

ABSTRACT
Ribonucleotidyl transferases (rNTases) add non-templated ribonucleotides to diverse RNAs. We developed a screening strategy in S. cerevisiae to identify sequences added by candidate enzymes from different organisms at single-nucleotide resolution. The rNTase activities of 19 previously unexplored enzymes were determined. In addition to poly(A)- and poly(U)-adding enzymes, we identified a C-adding enzyme that is likely part of a two-enzyme system that adds CCA to tRNAs in a eukaryote; a nucleotidyl transferase that adds nucleotides to RNA without apparent nucleotide preference; and a poly(UG) polymerase, C. elegans MUT-2, which adds alternating U and G nucleotides to form poly(UG) tails. MUT-2 is known to be required for certain forms of RNA silencing, and mutations in the enzyme that are defective in silencing also fail to add poly(UG) tails in our assay. We propose that MUT-2 poly(UG) polymerase activity is required to promote genome integrity and RNA silencing.
INTRODUCTION
Covalent modifications pervade biological regulation, and the discovery of enzymes that modify proteins and DNA have led to breakthroughs in metabolism, transcription and drug design. RNAs are extensively modified: 5’ termini are often capped, internal positions are altered both on ribose rings and bases, and 3’ termini receive untemplated nucleotides, referred to as “tails”. In eukaryotes, these 3’ tails control RNA stability, transport, processing and function, and affect virtually all classes of RNA, including mRNAs, snRNAs, tRNAs, lncRNAs and miRNAs. Tails, and the enzymes that add them, are critical in a wide spectrum of biological events. For example, uridylation is implicated in tumorigenesis, proliferation, stem cell maintenance, and the immune response1-10 and regulated poly(A) addition in early development, cancer, and memory11-17. Unbiased and global approaches with single-nucleotide resolution are needed to uncover new types of tails and alternate modification systems that may have gone unnoticed.
Members of the DNA polymerase β-like superfamily of nucleotidyl transferases catalyze non-templated addition of nucleotides18,19. Nucleotidyl transferases are related in amino acid sequence, but add nucleotides to divergent substrates, including RNAs, nucleotides, and antibiotics19. Nucleotidyl transferases that act on RNAs are referred to as ribonucleotidyl transferases (rNTases). rNTases include poly(A) polymerases (PAPs), poly(U) polymerases (PUPs; aka TUTases), and CCA-adding enzymes that add CCA tails to the 3’ end of tRNAs20. PAPs and PUPs cannot be distinguished unambiguously by inspection of their protein sequences.
Current methods to assay rNTase activity and nucleotide specificity generally are low-throughput and may not recapitulate rNTase specificities in living cells. In vitro approaches, which involve expression of recombinant protein or immunopurification, are dependent on assay conditions. Small molecules present in vivo can alter the specificities of tailing enzymes dramatically, complicating interpretation of in vitro studies21. Expression of candidate rNTases in Xenopus oocytes has enabled identification of multiple rNTases, but is low-throughput and not readily suitable for genome-wide analysis16,22,23.
We suspected that other tailing enzymes and forms of tails exist but have escaped detection. Powerful sequencing methods have been developed to identify tails on RNAs extracted from cells24-26. However, some tails may be added only at specific times or in certain cell types, occur on novel RNAs not commonly analyzed, or exist only transiently, perhaps triggering the RNA’s destruction. The challenge is to uncover all forms of tails, and identify the enzymes responsible, at a genome-wide scale.
We developed a screening approach to identify enzymes that add non-templated nucleotides to RNAs. Candidate rNTases were tethered in vivo to a reporter RNA in S. cerevisiae, and the number and identity of nucleotides they added were determined at single-nucleotide resolution using high-throughput sequencing. Our studies reveal previously undetected enzymes and tails, including a eukaryotic system with separate enzymes that add CC and A to form the ends of tRNAs, and a previously unknown enzymatic activity that adds alternating U and G residues to RNA 3’ termini. Mutations in the gene that encodes this poly(UG) polymerase are known to elevate transposition frequency27-29, disrupt silencing in the germline30-34, and impair RNA interference elicited by double-stranded RNA (RNAi)35-39. The same mutations abolish the enzyme’s poly(UG) addition activity. The poly(UG) polymerase, and likely poly(UG) tails, are required for these diverse RNA-dependent forms of regulation.
RESULTS
An in vivo tethering assay identifies rNTase activities
We devised an approach to classify rNTases by identification of the sequences they add to RNA (Fig. 1, Fig. S1). Candidate rNTases were fused to MS2 coat protein and an epitope tag (RGS-H6), and co-expressed in yeast with a reporter RNA that contained high-affinity MS2 binding sites. The interaction of MS2 coat protein with MS2 binding sites tethers the candidate protein to the reporter RNA, and circumvents RNA-binding proteins that might be required in a natural context40. We identified the nucleotides added by candidate enzymes by RT-PCR and high-throughput sequencing of whole-cell RNA extracted from yeast.

