also identified 4,029 genes (13% of expressed autosomal genes) with a strain bias greater than two-fold (5% FDR, Fig. 3B and Additional File 1), evenly split between B6 dominance (2,027 genes) and Cast dominance (2,002).
Known imprinting control regions are observable by nanopore sequencing
We combined the methylation and haplotyping data from the nanopore reads to compare methylation between the parental alleles. To highlight the linkage information of the methylation data available for nanopore reads, as well as the per-site per-read data, we plotted the loess fit of the cytosine methylation levels for each read in the region of interest (Fig. 4A and B).

A. Allelic methylation plot of maternally imprinted gene Impact displays a clear DMR at its imprinting control region (ICR). Haplotyped RRBS data shows concordance with nanopore allelic methylation. Allele-specific RNA-seq coverage plots show monoallelic paternal expression. CGI: CpG Islands are displayed in black, with CpG shores in dark grey and CpG Shelves in light grey. Nanopore: Vertical bars at the base of the B6Cast track denote CpG sites used for methylation calling, while ‘+’ signs at the base of the CastB6 track denote SNPs used for haplotyping. Highlighted red regions indicate DMRs detected by DSS. The maternal allele is shown in red and the paternal allele in cyan for all plots. B. Allelic methylation plot as in A. for the reciprocally imprinted genes Nespas and Gnas. RNA-seq gives very low expression and is not shown. C. Heatmap of differences (maternal − paternal) in allelic methylation in relative-width bins along known ICRs. Regions are sorted in order of average methylation difference, with regions in the same imprinting cluster placed adjacent to each other. Regions without haplotyped calls for both alleles are shown in gray.
Differentially methylated regions at known imprinting control regions (34) were readily visible and concordant with matched allele-specific RNA-seq and RRBS data (Fig. 4A and B). Nanopore data recapitulated methylation differences at most known imprinting control regions (Fig. 4C), often showing extended differential methylation past the annotated ICR borders.
Nanopore sequencing reveals novel differentially methylated regions
Next, we sought to define DMRs between parental alleles as well as between strains de novo, using the differential methylation tool DSS (36). We ranked putative DMRs based on the area statistic. Using the DSS default threshold of 10−5, we obtained a total of 933 DMRs, of which 309 were explained by parent-of-origin differences, and the remainder by strain-specific effects (Additional File 2). We then examined these DMRs, in conjunction with haplotyped RRBS and RNA sequencing data for corroborating evidence of differential methylation and differential expression respectively, in order to find putative DMRs of interest at imprinted genes.
Of the 20 highest ranking DMRs, 15 corresponded to known ICRs. Although many of the lower-ranking DMRs are potential false-positives, they also included regions of known imprinted expression (for instance two small detected DMRs immediately adjacent to known DMRs at the IMPACT and NESP ICRs, shown in Figures 4A and B respectively.) Thus in the absence of statistically robust DMR-finding methods for nanopore data we kept this permissive threshold.
Five ICRs annotated in the WAMIDEX database (34) were not detected de novo (Table 1). INPP5F_V2 and GRB10 simply lacked coverage in the B6 × Cast sample but showed clear differential allelic methylation in the Cast × B6 sample; GNAS-EXON1A also lacked coverage in B6 × Cast but the reciprocal sample and the RRBS did not suggest differential methylation, while NDN lacked coverage in both samples. The last undetected region was GPR1-ZBDF2; however Duffié et al. have shown that this region lacks important features of a bona fide ICR, and that a neighbouring maternally hyper-methylated region is the true ICR (17). The region in question was readily detected as differentially methylated from the nanopore data (DMR #229, Table 1).
List of known and proposed DMRs associated with imprinted genes. A check mark is shown where RRBS and RNA-seq evidence supports the DMR. The DMR rank is shown when supporting nanopore data exists.
In addition to ICRs, we detected numerous DMRs at imprinted genes that do not appear to be present in gametes (37–39), and which therefore likely constitute secondary DMRs (Table 1). When RRBS coverage was present, the bisulfite data corroborated the de novo DMR identification. Five of the secondary DMRs have been described previously, although they are not currently compiled in a database: maternal hypermethylation at the Igf2 promoter (40), maternal hypermethylation at the placental-specific promoter of Gab1 (41), paternal hypermethylation of the Meg3 transcriptional start site (TSS) (42), maternal hypermethylation at the Slc38a4 TSS (43), and paternal hypermethylation at the Igf2r promoter (2).
