

Abstract
Commensal microbes in the gut do not act alone but instead as cooperative consortia to conduct their myriad functions. Cooperative interactions and feedback mechanisms are key to consortia performance, yet are often ignored in current synthetic biology efforts to engineer the microbiota. To this end, we engineered mutual metabolic dependencies between four heterogeneous gut-dwelling bacterial species. Each species was made auxotrophic for three amino acids and an overproducer for one amino acid to share with the other species. By performing dynamical systems inference from time-series measurements, we show that our engineering introduced positive interactions that either reversed or neutralized pre-existing competitive interactions and improved stability of the consortium. We further demonstrate that we can induce population balance in the engineered consortia, both in vitro and in the mouse gut, through nutrient and dietary manipulations. Our findings indicate that induced cooperation can introduce evenness and stability in a synthetic microbial ecosystem, and have implications for development of synthetic approaches to manipulate the microbiome.
Introduction
In nature, microbes occur as conglomerates of various species with diverse sets of genomes and metabolic capabilities, allowing for division of labor and increased robustness (Hays et al., 2015a). For example, microbial consortia have been shown to withstand external perturbations such as invasion of other species, toxic compounds and nutrient sparseness (Burmølle et al., 2006; Lapara et al., 2002). A major driver for microbial consortia robustness is cooperative behavior through production of public goods and metabolic cross-feeding (Cavaliere et al., 2017). Consortia robustness is correlated with population balance among the microbes and an extensive network of interactions between species (Stelling et al., 2004; Stenuit and Agathos, 2015).
Metagenomic analyses reveal nutrient auxotrophies as a prevalent feature of microbial communities, suggesting that cross-feeding might be a common mode of interaction in natural consortia (Mee et al., 2014; Pande et al., 2014). Examples of such natural microbial consortia include metabolically interacting communities in soil (Venail and Vives, 2013) and in the mammalian gut (Rakoff-Nahoum et al., 2016). Distributing metabolic capabilities over multiple species, a form of functional complementarity, can increase productivity of the consortium through more efficient resource utilization ((Pande et al., 2014; Savage et al., 2007).
Amino acid cross-feeding is an attractive means to introduce cooperation into synthetic microbial consortia. These metabolites are more readily secreted than others, e.g., E. coli secretes certain amino acids upon starvation (Burkovski et al., 1995; Kaderbhai et al., 2003; Valle et al., 2008). Indeed, amino acids have been shown to play important roles in inter-species communication in natural systems (McCutcheon and Von Dohlen, 2011). For instance, amino acids are used by S. cerevisiae to regulate nitrogen overflow, which leads to natural cross-feeding to lactic acid bacteria (Ponomarova et al., 2017). Numerous studies have engineered pairwise amino acid cross-feeding in E. coli producing normal amino acid levels and generated quantitative models to describe their behavior (Estrela and Gudelj, 2010; Kerner et al., 2012; Pande et al., 2014; Stolyar et al., 2007; Wintermute and Silver, 2010a).
Engineering cooperative microbial consortia has been of longstanding interest in synthetic biology; studies were performed to gain basic scientific insights, or as engineering proofs-of-principles (Mee et al., 2014; Wintermute and Silver, 2010b). With recent advances in our understanding of the human microbiota, there is increasing interest in applying synthetic biology approaches to construct a well-defined gut microbiome, living bacterial diagnostics and therapeutics (Riglar and Silver, 2018). Creating such diagnostics and treatments in the context of cooperative consortia has numerous potential advantages, including the aforementioned capability of consortia to carry out more complex functions in a more stable manner (Cavaliere et al., 2017), with stability in this context defined as the ability to withstand external disturbances. Additionally, consortia can potentially allow for greater safety and control. For instance, a consortium that maintains population balance through cooperativity could be used to control dosing of a therapeutic compound in the gut.
However, almost all prior synthetic biology studies that have engineered cooperativity in bacterial communities have used a single species (Kong et al., 2018; Mee et al., 2014; Wintermute and Silver, 2010b). However, natural microbial ecosystems contain a diversity of interacting species. In advancing synthetic biology to real applications in complex environments, it will be essential to expand engineering capabilities to diverse, multi-species consortia. Importantly, bacteria from naturally occurring ecosystems are likely to have pre-existing interactions, which are often competitive. These interactions must be considered and often overcome to achieve cooperative consortia.
As a step toward developing multi-species consortia, we have constructed a synthetic consortium of four different bacterial species, each derived from the mammalian gut, and engineered mutual interactions by cross-feeding of four amino acids. Using several experimental approaches, combined with statistical inference from data and computational modeling, we demonstrate our ability to engineer cooperativity in the consortium that overcomes pre-existing competitive interactions. Further, we show that this cooperativity is inducible through nutrient or dietary manipulations, and that the engineered consortium exhibits population balance that is stable when subjected to perturbations in vitro and when introduced into the mammalian gut.
Results
Cooperative Consortia Design and Engineering
To gain intuition into synthetic consortia designs, we simulated behavior of non-interacting collections of four bacterial species versus consortia linked by positive/cooperative interactions (Figure 1A). Our simulations demonstrate that while a collection of non-interacting bacteria can exhibit population balance, it is highly susceptible to external disturbances that can drastically change the composition of the community. In particular, disturbances can readily cause a species to die out in the community. However, when we linked bacterial species through cooperative interactions, the resulting consortia can withstand much higher levels of external disturbances without dramatically altering its composition. Thus, our simulations suggest that engineering a network of positive interactions within a bacterial consortium could introduce stability towards environmental disturbances.