(a, b) TRAID-Seq strategy. (a) tRNASer(AGA) variable arm (gray) is mutated to an MS2 stem loop (cyan) in the tRNA reporter. (b) Left, tRNA reporter is co-expressed with an MS2 coat protein-rNTase fusion in the yeast S. cerevisiae, and the tethered rNTase adds nucleotides to the 3’ end of the tRNA. Right, RT-PCR analysis to detect A tails or U tails added to the reporter tRNA by control rNTases, relative to empty vector or to a no-reporter control. Lanes marked with a dash indicate reactions performed without reverse transcriptase. (c) Schematic of sample processing. DNA adapter (dark blue) with a randomized nucleotide heptamer (N7, orange) is ligated to total RNA extracted from samples, followed by reverse transcription, PCR, and high-throughput sequencing. (d) Computational pipeline. (e-g) Tail-o-grams of nucleotides added by control rNTases. Data are shown for C. elegans PUP-2 (e), S. pombe Cid1 (f), and C. elegans GLD-2 (g). Percent of each nucleotide at each tail length is plotted on the y-axis. Each nucleotide is indicated by a different color: U (green), C (yellow), G (purple), A (brown). Percentages are given on the left y-axis. The number of tails detected per million heptamers (TPMH) are indicated by black diamonds, and correspond to the log scale on the right y-axis.
We identified an appropriate reporter RNA to serve as a substrate for rNTase enzymes. We first tested an RNase P-derived RNA that contained two MS2 binding sites41,42 (Fig. S1a,b). In cells expressing this RNA and an MS2 coat protein fusion with a known PUP (C. elegans PUP-2 or S. pombe Cid1), we detected addition of U tails to the reporter RNA, indicative of PUP activity (Fig. S1c); and this activity was not detected with a catalytically inactive form of the PUP. However, high background polyadenylation activity in yeast, observed in the absence of expressed rNTase enzymes, complicated analysis (Fig. S1c). We therefore created an alternative RNA substrate based on S. cerevisiae tRNASer(AGA), a class II tRNA with a four base-pair variable arm. We replaced the variable arm with an MS2 binding site (Fig. 1a). Use of this substrate significantly reduced endogenous polyadenylation of the reporter RNA alone, as judged by reverse transcription and PCR (Fig. 1b), and enabled us to analyze MS2-rNTase fusion proteins unambiguously. We used this RNA in subsequent studies, and refer to it as “reporter tRNA” for simplicity.
Our approach accurately identified the activities of well-characterized rNTases. As proof-of-principle, we analyzed two known PUPs, C. elegans PUP-222 (Ce PUP-2) and S. pombe Cid122,43 (Sp Cid1), and a known PAP, C. elegans GLD-244 (Ce GLD-2). The tails added in vivo to the reporter tRNA by each enzyme were analyzed using RT-PCR assays designed to detect U or A tails (Fig. 1a, Fig S1b). The U tail-specific primer yielded products with Ce PUP-2 and Sp Cid1 samples, while the A tail-specific primer yielded products only with Ce GLD-2. Tails were not detected when the active sites of Ce PUP-2 or Sp Cid1 were inactivated by point mutations (Ce PUP-2 mut, Sp Cid1 mut), nor when the reporter tRNA was expressed alone (Fig. 1b).
To identify tails of any nucleotide composition and length, we used high-throughput sequencing (Fig. 1c). Total RNA from each sample was ligated to a DNA adapter, such that the adapter was linked to the 3’ end of all RNAs in the sample. The presence of the adapter enabled detection of any nucleotides added and introduced a seven-nucleotide randomized sequence (random heptamer) that enabled us to remove PCR duplicates computationally. These features allowed us to analyze RNA molecules at single-nucleotide resolution. Following reverse transcription, samples were PCR-amplified using primers specific for the tRNA/MS2 stem loop and 3’ adapter sequences, and gel-purified products were subjected to paired-end sequencing on an Illumina platform.
To analyze the data, added tails first were extracted computationally, as defined by nucleotides between the 3’ end of the mature tRNA reporter (including the CCA sequence) and the random heptamer (Fig. 1d). We removed PCR duplicates and quantitated the number of unique tails and the composition of each nucleotide in the population of tails at each detected length (Fig 1d). Tail length, nucleotide composition, and the number of unique tails are plotted in “tail-o-grams” (Fig. 1e-g). In these plots, each tail length is assessed as a population to determine the percent of each nucleotide added among all tails of that length. Tails shorter than five nucleotides were discarded, as they were detected in the absence of the tethered enzymes and were random in sequence. To visualize the data, A, C, G and U are color-coded, and nucleotide compositions of tails are plotted relative to the length of tail added. Numbers of reads were normalized to the number of unique random heptamers (TPMH, tails per million heptamers) at each tail length, and displayed on a log scale.