The remaining secondary DMRs have not been previously characterised. Six of them overlapped the transcriptional start sites of imprinted genes: Sfmbt2, Jade1, Ascl2, Cd81/R74862, Tssc4, and AC158554.1.
Other novel secondary DMRs overlapped introns rather than transcriptional start sites. Park2, a recently identified maternally-biased gene (13), had seven intronic DMRs, all displaying hypermethylation of the maternal allele. Rian displayed a DMR that had not been previously reported in mice, although its human ortholog also presents an intronic imprinted DMR (44).
In some cases, inspection of the parent-specific DMRs revealed unannotated imprinted transcription nearby, for example in the Kcnq1 and Igf2r clusters (Table 1). These RNA-seq reads may be part of imprinted long non-coding RNAs, frequent at imprinted clusters.
While we have collated all the imprinted DMRs that we found to directly overlap with imprinted expression -in addition to the WAMIDEX ICRs- in Table 1, we note that other imprinted DMRs may be associated with the imprinted expression of more distant genes, or with genes that are only expressed or imprinted in specific tissues. For example we found imprinted DMRs in the promoters of Smoc2 (DMR #224) and Arid1b (DMR #863), two genes recently identified as being imprinted (and also imprinted in our data). The strong DMR #110 overlapped the transcriptional start site of Gtsf2, which was poorly expressed in placenta but may be imprinted in the tissues where it is expressed (in gonocytes and spermatids (45)). In the absence of chromatin conformation data or functional validation, we did not attempt to formally assign these DMRs to specific genes.
We however used the top, most reliable 400 DMRs to calculate the distance of genes to their nearest DMR depending on their expression status. We found that parentally biased genes were more likely to be proximal to parent-of-origin DMRs than unbiased genes (median distance 0.9 Mbp compared to 7.4 Mbp), whereas strain-biased genes and DMRs did not show this relationship (median distance 2.9 Mbp compared to 3.1 Mbp) The distributions of distances to the nearest DMR are shown in Fig. 5, which shows a striking relationship between parentally biased genes and parent-of-origin DMRs. This result is consistent with parental bias in expression being driven necessarily by epigenetic differences, whereas differential expression between strains is mainly driven by genetic differences.

A. Distribution of distance from genes to imprinted DMRs, shown on a log-scale. Inset shows distances from 0 to 10,000bp on a linear scale. Imprinted genes are much more frequently located within 100-100,000 bp of an imprinted DMR. B. Distribution of distance from genes to strain-specific DMRs. Strain-biased genes are for the most part located no closer to a strain-specific DMR than non-differentially expressed genes, indicating the strain-specific differential expression is likely caused by other factors, such as genomic differences. In both cases, we use only DMRs ranked in the top 400.
Long reads provide advantages in differential methylation analysis
Inspection of the DMRs revealed multiple advantages of our nanopore-based method of methylome haplotyping over traditional bisulfite sequencing (Fig. 6).