(A) Simulations show increased stability in consortia with positive interactions. In a non-interacting consortia, disturbances can lead to dramatic changes in consortia composition; in this example, the disturbance causes one of the strains to die out. Introducing positive interactions among the consortia strains increases stability in the presence of disturbances. (B) Our engineering design, which introduces mutual positive interactions by cross-feeding metabolites. Each strain was knocked out for three amino acid biosynthesis pathways and mutated to overproduce one amino acid. Thus, four amino acids L-methionine (Met), L-histidine (His), L- tryptophan (Trp) and L-arginine (Arg) are cross-fed between the four strains.
To construct our synthetic consortium, we selected four bacterial species, Escherichia coli NGF-1, Salmonella enterica subsp enterica serovar Typhimurium LT2, Bacteroides thetaiotaomicron VPI-5482, and Bacteroides fragilis 638R. These species are not only genetically tractable, but also able to survive in the mammalian gut in diverse niches and have varied abundances within the total microbiota. These characteristics allow us to investigate key synthetic biology engineering principles in a controlled, but more realistic context, and also maximize potential for downstream applications, for example for bacterial therapeutics and diagnostics in the gut.
E. coli NGF-1 was isolated from BALB/c mice, has been shown to stably colonize the mouse gut, and can be engineered with standard genetic tools (Kotula et al., 2014; Riglar et al., 2017). S. Typhimurium LT2 was further attenuated by removing the pathogenicity islands SPI1 and SPI2, and thus did not cause any disease phenotype when administered to mice. The two Bacteroides species, Bacteroides thetaiotaomicron and Bacteroides fragilis, are human commensals that can achieve high abundance in the mammalian gut, and are also genetically tractable.
We engineered each of the constituent species to depend on the other three by crossfeeding of the four metabolites L-methionine, L-histidine, L-tryptophan and L-arginine (hereafter referred to as Met, His, Trp and Arg) (Figure 1B). Auxotrophies for three of these amino acids were generated in each strain (E. coli: His, Trp and Arg; S.Typhimurium: Met, Trp, Arg; B. theta: Met, His, Arg; B.fragilis: Met, His, Trp), along with the ability to overproduce one amino acid in each strain (E. coli: Met; S. Typhimurium: His; B. theta: Trp; B. fragilis: Arg) (Table 1). E. coli and S. Typhimurium were engineered by sequential phage transduction from three single auxotroph strains. E. coli was transduced with genome fragments from BW25113 that contained insertions in argA, trpC, hisA (see Methods) and S. Typhimurium with genome fragments of the same parent strain with insertions in argA, trpC, metA. Bacteroides spp. triple knockout generation utilized the pExchange-tdk vector to precisely delete metA, hisG and argF in B. theta and metA, hisG and trpC in B. fragilis. To engineer overproduction of amino acids, we selected for bacterial strains that showed resistance to specific antimetabolites.
a)prevents feedback inhibition (Veeravalli et al., 2014), b)decouples from histidine feedback inhibition (Malykh et al., 2018); c)trpE, removes feedback inhibition (Fang et al., 2015); d)arginine repressor, nonfunctional (Ginesy et al., 2015)
Characterization of Auxotrophies and Overproduction
To assess the auxotrophic strains’ amino acid requirements, we measured growth on varying concentrations of each metabolite in the presence of non-limiting concentrations of all the other metabolites (Figure 2A). Each strain had a requirement for specific and differing levels of the amino acids. Overproduction of metabolites was measured in comparison to a defined amino acid standard using LC-MS (Figure 2A, horizontal bars). In order to compare overproduction with each species’ amino acid requirements, we fit a sigmoidal curve to the growth response data (Figure 2A), to produce an expected concentration (OD600) for the species for a given overproduction rate. This comparison of requirements and overproduction levels provides information about the expected relative strengths of the engineered interactions.
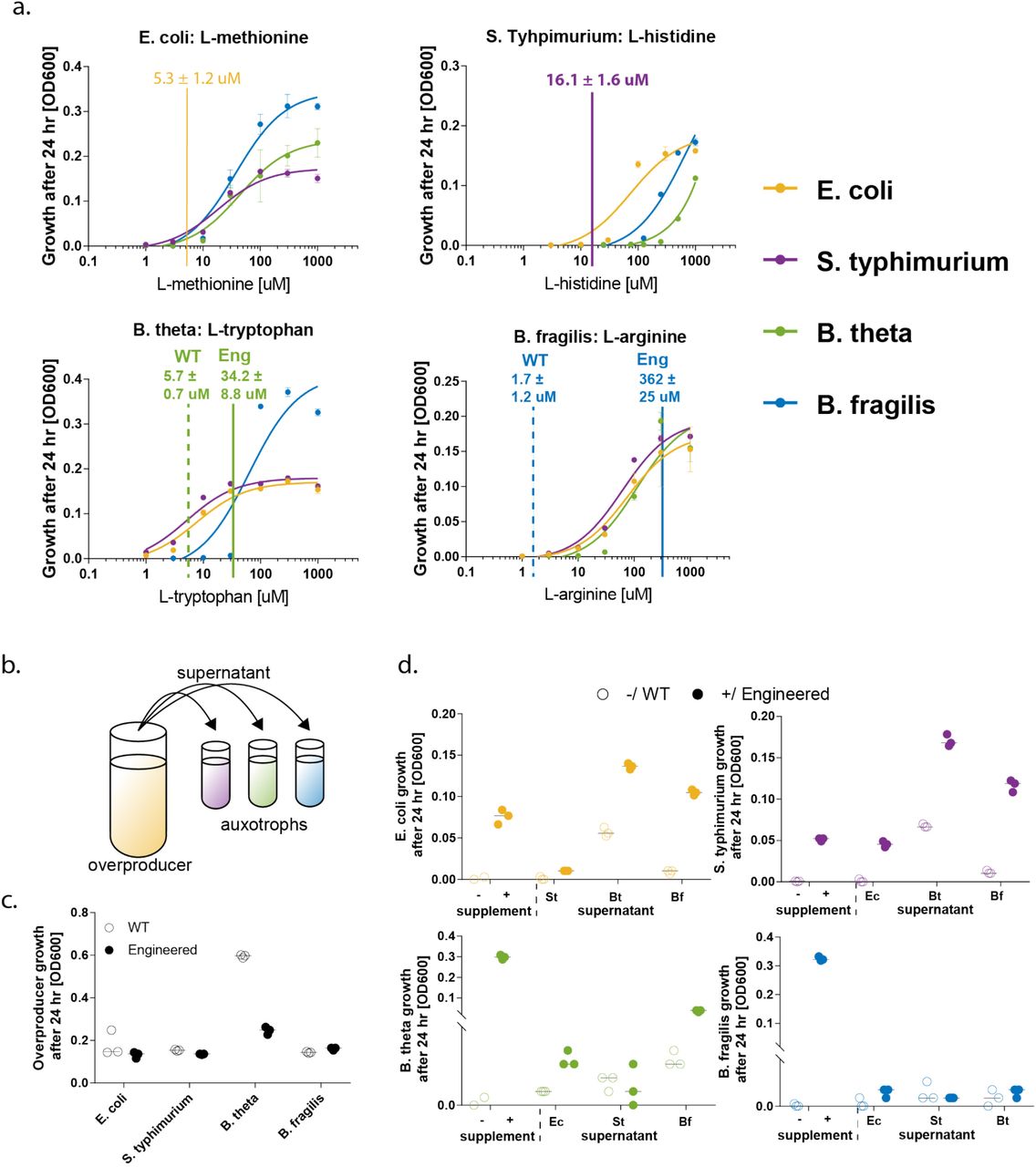
a. Growth response with indicated overproduction. Each auxotroph was grown in media supplemented with varying concentrations of one amino acid and saturating concentration of the two others. Depicted is the average of three biological replicates; error bars indicate standard deviation. A sigmoidal curve was fit using GraphPad Prism 7. Overproduction (horizontal lines) falls largely within requirement values. b. Cross-feeding capabilities of each strain were assessed by testing for rescue of auxotrophs in supernatants obtained from overproducers. c. Growth of overproducers after 24 hr, at which point supernatant was collected for cross-feeding experiment. Overproduction does not affect growth with the exception of B. theta (3-fold reduction). Shown are three biological replicates with median indicated as horizontal line. d. Growth of auxotrophs with and without amino acid supplementation and in supernatant of engineered overproducers and WT equivalents. Shown are three biological replicates with median indicated as horizontal lines. Except for B. fragilis, all auxotrophs can be rescued to varying degrees.
B. fragilis was the highest overproducer (Arg at 362 uM). Corresponding supplementation would allow growth of E. coli to OD 0.144, S. Typhimurium to OD 0.166 and B. theta to OD 0.154. B. theta overproduced Trp at 34 uM, allowing expected growth for E. coli to OD 0.411, S. Typhimurium to OD 0.156 and B. fragilis to OD 0.128. E. coli overproduced Met at 5.3 uM, allowing expected growth of S. Typhimurium to OD 0.032, B. theta to 0.017 and B. fragilis to OD 0.027. S. Typhimurium overproduces His at 16 uM, allowing for expected growth of E. coli to OD 0.028, but not supporting Bacteroides spp. growth, which required concentrations higher than 100 uM. Interestingly, the unengineered Bacteroides spp. also produced detectable amounts of some amino acids, whereas the other wild-type species did not. In the case of B. theta, the detected levels of Trp would, in principle, be high enough to support growth of other consortium members. Overall, our findings suggest relatively strong engineered cooperation from Bacteroides spp to other strains, moderate cooperation from E. coli to other strains, and the weakest cooperation from S. Typhimurium to other strains.
Amino Acid Overproducers can Rescue Growth Defects of Corresponding Auxotrophs
Having established amino acid requirements and overproduction levels for each strain, we assessed pairwise cross-feeding using culture supernatants from the overproducing and wild-type strains to test for growth of the corresponding auxotrophs (Figure 2b). Cells were grown for 24 hr before supernatant was collected (Figure 2c). Notably, three out of the four engineered species did not show any growth defect compared to the WT; B. theta growth was decreased by 3-fold. Extent of rescue was determined by OD600 values after 24 hr of growth in supernatant that was diluted 1:1 with fresh media lacking the tested amino acid (Figure 2d). As another comparator, we grew the auxotrophic strains without amino acid supplementation and with full supplementation (1 mM of each amino acid). As expected, we detected no growth in any of the auxotrophs when no amino acid was supplied, and growth with full supplementation.
Consistent with our design, and our amino acid requirement and overproduction data, E. coli grew well in supernatant from engineered B. theta (180% of fully supplemented growth) and B. fragilis (130%), and somewhat in supernatant from engineered S. Typhimurium (13%). Interestingly, E. coli grew better in Bacteroides spp. supernatant than in fully supplemented media, suggesting that these Bacteroides spp. may produce other beneficial metabolites for E. coli. S. Typhimurium grew relatively well in E. coli supernatant (88%), and also showed enhanced growth in the Bacteroides spp. supernatants (330% in B. theta supernatant, and 227% in B. fragilis supernatant). As expected from our requirement and overproduction data, B. theta is only marginally rescued by engineered E. coli (3%). Overall, these experiments indicate cross-feeding between the engineered bacterial strains.