The assay was accurate and sensitive, as judged by analyses of Ce PUP-2, Sp Cid1 and Ce GLD-2 enzymes. Ce PUP-2 and Sp Cid1 added tails primarily of uridines, and Ce GLD-2 added tails of adenosines (Fig. 1e-g), consistent with each of their known nucleotide specificities. Furthermore, the high sensitivity of the assay also enabled detection of secondary nucleotide addition preferences. For example, Sp Cid1 added uridine tails with 8.6% adenosine (Fig. 1f, Fig. 2c), consistent with its ability to add both A and U in vitro43,45.

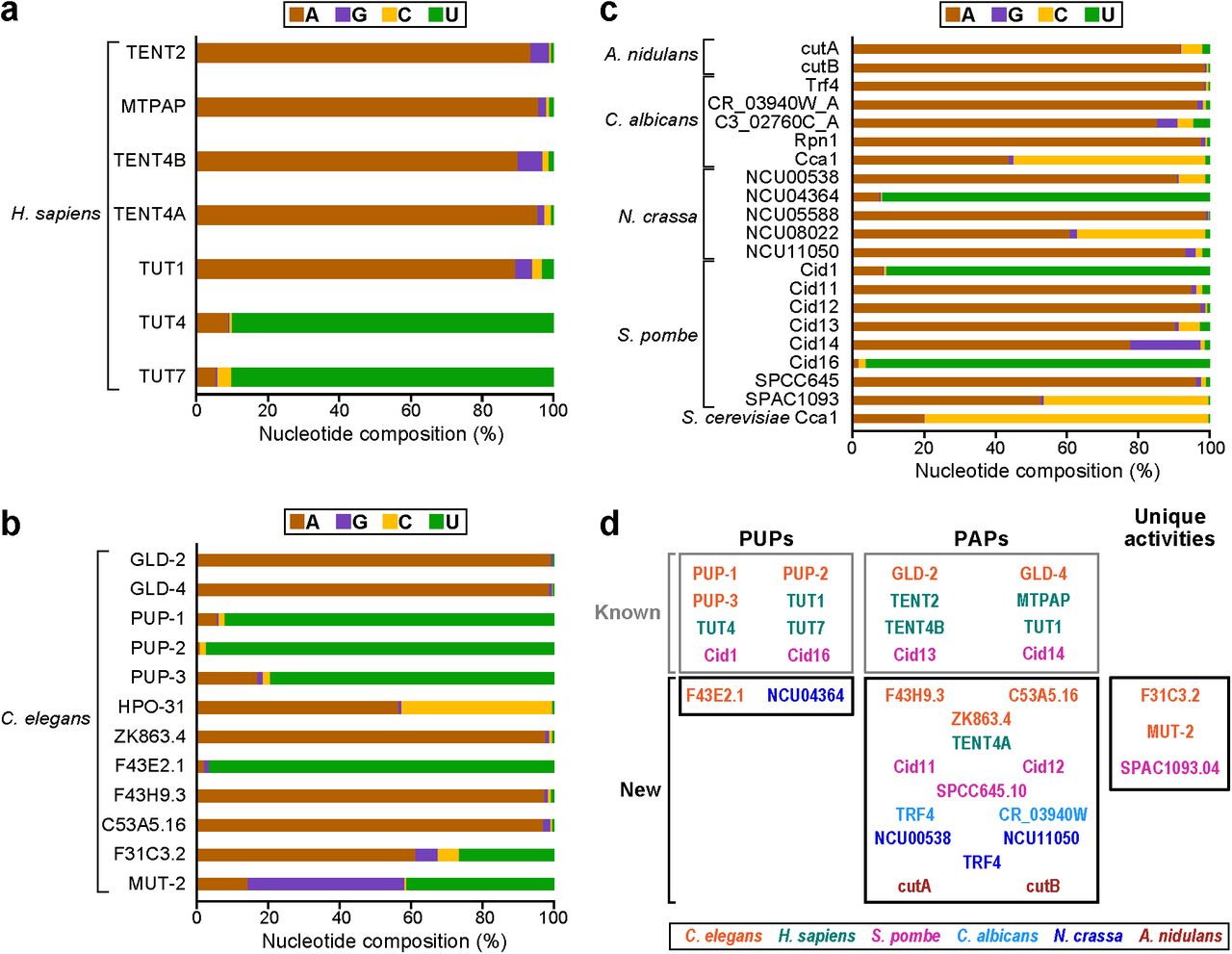
Overall percentages of each nucleotide added by (a) H. sapiens, (b) C. elegans, and (c) fungal rNTases. (d) Categorization of rNTases as PUPs, PAPs, or those of unique activity. rNTases are color-coded by organism. Gray boxes (top) indicate previously characterized (known) enzymes, and black boxes (bottom) indicate new enzymes.