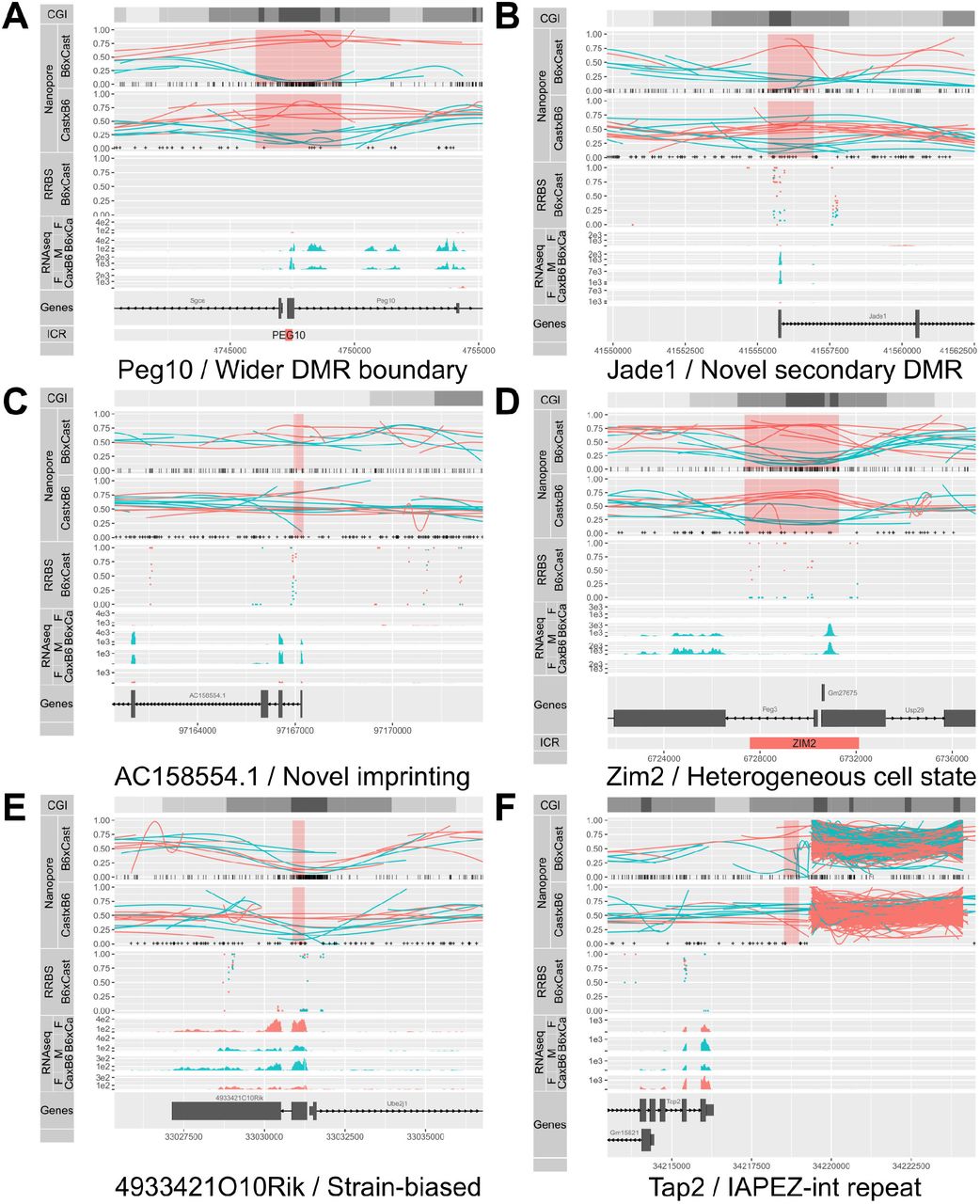
A. Allelic methylation (as in Figure 4) plot of maternally imprinted gene Peg10 displayed a clear DMR at its ICR which was much wider than the previously annotated DMR (bottom). B. Previously uncharacterised secondary DMR at the TSS of maternally imprinted gene Jade1. C. Novel maternally imprinted gene AC158554.1, with imprinted methylation at its TSS. D. Allelic methylation plot of maternally imprinted gene Peg3 showed consistently high methylation across some maternal reads, and consistently low methylation across others, a conclusion that could not be drawn from the middling bisulfite methylation values. E. Strain-of-origin DMR associated with the strain-biased expression of 493342110Rik. F. DMR associated with the omission of a IAPEZ repeat from the Cast genome, suggesting that the methylation in the flanking region was affected by the presence or absence of the repeat.
We were able to resolve DMRs in regions of low SNP density, where there were no haplotyped RRBS reads despite the presence of a CpG island (Fig. 6A). The particular DMR in Fig. 6A encompassed the transcriptional start site of the imprinted gene Peg10, and was much wider than the previously annotated ICR. The increased DMR width was a regular occurrence at ICRs (Table 1).
Our method also uncovered novel secondary DMRs at known imprinted genes such as Jade1 (Fig. 6B), as well as at previously uncharacterised imprinted transcripts such as AC158554.1, annotated as a lincRNA (ENS-MUSG00000116295, Fig. 6C).