However, in some cases the supernatants did not perform as well as predicted or in comparison to co-cultures. B. fragilis rescued B. theta growth much less than expected (12%) from our overproduction data. This finding suggests that B. fragilis may secrete factors that inhibit B. theta growth, but not E. coli or S. Typhimurium. Indeed, competitive interactions among more closely related species have previously been reported, possibly due to competition for similar niches (Bauer et al., 2018). No growth by B. theta in S. Typhimurium supernatant was evident. B. fragilis was not rescued well by any of the strains (Figure 2d, blue panel). Taken together, these results may reflect production of toxic compounds that are enriched in supernatants of grown cultures but might play a lesser role in co-cultures.
Cooperation and Population Balance of the Consortium in vitro is Inducible Based on Amino Acid Abundance
Having investigated pairwise interactions in our consortium, we next sought to characterize properties of the entire consortium versus individual members. Using a medium that we specifically designed to accommodate the four bacterial species in a single batch culture, and without amino acid supplementation, we grew monocultures and co-cultures of WT and the engineered consortia and estimated bacterial abundance (cfu/mL) via qPCR after 24 hr (Figure 3A,B).
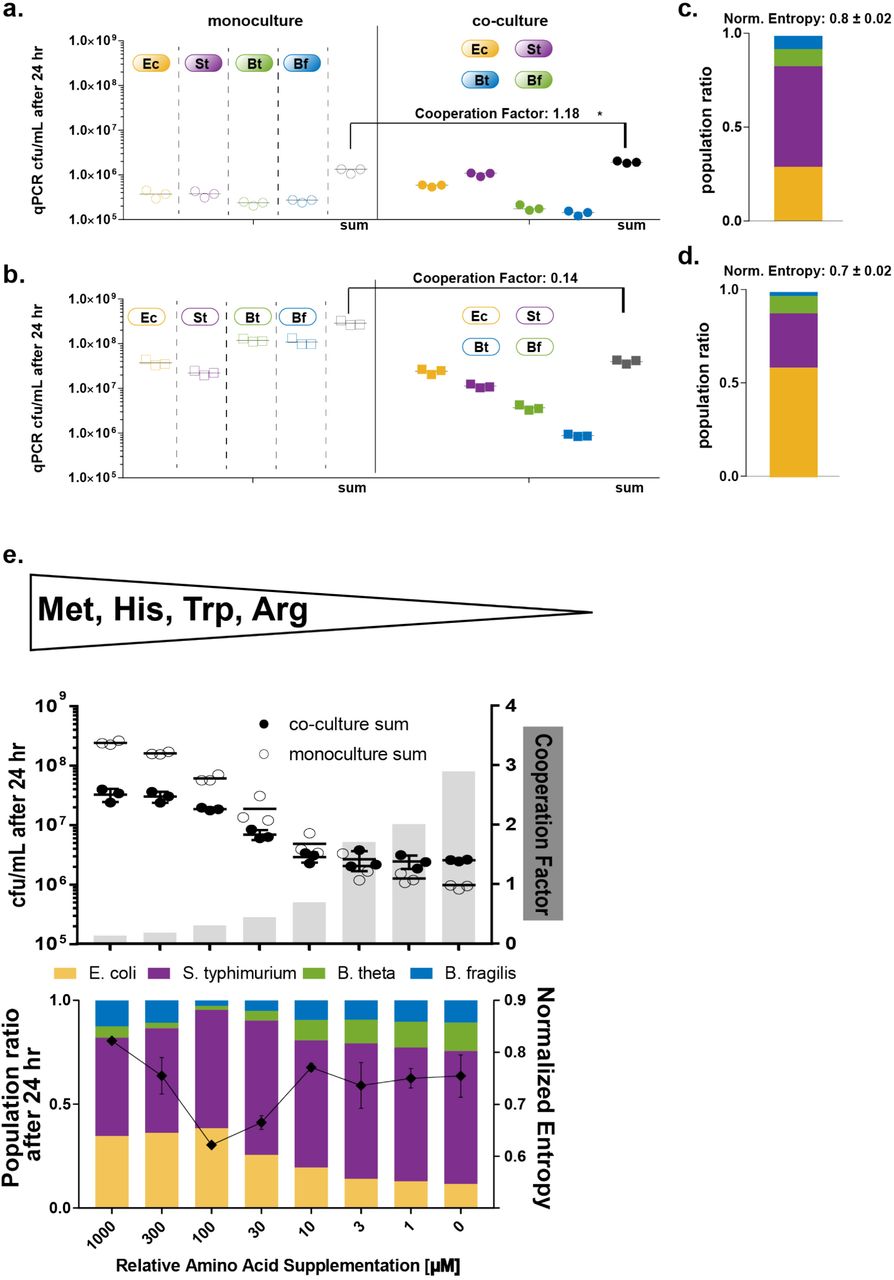
a./b. Growth of engineered (a.) and WT (b.) mono cultures and co-culture grown without supplementation was recorded after 24 hr as qPCR cfu/mL estimates. Shown are three replicates with median indicated. For engineered strains co-culture reaches higher cfu/mL than sum of monocultures quantified by cooperation factor. ∗sum in co-culture in engineered strains is significantly larger than sum in monocultures (Mann-Whitney test; p-value: 0.0071) c./d. Population ratios and calculated balance factor for engineered and WT co-cultures. Balance for engineered strains is higher than for WT strains. e. Supplementation titration experiment. Engineered consortia co-culture and mono cultures were subjected to a range of amino acid supplementation and was analyzed after 24 hr via qPCR. Upper panel: sum of monocultures growth (empty circles) and co-culture growth (filled circles) and calculated cooperation factor (grey bars). Shown are three replicas with median indicated as black line. Cooperation increases with decreasing supplementation. Lower Panel: Population ratios of cocultures as function of supplementation. Shown is the mean of three replicas. At high supplementation, evenness is high, then drops at intermediate levels and rises again at lower levels. Cooperation and evenness co-vary.
By comparing growth of strains in monoculture to the full consortium co-culture, we can assess the degree of cooperativity in the bacterial community. We quantify this behavior using a cooperation factor, defined as the total concentration of the co-culture divided by the sum of the concentrations of the monocultures. According to this definition, a cooperation factor <1 indicates competitive behavior, whereas a cooperation factor of >1 indicates cooperation. For the WT consortium, each of the strains grew better in monoculture than in co-culture (Figure 3B), with a cooperation factor of 0.14. This finding suggests pre-existing negative interactions among the WT species, e.g., competition for nutrients or production of compounds toxic to the other species. For the engineered consortia, the cooperation factor is 1.18, indicating that our engineering strategy has led to a net growth improvement when the complete consortium is able to interact. Note that much of the growth improvement is due to E. coli and S. Typhimurium growth, whereas B. theta and B. fragilis growth is essentially unchanged. This suggests that our engineering introduced net positive interactions for E. coli and S. Typhimurium, while neutralizing competitive effects on B. theta and B. fragilis present in the WT consortium.
Since our engineering design is based on amino-acid cross-feeding, we hypothesized that by varying the concentrations of amino acids in the medium, we could control the degree of cooperativity among the consortium members. To test this hypothesis, we subjected the engineered bacteria in monoculture and co-culture to different concentrations of relative amino acid supplementation (Figure 3C). We measured bacterial abundance (cfu/mL) via qPCR after 24 hr and calculated cooperation factors for each condition. Overall, bacterial growth decreases for both monoculture and coculture conditions with decreasing supplementation, as expected. Interestingly, we were effectively able to ablate cooperativity with high amino acid supplementation; in this regime, the engineered consortium behaves like the WT consortium, with monoculture growth exceeding co-culture growth. As supplementation decreased, we found that the cooperativity factor consistently increased, with the factor exceeding 1 at a supplementation level of 3 uM. These findings are consistent with cross-feeding behavior in naturally occurring microbial consortia, in which cooperativity only occurs during nutrient scarcity (Carlson et al., 2018).
Our simulation studies showed that our engineering design could result in a consortium with a balanced population less susceptible to environmental changes. Thus, we were interested in how varying amino acid supplementation would affect population balance. To assess this, we measured relative abundances of each species in the engineered consortium (Figure 3C, lower panel) via strain-specific qPCR. We quantitated population balance using the normalized entropy measure (also called the evenness index) for the consortium. With this measure, a completely even (balanced) community would have a relative entropy of one.
We found that the highest population evenness occurred with the highest supplementation (1000 uM.) This is likely because all the required amino acids are supplied, which reduces competition that would lower population evenness. When supplementation is decreased to intermediate levels (30 and 100 uM), the relative abundances of bacteria become less even, i.e. entropy decreases. Specifically, Bacteroides spp. abundance decreases dramatically, and E. coli and S. Typhimurium dominate the culture. This level of supplementation represents a “mismatched” regime, in which amino acid concentrations are high enough to support some of the species (S. Typhimurium and E. coli, which have the lowest amino acid requirements), but not the others (the Bacteroides spp., which have higher requirements.) When amino acid supplementation is reduced further (particularly below 20 uM), the ecosystem enters a low nutrient regime characterized by increased cooperativity as described above, and population evenness increases again, almost to levels seen with the highest levels of supplementation.
The Engineered Consortium Exhibits Greater Stability
Our amino acid supplementation experiments demonstrated that a microbial consortium with low cooperativity can exhibit high population evenness, but can also be less stable. One measure of stability is the extent to which a system tends to reach the same endpoint or steady-state, even if it starts in a different initial state. Indeed, this behavior is a necessary condition for common microbial dynamical systems models to exhibit asymptotic stability (Gibson et al., 2017). To assess the stability of our consortium according to this criteria, we inoculated both WT and engineered consortia at five different starting concentrations each (Figure 4). In condition 1, all bacterial species were inoculated at the same ratios, and in conditions 2-5, we reduced one of the species’ inocula by a factor of 10. We then assessed growth of each strain in co-culture over 12 hours (Figure 4A, B) via strain-specfic qPCR. Both consortia reached consistent total concentrations at 12 hrs (approximately 5 × 107 cfu/mL, 1 × 106 cfu/mL for engineered.) However, the end-point abundances of the WT consortium members differed markedly, depending on the starting condition. In particular, for conditions 2-4, the low inoculum species remained low at 12 hrs. In contrast, the engineered consortium exhibited significantly greater consistency (p-value 0.0355) in end-point concentrations of the consortium members, regardless of the starting condition. These results demonstrate increased stability, a dynamical systems property, of the engineered consortium.