Another advantage provided by the long reads was apparent at the ZIM2-PEG3 ICR (Fig. 6D). Intermediate methylation values for the maternal allele in the RRBS data (from the B6Cast F1) suggested that either certain CG dinucleotides were variable in their methylation across otherwise similar DNA molecules, or that there existed two classes of DNA molecules: some highly methylated throughout, and others with consistently low methylation. The long nanopore reads revealed that it was the latter: some of the maternal alleles show a contiguous loss of methylation. Although this result is well known to those who have practiced Sanger bisulfite sequencing, the haplotyped methylome derived from nanopore sequencing allowed investigation of this variability more accurately (no PCR bias) and across the whole genome.
Eight strain-specific DMRs were also found within 5 kb of a gene with strain-biased expression (Fig. 6E). Most of these exhibit structural variation proximal to the DMR, although a small number exhibited changes in expression seemingly not associated with any structural variant.
One example of a structural variant between B6 and Cast associated with differential methylation can be found on Fig. 6F. Upstream of the Tap2 gene, an IAPEZ retrotransposon in the B6 genome is absent from the Cast genome (Cast reads are truncated upstream of the repeat and absent downstream), and the gene-proximal border is more highly methylated on B6 alleles than on Cast alleles. This differential methylation would be consistent with the insertion of the transposon attracting methylation that spreads to adjacent regions. We could also see at this repeat region a lot of spuriously mapping reads from both strains, suggesting that the repeat is present in multiple copies that the current assembly fails to account for.
Discussion
Determining allele-specific methylation patterns in diploid or polyploid cells with short-read sequencing is hampered by the dependence on a high SNP density and the reduction in sequence complexity inherent to bisulfite treatment. In this study we demonstrate the use of long-read nanopore sequencing to derive haplotyped methylomes of the embryonic portion of mouse placentae. Methylation estimates from nanopore reads are consistent with previous knowledge (Fig. 1). The longer read lengths allowed most reads to overlap multiple SNPs, resulting in accurate haplotyping of 75% of the reads, a much higher proportion than comparable short-read data (Fig. 2). Sequencing of native DNA not only maintains the sequence complexity that is lost in bisulfite treatment, but also has the potential to detect a variety of base modifications outside 5mC, bypassing the need for specialised chemistries such as bisulfite (for 5mC) or oxidative-bisulfite (for 5hmC) treatments. Furthermore, we are able to characterise allele-specific methylation at a relatively shallow level of genomic coverage (~10X), which is substantially lower than the coverage required by Pacific Biosciences single-molecule sequencing to ascertain any native base modification (25X) or 5mC in particular (250X) (26).
Recent increases in throughput of nanopore sequencing instruments make this approach a cost-effective way of obtaining genome-wide allele-specific methylation for mammaliansized genomes, compared to the alternatives of short-read whole-genome bisulfite sequencing, or PacBio SMRT sequencing. Thus the approach we present is unique in its ability to characterise allele-specific single-molecule cytosine methylation state in eukaryotes, in which the 5mC modification is both common and highly relevant to transcriptional regulation.
The haplotyped methylomes for reciprocal B6 × Cast F1 samples confirm the parent-of-origin specific methylation of ICRs and provide an improved definition of their boundaries (Fig. 4). By integrating the haplotyped methylomes with allele-specific expression data, we identified novel DMRs linked to imprinted genes. These are likely to constitute secondary DMRs, whose role and origin are unclear. We confirm a large number (70) of previously identified imprinted genes and propose another 65 as new candidates (Fig. 3 and Additional File 1). This suggests that although the monoallelically expressed genes are now well characterised, sensitive analyses can still uncover parentally biased genes. Interestingly, though we find more maternal-dominant genes than paternal-dominant ones (88 and 47, respectively), the imbalance is much less pronounced than in Finn et al. (2014) (12) (96% maternal dominance). Applying long-read sequencing to the transcriptome also promises improvements in the percentage of usable data, the detection of allele-specific as well as isoform-specific differential expression, and even the detection of RNA base modifications.
Our allele-specific methylation and expression data can also be used to reveal strain-biased expression of genes linked to strain-specific DMRs. The genetic divergence between the two strains accounts for most of the differences in expression, however the presence of DMRs could suggest an epigenetic component to the regulation of a subset of genes.