a. We inoculated WT and engineered consortia in media without amino acid supplementation and followed growth over time. Starting inocula varied to mimic external perturbations. Each strain was inoculated at the same ratio (condition 1) and then each one dropped down by 1:10 (condition 2-5). Total bacterial abundance (black dots) and relative abundances (colored bars) for each starting condition for WT and engineered strains in sequence. Shown are data from one representative experiment. WT consortia grow to about 100-fold higher cfu/mL. c. Hellinger distance between population ratios of all conditions at 12 hr time point calculated for WT and engineered consortia. The Hellinger distance for engineered consortia is lower than for WT consortia (Mann-Whitney p-value: 0.0355). d. Inferred growth rates and Jacobians of the inferred dynamics for both WT and engineered consortia (only interactions with Bayes factors greater than 1 shown). The Jacobians provide normalized measures of interaction strengths. e. Difference between engineered and WT Jacobians identifies 5 net positive, 7 neutral interactions.
Computational Analysis of in vitro Growth Dynamics of the Engineered Consortia over Time Elucidate a Net Positive Interaction Network
Our design created a mutually coupled bacterial consortium designed to function together, suggesting that consortium behavior is best assessed in the full assemblage, rather than through pairwise co-culture experiments. In order to investigate such behavior, we analyzed the densely sampled time-series data from our experiments using the entire consortium with different initial starting conditions (Figures 4A,4B; described in the previous section) with a dynamical systems inference approach.
Our dynamical systems approach uses a tailored model based on stochastic generalized Lotka-Volterra (gLV) dynamics and an associated fully Bayesian machine- learning/statistical inference algorithm (Methods, Gibson and Gerber, 2018). Continuous time stochastic generalized Lotka-Volterra (gLV) dynamics can be expressed as:
 where xt,i,l is the abundance of microbe i at time t in experiment l. The parameter ri denotes the growth rate of microbe i, and ai,j is the effect that microbe j has on microbe i. When i = j the expression aj,i is a self-limiting term and together with ri determines the carrying capacity,
where xt,i,l is the abundance of microbe i at time t in experiment l. The parameter ri denotes the growth rate of microbe i, and ai,j is the effect that microbe j has on microbe i. When i = j the expression aj,i is a self-limiting term and together with ri determines the carrying capacity,  of microbe i if no other microbes were present. Finally, w is the process disturbance term, which we assume is a Brownian motion. For inference, we discretize the continuous dynamics as described fully in Methods.
of microbe i if no other microbes were present. Finally, w is the process disturbance term, which we assume is a Brownian motion. For inference, we discretize the continuous dynamics as described fully in Methods.
We used our method to infer growth rates and microbe-microbe interaction strengths from our time-series data (Figure 4E,F). As we described above, the WT consortium achieves an overall higher concentration of approximately 5 × 107 versus 1 × 106 cfu/mL for the engineered consortium. This results in different scales for the self-interaction and interaction parameters across the two consortia, so we normalized the interaction matrices by steady-state dynamics (see Methods) to render the two consortia comparable. We see that the WT consortium has several strong aggregate negative interactions, e.g., mutual negative interactions between E. coli and S. Typhimurium. In contrast, the engineered consortium has aggregate neutral interactions, aside from one strong positive interaction from B. fragilis to S. Typhimurium.
We can gain insight into the net changes in the quantitative structure of the synthetic microbial interaction network introduced by engineering, by subtracting the normalized WT network from the normalized engineered network, and keeping only interactions deemed significant with our inference method in at least one network. Using this analysis, we found 5 net positive and 7 net neutral interactions, confirming the ability of our engineering approach to promote cooperation in the consortium. Interestingly, since the WT interaction network shows strong competitive interactions, our model suggests that our engineering approach mostly promotes cooperatively by significantly weakening the naturally occurring competitive interactions.
The inferred network is generally consistent with our mono-culture and co-culture results. For instance, the strongest positive interaction in the engineered consortium is from B. fragilis to S. Typhimurium. Concordantly, B. fragilis overproduces the highest amount of its crossfed metabolite Arg, and S. Typhimurium is the strain that benefits most in co-culture. As another example, we did not infer any incoming positive interactions for B. fragilis, which is consistent with our finding that B. fragilis does not show improved growth in co-culture compared to monoculture. Our model inferred mutual net positive interactions between E. coli to S. Typhimurium, which are consistent with our supernatant complementation experiments.
Some aspects of the inferred model are inconsistent with the mono- and co-culture experiments, however. For instance, these experiments suggest a positive interaction from B. fragilis to E. coli, but this interaction does not appear in the inferred network. Because our model is inferred from longitudinal data, which is relatively sparse and noisy, it is not surprising that some interactions may not be detected. Further, our approach is fully Bayesian and takes into account uncertainty in both the model and measurements. This approach is by design conservative, meaning that it requires strong evidence from the data to formally detect an interaction. Thus, our approach will tend to report the strongest pre-existing or engineering induced interactions, and may miss weaker but still present interactions. Although weak interactions were enhanced by removing any amino acid supplementation in our experiments, these interactions may still fall below our threshold of detection.
Simulations Based on Realistic Design Constraints Reveal Principles for Stability in Metabolically Cooperative Microbial Consortia
We used information from our experiments and data-derived microbial interaction networks to systematically study consortia stability, and gain insight into general design principles. We present these simulations with increasingly realistic design constraints and demonstrate how such constraints lead to different cooperativity regimes. For simplicity of exposition, we assume the four species have identical growth rates ρ, selfinteraction terms δ (which is always negative), and identical interactions coefficients α (Figure 5A). In our first, and least realistic design (Figure 5B), the growth rate and selfinteraction terms are kept constant, and only the positive interaction strength is increased. Under these constraints, the carrying capacity and stability of the system increases as the interaction strengths increase, up to the point of system instability. However, such a scenario is unrealistic, because on theoretical grounds, it would allow for the overall carrying capacity of the ecosystem to be arbitrarily increased. Moreover, it is inconsistent with our experimental evidence and data-derived model, which both show a lower overall carrying capacity for the engineered consortia relative to WT. This lowered carrying capacity is driven by both lower growth rates as well as increased negative autoregulation in each species, likely due to the dual burden of auxotrophy and overproduction.

a. Baseline model of Lotka-Volterra dynamical system parameterized by growth rate ρ, self-interaction δ (which is negative), and identical interactions coefficients α, for all species. b. Scenario 1: as the interactions coefficient is increased, the system has an increased carrying capacity and robustness up to the point of system instability. c. Scenario 2: interactions coefficient is increased as the magnitude of the self-limiting term is increased, and the growth rate is decreased. This results in a more realistic scenario and a trade-off between cooperativity and self-interest emerges. d. Scenario 3: similar to Scenario 2, but instead of the interactions coefficient starting at zero, it begins with a negative value (pre-existing competitive interactions), mimicking what we observed in our engineered consortia. This also results in a trade-off between cooperativity and self-interest, with a higher optimal interactions coefficient than c.
To match these realistic design constrains, we ran simulations in which increases in cooperative interactions were always accompanied by decreases in intrinsic growth rates and increases in the magnitude of negative autoregulation (Figure 5C.) We further assumed no interactions prior to engineering. In this scenario, the stability margin cannot be arbitrarily increased, and an engineering design arises, with intermediate cooperation strengths, that optimizes consortium robustness. If cooperation strengths are increased beyond this level, the consortium becomes less stable. Thus, we see that under realistic constraints, there is a trade-off between cooperativity and self-interest in the consortium, with an optimal intermediate.
We next investigated the impact of pre-existing interactions, an important feature of naturally occurring heterogeneous bacterial species, on stability (Figure 5D.) In this case, the robustness of the consortium again cannot be arbitrarily increased, and an engineering design that optimizes consortium stability arises. However, in contrast to the case with no interactions prior to engineering (Figure 5C), the optimal cooperativity strength is higher. This reflects the fact that engineering must first push pre-existing competitive interactions toward neutrality before pushing interactions into the optimal regime for consortium stability. These results suggest qualitative design principles for engineering cooperative bacterial consortia, and provide tools for future analyses of specific designs.
Consortia Engineering Increases Population Calance in the Mammalian Gut in a Diet Dependent Manner
We investigated the behavior of our consortium in the mammalian gut, using gnotobiotic mice as a controlled yet sufficiently complex environment for evaluation. To investigate the role of amino acid cross-feeding in vivo, we altered amino acid levels in the gut by changing the animal’s diet (Ravindran et al., 2016). Groups of five germfree mice were fed standard or low protein (3%) chow and gavaged with either the WT or engineered consortium (Figure 6). The consortia were allowed to colonize for 10 days, and then stool samples were collected and interrogated via qPCR with species-specific primers. The engineered consortium consistently exhibited greater population evenness in mice that were fed low protein diet compared to the three other groups (Mann-Whitney test; p-values: 0.02, 0.03, 0.02).