We foresee a number of improvements that will make the determination of haplotyped methylomes by nanopore sequencing more efficient and comprehensive in the future. Firstly, we expect to see an expansion in the types and nucleotide contexts of base modifications characterised. Our analysis is based on Simpson et al. (2017) (21), and is limited to 5mC at CpG sites. However that is not a limitation of the technology, as has been demonstrated by others (22–24). Secondly, improvements can be made in reaching true nucleotide-resolution methylation calls. Where multiple CpG sites occur within less than twice the K-mer length (here 6) all these sites are considered to have the same methylation state. Again, this is not a limitation of the technology, as more complete training data will allow resolution of mixed methylation states. Finally, we see opportunities for improvement in the analysis of nanopore methylation data. Instead of binary calls from bisulfite sequencing, the output of nanopore sequencing is a likelihood ratio that the site is methylated vs. unmethylated. Currently there is no method for the detection of differential methylation that accepts these continuous values as input. Additionally, the DMR detection algorithm that we used was designed originally for bisulfite data, and we expect that algorithms designed specifically to incorporate long reads and probabilistic methylation assignment would achieve greater levels of accuracy.
Conclusions
We demonstrate that long-read sequencing using nanopore technology can efficiently generate haplotyped mammalian methylomes. With no additional sample preparation than that routinely used for basic sequencing and with only a mean coverage of about 10X, we identify differential allelic methylation throughout the genome. Combined with expression data, this improves the resolution of imprinting analyses. Our approach is widely applicable to other systems, for instance with more complex genetics, or to phase cancer mutations with methylation state, and to determine the effects of structural variation on methylation.
Methods
Animal Strains and Husbandry
All mice were maintained and treated in accordance with Walter and Eliza Hall Institute Animal Ethics Committee approved protocols under approval number WEHI AEC 2014.026. Mus musculus castaneus mice were obtained from Jackson Labs. Note that due to prior inter-crossing for transgene transmission the female Mus musculus domesticus C57BL/6 mice that served as dams for our study comprise 12.5% FVB/NJ genome, however for simplicity we will refer to this mouse as B6. Wild-type B6 were reciprocally mated to wild type Mus musculus castaneus (Cast).
DNA and RNA Extraction
Pregnant females were sacrificed at E14.5 by CO2 asphyxiation and the embryonic portion of each placenta was dissected from the maternal portion in PBS, as we have done previously (46). Samples were snap frozen in buffer RLT plus (Qiagen) and DNA and RNA were later extracted from the same sample using the AllPrep DNA/RNA Mini Kit (Qiagen), according to manufacturer’s instructions. Samples were sexed by PCR using primers for Otc (X-linked gene) and Zfy (Y-linked gene) as previously described (47), and male samples were selected for further analysis.
Illumina Sequencing
RRBS libraries were made from 100 ng of DNA purified from the embryonic layer of a male B6 × Cast E14.5 placenta using the Ovation RRBS Methyl-Seq System (NuGEN), according to the manufacturer's recommendations, which include use of the Qiagen Epitect kit for bisulfite conversion. The resultant library was sequenced on a HiSeq 2500 (Illumina) using 100 bp paired end reads and analysed as previously described (47, 48).
RNA-seq libraries were prepared from 1 μg of RNA from four B6 × Cast and four Cast × B6 samples, including the same sample as the RRBS library, using the TruSeq RNA sample preparation kit (Illumina). 75-bp paired-end sequencing was performed on a NextSeq 500 (Illumina). Reads were trimmed with Trim Galore v0.4.2 and mapped with HISAT2 v2.0.5 (49) with option --no-softclip to the GRCm38 (mm10) mouse genome with N-masked castaneus SNPs. Mapped reads were haplotyped with SNPsplit v0.3.2 (30), and gene counts obtained by running featureCounts (50) on the GRCm38_v90 Ensembl annotation. Differential analysis was performed with edgeR (51, 52) using quasi-likelihood fits (53) and controlling the false discovery rate (FDR) at 10% (54).
Nanopore Sequencing
The B6 × Cast F1 sample was sequenced on three MinION flow cells with the 1D Sequencing Genomic Ligation (LSK108) protocol from ONT with minor adjustments: 4 μg of starting material were used for each library preparation, and for two libraries DNA was sheared to 10 kb with a Covaris G-Tube, whereas no shearing was done for the third library (resulting in longer read lengths). Reads were basecalled with Albacore 1.2.2. The Cast × B6 F1 was sequenced on one PromethION flow cell with the 1D Sequencing Genomic Ligation (LSK109) protocol without shearing, and basecalled with Albacore 2.2.7. Nanopore reads were aligned to the same SNP-masked genome as before, using BWA-MEM (55).