a. Four groups of 5 germfree mice were either fed low protein diet or standard diet, and inoculated with either the engineered or WT bacterial consortia. Fecal samples 10 days post-inoculation were analyzed via strain- specific qPCR to assess concentrations of each consortia species. b. Population balance or evenness of consortia, expressed as normalized entropy of the population proportions. Bar indicates Median. Mann-Whitney test showed significantly increased population of the engineered consortia in mice that were fed low protein diet compared to the consortia in the three other groups (p-values: 0.024, 0.032, 0.015). Ec: E. coli; St: S. Typhimurium; Bt: B. theta; Bf: B. fragilis.
Our results show that diet influences total bacterial concentrations in both engineered and WT consortia, and each species in the consortium is affected to a different extent. For the engineered consortium, species abundances were higher by a factor of approximately 3 for E. coli, 8 for S. Typhimurium, 16 for B. theta, and 11 for B. fragilis in mice fed a standard versus a low protein diet. In the case of the WT consortium, S. Typhimurium, B. theta and B. fragilis concentrations were similarly higher on standard chow (fold changes of approximately: 10, 22, 13 respectively). WT E. coli concentrations were dramatically higher (fold change of approximately 51), and partially account for the greater population imbalance in the WT consortium. Interestingly, in the mice on a low protein diet, S. Typhimurium grew about 8-fold better in the engineered consortium compared to the WT consortium. This finding is consistent with our in vitro results, which indicated that engineered S. Typhimurium benefits most from growing in co-culture. The same trend can be observed in mice that were fed standard diet, albeit to a lesser extent.
Discussion
We have engineered a heterogeneous synthetic bacterial consortium from four different gut-derived species, and demonstrated that this consortium exhibits inducible cooperativity, with increased stability and population balance. Using data-driven dynamical systems models, we have elucidated the interaction network among consortia members and shown that our engineering strategy acts to increase cooperativity largely by neutralizing pre-existing competitive interactions. Simulations based on the derived model provide further insights into general synthetic design strategies for these systems, showing a regime of optimal cooperativity that maximizes consortium stability. Finally, we demonstrate that our engineered consortium exhibits increased population balance in the complex mammalian gut environment when induced to cooperate, with this behavior alterable by the host diet.
This work differs from previous approaches in two key aspects. First, while there have been reports of engineered multi-strain consortia (Minty et al., 2013; Zhou et al., 2015), we have engineered four different species that have relevance to gut applications. Second, previous efforts to engineer interactions via metabolite cross-feeding have relied on metabolite auxotrophies without corresponding overproduction (Mee et al., 2014; Wintermute and Silver, 2010a). Here, we selected for overproducing strains for each of the four species, applying knowledge from industrial amino acid overproduction (Becker and Wittmann, 2012).
We found that there are strong pre-existing negative interactions between WT strains (Figure 3b), which is not altogether surprising given the natural history of gut commensal bacteria. Negative interactions may include acidification (Ratzke et al., 2018), scavenging of metals or other micronutrients (Hider and Kong, 2010), and competition for carbohydrate sources, among others. In some instances, we achieved measurable positive interactions, whereas in other cases the negative interactions were neutralized. Of note, our engineered consortia growth is reduced by about 100-fold compared to the WT consortia (Figure 3 a,b). In essence, we have introduced improved cooperativity into the consortium with concomitant gains in stability and population balance, at the expense of strains’ individual fitness. Loss of fitness in this case is likely due to insufficient complementation by the overproducers as shown in our initial system characterization (Figure 2). Complementation could be improved in two ways: first directed evolution approaches could render a more efficient consortium, and second, we could apply rational engineering (e.g. introducing transporters or increasing overproduction) to improve cross-feeding. Genome sequencing of our mutated strains could aid in such approaches in future work.
We have created a model ecosystem that allows us to study the effects that cooperation has on microbial consortia. While microbial cooperation is found in natural habitats (Hays et al., 2015b; Ponomarova et al., 2017; Rakoff-Nahoum et al., 2016), and there are many ecological theories that attempt to explain its evolution (Nowak, 2006; West et al., 2006; Zomorrodi and Segré, 2016), there is a dearth of experimental systems to test such hypotheses. In our system, cooperation leads to improved growth of the overall synthetic consortia, but it also promotes continuing survival of each single species through improved population evenness. These two characteristics of our system make it an attractive test-bed to address questions about the evolution of cooperation, which serves both the consortia as a whole and each single species’ survival.
We chose cross-feeding of amino acids as a model of cooperativity, because this approach has been well established and could readily be applied to disparate bacterial species. However, amino acids are also abundant in the animal gut and other environments, raising the possibility that background amino acids levels would simply saturate our synthetic consortium. Remarkably, our consortium demonstrated inducible cooperativity with increased population balance in our gnotobiotic mouse model (Figure 6), despite the fact that the germ-free mouse gut contains amino acids from both diet and host sources. This experimental system provides a relatively controlled, but still complex environment, and allows us to readily interpret behavior of the consortia alone without having to consider interactions with a pre-existing microbiota. Of note, all of the strains we used have been shown to individually colonize conventional mice, in some cases without the need for prior antibiotic treatment (Kotula et al., 2014; Riglar et al., 2017). However, our approach using amino acid crossfeeding may not translate to a mammalian gut colonized with a pre-existing microbiota. Thus, when extending our results to more complex systems, more orthologous approaches to cooperativity are likely to be necessary to avoid host interference.
Our computational approach is data-driven and phenomenological, abstracting various types of possible biological interactions (e.g., competition for nutrients, bacteriocin production, syntrophy, etc.) into quantitative pairwise interaction coefficients. This is a different modeling approach than that of many prior auxotroph-overproducer studies, which built detailed metabolic models. These models have not been shown to be reliable for diverse bacteria, such as the Bacteroides spp. in our consortium, in part due to limited knowledge of bacterial metabolism outside of a small number of model organisms. Moreover, as discussed, in a consortium with heterogeneous commensal gut bacteria as members, we expect there to be pre-existing interactions, many of which may not be metabolic. Thus, to gain insights into our consortium, rather than build a bottom-up metabolic model, we infer a phenomenological model from data. Our model is based on stochastic generalized Volterra-Lotka (gLV) dynamics, which is a relatively simple model. More sophisticated models incorporate higher-order interactions or nonlinearities such as saturation effects. However, gLV models have been shown to accurately forecast dynamics in complex host-microbial ecosystems (Bucci et al., 2016), suggesting that the relatively simple assumption of pairwise quadratic interactions may dominate higher-order and more nonlinear effects. Moreover, reliable inference from data for more complicated models is not possible given the amount of densely sampled time-series data that we could feasibly collect for the present study.
Overall, we have demonstrated a design, build and test cycle, applicable to engineering microbial consortia that function in the mammalian gut. There is currently interest in developing living bacterial diagnostics and therapeutics for human diseases. The first approach has been to attempt to transfer an uncharacterized microbiota, i.e., fecal microbiota transplants (Boyle, 2015). Subsequently, there have been efforts to assemble defined collections of naturally occurring commensal bacteria (Atarashi et al.). While these approaches may work for some diseases, we could anticipate a need for precise and controllable behavior of therapeutics, which will require synthetic biology. Our results help to close this gap, providing new insights into the design principles needed to engineer robust and heterogeneous bacterial consortia.
Author Contributions
Conceptualization, J.C.W, P.A.S, G.K.G; Methodology, M.Z., T.G., J.O., G.K.G.; Software, T.G.; Formal Analysis, M.Z., T.G.; Investigation, M.Z., J.O., A.S., T.G., N.D., K.L.; Resources, L.B., P.A.S., G.G.; Writing – Original Draft, M.Z., T.G.; Writing – Review & Editing, B.B.H., D.T.R, P.A.S., G.K.G.; Visualization, D.T.R., B.B.H.; Supervision, D.T.R., B.B.H., L.B., J.C.W., G.K.G., P.A.S.; Funding Acquisition, D.T.R., J.C.W., P.A.S., G.K.G.
Financial Interests
G.K.G. is a shareholder and member of the Strategic Advisory Board of Kaleido Biosciences, and shareholder and member of the Scientific Advisory Board of Consortia Rx; neither company provided funding for this work.
Methods
Auxotroph engineering
For auxotroph generation in the E. coli NGF-1 strain we introduced multiple knockouts using sequential P1 transduction (Thomason et al., 2007) from the Keio knockout collection (Baba et al., 2006). Flip-out of kanamycin cassettes was done using pCP20 (Cherepanov and Wackernagel, 1995). In brief, for P1 transduction we prepared phage by diluting an overnight culture of the donor strain 1:100 LB with 0.2% glucose, 5 mM CaCl2 and 25 mM MgCl2 and incubated for 1-2 hours at 37 °C until slightly turbid. We then added 40 μL P1 lysate and continued growth for 1-3 h at 37 °C while shaking until lysed. Lysate was then filtered with a 20 μm sterile filter and stored in the fridge. For transduction, we harvested 2 mL overnight culture of recipient strain and re-suspended in 2 mL LB with 5 mM CaCl2 and 100 mM MgSO4. We then mixed 100 μL donor lysate with 100 μL recipient, incubated 30 min at 37 °C and added 200 μL sodium citrate (1 M, pH 5.5) and 1 mL LB and incubated for another 1 hr at 37 °C. Cells were harvested, resuspended in 100 μL LB with 100 mM sodium citrate and plated on LB Kan plates (75 μg/mL). The transduced kanamycin cassette was then removed using pCP20 according to protocol. We transformed pCP20 via electroporation and transformants were selected on LB agar plates supplemented with 100 μg/mL carbenicillin grown at 30 °C. Single colonies were re-streaked on LB without drugs and incubated for 10 hours at 42 °C. From there, single colonies were re-streaked on LB plates without drugs and grown overnight at 37 °C. Colonies were checked for Carbenicillin, and Kanamycin sensitivity and further confirmed via PCR at respective loci. This procedure was repeated until all knockouts were introduced.
Engineering of S. Typhimurium LT2 required generation of single knockout strains that contained pKD46 integrated into the genome, which allowed for linear DNA integration using lambda red recombination (Cherepanov and Wackernagel, 1995). We then introduced the knockouts into the S. Typhimurium strain through sequential P22 transduction and pCP20 flipout analogous to E. coli engineering. Single knockout strains were generated by PCR amplifying a Kanamycin resistance cassette from pKD13 generating linear fragments that contained upstream and downstream homology to the gene of interest and the kanamycin cassette with FRT sequences. Fragments were introduced via electroporation and selected on LB agar plates supplemented with 50 μg/mL Kanamycin. Sequential P22 transduction and pCP20 flip-out was essentially performed as described above for P1 transduction but lysis was done overnight.
For knockout generation of both B. theta and B. fragilis, we used pExchange KO vectors as described (Mimee et al., 2015). Briefly, we introduced 750 bp flanking regions for genes of interest adjacent to each other into the vector. The vector contains an erythromycin resistance positive marker and a thymidine kinase as counter selection marker. Cloning was done in pir+ E. coli strains and vectors were transferred to MFDpir for conjugation (Ferrières et al., 2010). Conjugation was done according to protocol with minor changes. In brief, five drops of overnight culture of E. coli donor was inoculated in LB supplemented with 300 μM Diamino pimelic acid (DAP) and five drops of recipient overnight culture was inoculated in 50 mL basal media. Both cultures were grown for about 2 hr (E. coli aerobically, Bacteroides spp. anaerobically) until E. coli culture was well turbid and Bacteroides culture just slightly turbid. Subsequently, 9 mL recipient and 3 mL donor were combined and spun down for 10 min at 4000 rpm together. The pellet was re-suspended in 100 μL fresh basal media with 300 μM DAP and pipetted on basal media agar plates without cysteine and supplemented with 300 μM DAP. The cells were incubated at 37 °C aerobically face up for up to 20 hr, scraped off and re-suspended in 10% glycerol. Dilutions were plated on basal-agar plates supplemented with 10 μg/mL erythromycin and incubated at 37 °C anaerobically for 2-3 days. Single colonies were re-streaked in the presence of erythromycin and grown for another 2 days. 10 single colonies were inoculated in basal media without drug and grown overnight. 500 μL of each culture was mixed, spun down and re-suspended in 10% glycerol. We then plated different dilutions on basal media plates supplemented with 5-fluoro-2-deoxy-uridine (FuDR) (200 μg/mL) and incubated at 37 °C anaerobically for 3 days. Knockouts were verified via PCR. This procedure was repeated multiple times to obtain the multiple auxotroph strains.
Overproducer selection
Overproducers were generated by selecting for mutants that could grow on minimal media agar plates supplemented with anti-metabolites (E. coli: 5 mg/mL Norleucine for Met overproduction; S. Typhimurium: > 0.7 mg/mL beta-(2-thiazolyl)-DL-alanine for His overproduction; B. theta: 50 μg/mL 4-methyl tryptophan for Trp overproduction; B. fragilis: 80 μg/mL Canavinine for Arg overproduction). Single colonies that showed halos were re-streaked and overproduction was measured using a bioassay. In brief, for screening of overproducing mutants the isolated strains were grown overnight at 37 °C shaking aerobically (for E. coli and S. typhimurium) or anaerobically without agitation (for Bacteroides spp.). Supernatant was harvested, diluted 1:1 with fresh media, and E. coli auxotrophs were inoculated and their growth was recorded after 24 hr. For E. coli NGF-1 overproducers, we used a S. Typhimurium auxotroph instead, since its colicin production prevented the E. coli biosensor from growing. Confirmed overproducers were further quantified using LC-MS.
LC-MS for Overproduction Measurements
To quantitate amino acid levels in overproducer supernatants, a standard curve was obained using freshly prepared amino acid standards dissolved in growth media (1mM, 500 uM, 100 uM, 50 uM, 10 uM of L. Methionine, L/Histidine, L-Tryptophan, L-Arginine each). To prepare for HPLC-MS analysis, 0.5 mL sample or standard were added to 1.5 mL ice-cold methanol and incubated on ice for 10 min. The mixture was centrifuged for 5 min at 15,000 rpm and 500 μL supernatant was vacuum concentrated and re- suspended in 50 μL methanol. Samples were kept on ice or at 4°C. HPLC-MS analysis of standards and extracts was carried out using an Agilent 1260 Infinity HPLC system equipped with an Agilent Eclipse Plus C18 (100 × 4.6 mm, particle size 3.5 mm, flow rate: 0.3 mL/min, solvent A: dd.H2O/0.1% (v/v) formic acid, solvent B: acetonitrile, injection volume: 4 mL) connected to an Agilent 6530 Accurate-Mass Q-TOF instrument. The following gradient was used (time/min, %B): 0, 0; 0.5, 0; 14, 100; 19, 100; 20, 0, 25, 0. The mass spectrometer was operated in positive mode and the autosampler was kept at 4°C. After HPLC-MS analysis, extracted ion current (EIC) peaks were automatically integrated using the MassHunter Workstation Software (version: B.07.00). A plot of peak area versus amino acid concentration was used to generate a linear fit.
Sequencing
Bacterial cultures were prepared in rich media (basal for Bacteroides spp. and LB for E. coli and S. Typhimurium). Genomic DNA (gDNA) extraction was performed using the Wizard Genomic DNA Purification Kit (Promega) according to protocol. The extracted gDNA was sheared using Covaris DNA Shearing, and the library was prepared using Kapa Biosystem DNA Hyper Prep NGS Library (Dana Faber Core MBCFL Genomics). Sequencing was performed on the Illumina MiSeq instrument, with the 150 bp paired End (PE150) reagents. Sequences were analyzed for SNPs using Geneious software and published genome sequences (E. coli: CP016007.1; S. typhimurium: NC_003197; B. theta: AE015928; B. fragilis: NC_016776) (Table S1).
Growth and Media Conditions
All basal media and co-culture media was pre-incubated for at least 24 hr anaerobically before use. Bacteroides spp. were inoculated from glycerol stock into basal media, grown overnight and 400 μL was inoculated in 5 mL basal and grown 2 hr anaerobically. Cells were spun down, washed twice in PBS and diluted in growth media as described for each experiment in Results. E. coli and S. Typhimurium were inoculated from glycerol stock into LB and grown overnight at 37 °C while shaking. 100 μL of culture was then inoculated into pre-incubated LB and grown anaerobically for 2 hr, diluted, washed in PBS and diluted into co-culture media as described. Co-culture media consisted of modified M9 salts (0.2 g/L Na2HPO4, 90 mg/L KH2PO4, 30 mg/L NH4Cl, 15 mg/L NaCl), 1 mM MgSO4, 10 μg/mL heme, 0.1 mM CaCl2, 1 μg/mL Niacinamide, vitamin B12 and thiamine, 400 μg/mL L-cysteine, 0.3% bicarbonate buffer, 2.5 ng/mL vitamin K, 2 μg/mL FeSO4∗7H2O and carbon sources and amino acid supplementation as described in Results.
Multiplex qPCR
We designed strain specific primer/probe-fluorophore pairs according to IDT protocol (Table S2). We chose strain specific genes by multiple genome alignment between the strain of interest, the other consortia members and closely related strains using Mauve (Darling et al., 2004). Multiplex qPCR was used to quantify each strain in co-culture by using a standard curve obtained by plating late log phase cultures grown in rich media. In brief, each strain was grown from overnight culture for ~5 hours until about OD of 1. Cells were then counted by plating. Cultures were mixed, diluted and frozen at −80 °C for use as standard curve. Samples were diluted 1:10 in ddH2O and snap-frozen in liquid nitrogen and stored at least overnight at −80 °C. Growth curve and sample were both thawed together and prepared in a 5 μL Primetime Mastermix (IDT) with 1 μL Primer/Probe mixture (final concentrations: 100 nM for primers and 50 nM for probes). The qPCR was run with the following program: 20 min at 98 μC (to boil the cells and denature gDNA), followed by 40 cycles of 60 °C and 98 °C.
Calculation of Normalized Entropy (Pielou’s Evenness) and Hellinger Distance
Normalized entropy (Pielou’s evenness) was calculated according to the given formula:

Where pi refers to the population ratio of a given strain in the consortium of four strains. S is the number of species.
Hellinger distance was used as a metric for comparing consortia relative abundances. We computed Hellinger distances between different starting conditions in WT and engineered strains using the formula:

Where pi and qi are the ratios of each strain in the consortium in two different conditions.
Pairwise Supernatant Experiment
Overproducers and WT strains were grown overnight anaerobically at 37 °C in coculture media and supernatant was harvested and sterile filtered. Auxotrophs were prepared as described in Media and Growth conditions and inoculated in media that contained 50% fresh co-culture media and 50% spent supernatant. OD600 was measured after 24 hr growth anaerobically at 37 °C.
In vivo Experiments
Adult (6-8 weeks) male Swiss Webster germ free mice bred in house at the Massachusetts Host-Microbiome Center were used. Animals were fed either on standard chow in the facility for the entire experiment, or on low-protein diet (3% custom diet, envigo, doubly irradiated) beginning 10 days prior to the experiment and continuing for its duration. To prepare bacteria for gavage, we grew each strain to mid-log phase, plated for counting and snap-froze aliquots. For gavage, aliquots were thawed, spun down, combined to achieve concentrations of approximately 107 per bacteria per gavage, and re-suspended in 200 μL 1x PBS with 0.05% L-cysteine for gavage. After gavage, mice were transferred to Optimice cages and maintained gnotobiotic for 10 days. Fecal samples were collected prior gavage and at 10 days, and snap-frozen for storage at −80 °C.
Molecular Analysis of in vivo Samples
DNA was extracted from fecal samples using the Zymobiomic 96 DNA Kit with the following modification: we omitted the silicon-ATM-HRC wash. Cells were lysed in a bead beater at speed 20 for 10 min, plates were turned and lysed for another 10 min at the same speed. We added an additional 3 min incubation step for Binding Buffer and additional 5 min incubation steps when transferring to Zymo-Spin-I-96-Z plates. Elution was done in 50 μL ZymoBIOMIC DNase/RNase Free Water.
Direct multiplex probe-based qPCR was done on extracted DNA samples as described above. For standard curves, we used plated overnight cultures spiked into germfree fecal samples and extracted them as described above.
Synthetic simulations of deterministic microbial dynamics in the presence of a disturbance
For the synthetic results in Figure 1 and Figure 5, we used the following (deterministic) generalized Lotka-Volterra (gLV) dynamics with a constant disturbance d