Haplotyping
Haplotyping is achieved through the identification of SNPs that are unique to one or the other allele. Examining only the SNPs identified as passing all filters in Keane et al. (56), we combine two distinct methods to confidently haplotype each read.
Basecall Haplotyping
Where a read is aligned to a SNP position i on the reference genome, we assign a score Si if the aligned base agrees with the reference haplotype, or 1 − Si if the aligned base agrees with the alternate haplotype, where the score depends on the basecalling quality score qi of the base in question as
 where the coefficients of the above relationship were determined empirically on successfully haplotyped reads. Bases which match neither haplotype, or which exhibit a deletion at the SNP location are excluded from the analysis. Finally, the read is assigned an aggregate haplotype value h ∈ [0,1] across the n informative SNP calls as follows:
where the coefficients of the above relationship were determined empirically on successfully haplotyped reads. Bases which match neither haplotype, or which exhibit a deletion at the SNP location are excluded from the analysis. Finally, the read is assigned an aggregate haplotype value h ∈ [0,1] across the n informative SNP calls as follows:

Signal-level Haplotyping
For signal-level haplotyping, we use the HMM of Simpson et al., implemented in Nanopolish phase-reads (29). Briefly, the raw signal corresponding to the section of the read aligned to the reference at the SNP position is realigned using a Hidden Markov Model, and the likelihood of the sequence of 6-mers in this vicinity is maximised by choosing the more likely of the two possible alleles. Each read is then assigned scores according to the same rule as in Basecall Haplotyping, where Nanopolish quality scores are offset by −35 in order to exhibit a similar relationship to basecall quality scores.
Combining Haplotype Calls
For each read with nbase and nsignal associated SNP calls and associated haplotype values hbase and hsignal, we define the haplotype calls

The calls are then combined according to the following rules, applied in order:
 where H = 1 represents a read assigned to the reference haplotype, H = −1 represents a read assigned to the alternate haplotype, and H = 0 represents an unassigned read. This process is shown graphically as a flowchart in Fig. S3.
where H = 1 represents a read assigned to the reference haplotype, H = −1 represents a read assigned to the alternate haplotype, and H = 0 represents an unassigned read. This process is shown graphically as a flowchart in Fig. S3.
Resolution of Maternal Recombination
Owing to the cross of an FVB-strain into the maternal line in the grand-parental generation, it is necessary to resolve which section of the maternal genome was contributed by recombination from the FVB chromosome. We run the above haplotyping procedure with three possible outcomes, rather than two: mm10, FVB, and CAST, with variants called by Keane et al. (56). The proportion of maternal (non-CAST) reads within any 100Kb region was fitted to a recursive partition tree, which splits continuous data into a stepwise function, here representing the proportion of a contiguous section of chromosome haplotyped to FVB (Fig. S5). Fitting was performed using the R package rpart with parameters minsplit=5 and cp=0.1 (57). SNPs in sections of the chromosome with mean proportion of FVB greater than 50% were replaced with the FVB allele for further analysis.
Methylation Calling
We determined the methylation status of each CpG site on each read using Nanopolish call-methylation (21). Briefly, Nanopolish uses a 5-base alphabet, with 5-methylcytosine represented as M, to build a Gaussian mixture model representing every possible 6-mer with both methylated and unmethylated cytosine in a CpG context, excluding those 6-mers which contain both the methylated and unmethylated base. We ran Nanopolish separately on reads haplotyped to the maternal and paternal chromosome, using a SNP-masked version of each chromosome to decrease bias in reads with expected deviations from the mm10 reference.