The corresponding first order Euler integration of these dynamics with step size Afc is as follows

For the simulations to generate Figure 1, the growth rate vector, interaction matrix, and disturbance vector for the non-interacting consortia are

The growth rate vector, interaction matrix, and disturbance vector for the cooperative community are then as follows

Two simulation were performed for both the non-interacting and the cooperative community: one simulation without the disturbance present, and then the subsequent simulation with the disturbance present.
For the simulations to generate Figure 5, the parameterization and simulation parameters are as specified in the figure. The robustness margin was calculated as the largest value of κ for which the disturbance vector
 can be applied to the dynamics without any of the xi becoming <0.01 in abundance (i.e., without any of the species going extinct during the simulation). For all synthetic results shown in Figures 1 and 5, the step size was Δk = 0.005 for a total of 3000 steps. For generating Figure 5, the initial condition for all the species was an abundance of 2.
can be applied to the dynamics without any of the xi becoming <0.01 in abundance (i.e., without any of the species going extinct during the simulation). For all synthetic results shown in Figures 1 and 5, the step size was Δk = 0.005 for a total of 3000 steps. For generating Figure 5, the initial condition for all the species was an abundance of 2.
Bayesian dynamical systems inference
Our dynamical systems model and associated inference algorithm is a version of our previously published method (Gibson and Gerber, 2018) that we have customized for this study. Briefly, our model is based on continuous time stochastic gLV dynamics as described in Results. We approximate these dynamics with a first order Euler integration of step size Δk,l

Our model is fully Bayesian, with the inference goal being to learn the posterior probability distribution over both the model parameters as well as the qualitative interaction structure graph. To model the interaction structure graph, we use indicator variables zi,j, which indicate the presence or absence of the edge from species i to j in the graph. The resulting conditional distribution for dynamics is then:

Where  is the variance for the Brownian motion. Our measurements of microbial abundances are captured by the observed variable y. We introduce an auxiliary variable q that enables efficient inference of a relaxed system (see Gibson and Gerber, 2018 for details):
is the variance for the Brownian motion. Our measurements of microbial abundances are captured by the observed variable y. We introduce an auxiliary variable q that enables efficient inference of a relaxed system (see Gibson and Gerber, 2018 for details):

We estimate the measurement variance  directly from qPCR technical replicates, obtaining L = 1010, and νq = 106. The prior probability distributions for the indicator variables, the growth rates, and the interactions terms as defined below:
directly from qPCR technical replicates, obtaining L = 1010, and νq = 106. The prior probability distributions for the indicator variables, the growth rates, and the interactions terms as defined below:

Note that our prior for z, the indicator variables for presence or absence of interaction edges is set to indicate maximum uncertainty, e.g., no a priori assumption about the presence or absence of an interaction. The variance parameters in our model that are not a-priori fixed or estimated from technical replicates have the following conjugate priors

Our parameterization of the Inv − X2 is as follows
 which is sometimes referred to as the Scale − Inv − X2 distribution, because it has two parameters, number of degrees of freedom η, and scale parameter θ. In our model ηr, ηr, ηr = 1 with θr = 1, θa = 10−10, and θW = 106, specifying relatively diffuse priors. A compact representation of our model is shown in Supplemental Figure 1a with an accompanying graphical model with plate notation in Supplemental Figure 1b
which is sometimes referred to as the Scale − Inv − X2 distribution, because it has two parameters, number of degrees of freedom η, and scale parameter θ. In our model ηr, ηr, ηr = 1 with θr = 1, θa = 10−10, and θW = 106, specifying relatively diffuse priors. A compact representation of our model is shown in Supplemental Figure 1a with an accompanying graphical model with plate notation in Supplemental Figure 1b
We perform model inference using a custom Markov Chain Monte Carlo (MCMC) algorithm. Almost all variables in the model can be updated with Gibbs or collapsed Gibbs sampling, with the exception of q and x. Sampling of the auxiliary variables and latent trajectories require Metropolis-Hastings (MH) steps. For q, the MH proposal is based on a Generalized-Linear Model approximation. For x, we use a one time-step ahead proposal that is essentially the forward pass of a Kalman filter, see Supplemental Figure 1c and (Gibson and Gerber, 2018). Inference was performed with 6,000 MCMC steps where the first 1,000 steps are discarded (burn in).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
a. Description of key components of our Bayesian dynamical systems model for the consortia. Higher level priors not depicted for simplicity. b. Graphical Model depiction with plate notation. c. Portion of the Graphical Model unraveled in time, depicting a single time series experiment, and color coded to illustrate our inference method, using Metropolis-Hastings proposals, for filtering the latent state. The proposal uses information from the blue nodes to propose for the green node, but does not use future time information highlighted in red. The future information is however accounted for in the target distribution and thus the algorithm samples from the true posterior.
Acknowledgement
We thank Eli Bogart for computational advice; Laurie Comstock for providing B. fragilis 638R and helpful advice on Bacteroides handling. This project was funded by the Defense Advanced Research Program Agency (DARPA BRICS HR0011-15-C-0094), NIH T32 HL007627 (to T.G.) and The Wyss Institute for Biologically Inspired Engineering. D.T.R. is supported by a Human Frontier Science Program Long-Term Fellowship and a National Health and Medical Research Council (NHMRC) RG Menzies Early Career Fellowship from the Menzies Foundation Australia.
References