Nanopolish then assigns each section or “event” of nanopore current to a base on the reference genome and calculates the likelihood of each 6-mer containing the CpG site being either methylated (Lm (dij)) or unmethylated (LC (dij)) given the data dij for a call group i covered by a read j. Groups of consecutive CpG sites in which the distance between any two adjacent sites is less than 11 bases (therefore having overlap between 6-mers containing the cytosines in question) are chained into CpG call groups. All sites within the one CpG call group are assumed to have the same methylation status, such that each 6-mer is only considered once. We convert these likelihoods to probabilities as follows:

By Bayes’ Law,
 and since M and C are mutually exclusive and jointly exhaustive,
and since M and C are mutually exclusive and jointly exhaustive,

Then, defining the prior probability of methylation as P(M) = p0,
 and rearranging for P(M|dij),
and rearranging for P(M|dij),

Noting results from Decato et al. (28) showing methylation levels ranging from 0.433 to 0.538 for mouse placental tissue we set p0 = 0.5, so finally we define the single-read, single-site probability of methylation as

Comparison with RRBS Methylation Calls
Individual methylation calls on a single CpG call group are aggregated over the set of reads covering each group in order to compare with aggregate values provided by bisulfite sequencing. That is, for each CpG call group i covered by n reads, we define the call group average

In order to compare methylation calls between nanopore and reduced representation bisulfite sequencing, we must split CpG call groups defined by Nanopolish as CpG sites separated by less than 11 base pairs into individual sites, including GpC sites on the reverse strand, with each site retaining the same β value as the original call group. Only those CpG sites for which both RRBS and nanopore data exist are considered.
Identification of Differentially Methylated Regions
Following methylation detection and haplotype assignment of each read, it is possible to assign each call of methylation on the genome to one of the two haplotypes. The aggregated β methylation values for each CpG group are tested for DMRs using DSS (36). Briefly, DSS tests for differentially methylation at a single CpG-sites, using a Wald test on the coefficients of a beta-binomial regression of count data with an ‘arcsine’ link function. Then, using a default p-value threshold of 10−5, DSS aggregates differentially methylated sites into DMRs based on a maximum separation between sites and a minimum density and number of sites in each DMR.
To detect parent-of-origin DMRs, we perform DSS with the comparison B6♀ and Cast♀ vs. Cast♂ and B6♂; to detect strain-specific DMRs, we perform DSS a second time with the comparison B6♀ and B6♂ vs. Cast♂ and Cast♀.
Visualisation of Haplotyped Methylation
Owing to the noisy nature of nanopore methylation calls, we use a loess smoothing curve to visually represent the methylation of a single nanopore read (58). Here the smoothing parameter α is determined by
 where L is read length. This relationship was determined empirically to have minimal impact on visualisation while minimising computation time.
where L is read length. This relationship was determined empirically to have minimal impact on visualisation while minimising computation time.
Data availability
All sequencing data are available at ENA under study accession ERP109201. Processed data can be explored via a Genome browser and summary page available from bioinf.wehi.edu.au/haplotyped=methylome. All analysis scripts are available on Github at github.com/scottgigante/haplotyped-methylome.
AUTHOR CONTRIBUTIONS
Tissue collection was performed by TB and QG. ONT sequencing was performed by AL (with supervision from MT and LG) and QG. RRBS was performed by AK and TB, and RNA-seq by QG. Alignment, methylation calling and haplotyping of nanopore data was performed by SG. AK analysed the RRBS and QG the RNA-seq. Development of visualisation and DMR testing was performed by SG, CW and TPS. Figures were generated by SG, QG and AL. SG, QG, AK, TPS, MEB and MER interpreted the data. SG, QG, CW, MEB and MER wrote the manuscript. All authors read and approved the final version.
COMPETING FINANCIAL INTERESTS
The authors declare that they have no competing interests.
ACKNOWLEDGEMENTS
We would like to thank Jared Simpson for his help running Nanopolish and Stephen Wilcox for the Illumina sequencing. MEB was supported by Bellberry-Viertel Senior Medical Research Fellowship. MER was supported byanAustralian National Health and Medical Research Council (NHMRC) fellowship (GNT1104924). This work was made possible through NHMRC Project grants GNT1098290 and GNT1140976 (to MEB and MER), Victorian State Government Operational Infrastructure Support and NHMRC Research Institute Infrastructure Support Scheme.
List of abbreviations
- AUROC
- Area Under Receiver Operating Characteristic curve
- CpG
- 5’-C-phosphate-G-3’
- CGI
- CpG island
- DMR
- differentially methylated region
- DML
- differentially methylated locus
- F1
- first filial generation
- FDR
- false discovery rate
- ICR
- imprinting control region
- HMM
- Hidden Markov Model
- ONT
- Oxford Nanopore Technologies
- PB
- Pacific Biosciences
- RRBS
- reduced representation bisulfite sequencing
- SNP
- single nucleotide polymorphism
- TSS
- transcriptional start site





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}