Our assay, which we refer to as TRAID-Seq (tethered rNTase activity identified by high-throughput sequencing), thus detects known rNTase activities. It circumvents the need for purified enzymes, which can be problematic with this class of proteins, and precisely identifies many thousands of independently captured tail sequences, enabling a sensitive determination of their sequences and relative abundances.
New PUPs, PAPs, and CCA-adding enzymes
Using TRAID-Seq, we analyzed the nucleotide specificities of both characterized and previously untested rNTases. We tested 37 proteins from six species, including Homo sapiens (Hs), Candida albicans (Ca), Neurospora crassa (Nc), Aspergillus nidulans (An), Schizosaccharomyces pombe (Sp), and Caenorhabditis elegans (Ce) (Fig. 2, Table 1). Candidate rNTases were identified by the presence of a characteristic G(G/S) X7-13 DhDh motif and a downstream third aspartate20. To focus on noncanonical rNTases, we included putative rNTases with at least a partial type II nucleotide recognition motif (NRM)19,20, and excluded canonical rNTases, which are distinguished by the presence of a type I NRM20.
Nucleotide addition activities were classified first by the nucleotide composition of the tails added to the reporter tRNA (Fig. 2). For example, if tails added to the reporter tRNA consisted of primarily uridines, then the rNTase would be classified as a PUP. Through these analyses, we discovered 14 new PAPs and two new PUPs. We also identified likely CCA-adding enzymes in N. crassa (Nc), C. albicans (Ca) and C. elegans (Ce), consistent with homology predictions in each respective curated database. These enzymes exhibit a preference for both C and A in the tails they add (Fig. 2b,c) and show an enrichment for the repeating CCA pattern within the tails added to the reporter tRNA. The p-values of CCA occurrence among the tails added by each enzyme, determined using a one-sided Wald’s test, are highly significant (adjusted p-values less than 1.6 x 10-22 (see “Supplemental Information: Methods”).
Enabled by the sensitivity of TRAID-Seq, we confirmed nucleotide specificities of previously characterized rNTases16,21,22,43-53 and identified surprising secondary preferences in certain enzymes. Sp Cid13 and Sp Cid14 are exemplary. Both were previously identified as PAPs46, yet both added other nucleotides as well. Sp Cid13 added 90.3% adenosine (s.d. 0.3%; n=4), and so was classified as a PAP, yet also added 6.0% cytosine (s.d. 0.3%; n=4; Fig. 2c, Fig. S2a). Sp Cid14 added 77.9% adenosine (s.d. 1.2%; n=3) and 19.7% guanosine (s.d. 0.8%; n=3; Fig. 2c, Fig. S2b). Analysis of the patterns of nucleotides added by enzymes with secondary preferences revealed no specific sequence motifs, in contrast to the enriched CCA pattern yielded by the CCA-adding enzymes.
In addition to identifying new PAPs, new PUPs, and CCA-adding enzymes (Fig. 2d), and discovering previously unappreciated nucleotide flexibilities, our analyses also revealed enzymes with previously undetected activities, as discussed in the following sections.
C tails and a eukaryotic two-enzyme CCA-adding system
We identified an enzyme in S. pombe that primarily adds C nucleotides to RNAs. Based on sequence similarity, S. pombe SPAC1093.04 is predicted to be a CCA-adding enzyme, a highly conserved rNTase responsible for adding CCA to the 3’ end of virtually all tRNAs54. In TRAID-Seq with SPAC1093.04, we observed tails predominantly of oligo(C) or oligo(A) on reporter tRNAs with a CCA 3’ end (Fig. 2c; cytosine=46.0%, s.d. 6.0%; adenosine = 52.8%, s.d. 5.9%; n=5). Reporters with CC 3’ termini received almost exclusively oligo(C) (Fig. 3a, left, top). Tails added by S. pombe SPAC1093.04 and the S. cerevisiae CCA-adding enzyme (Cca1) clearly were distinct (Fig. 3a,b). The majority of tails added by S. cerevisiae Cca1 consist of repeating CCA motifs. In contrast, Sp SPAC1093.04 added long cytosine stretches (up to 19), which were often followed by a sequence of adenosines. The adenosines likely were added by endogenous PAPs in the TRAMP complex, which may recognize oligo(C)-tailed tRNAs as aberrant47,50. Differences between the activities of Sp SPAC1093.4 and S. cerevisiae Cca1 were manifest in computational analyses of sequence motifs of the tails they added. The trinucleotide CCA was highly enriched with the S. cerevisiae Cca1 but not S. pombe SPAC1093.04 (Fig. 3c, right). The products of both enzymes were significantly enriched for CC dinucleotides, as expected (Fig. 3c, left; for additional computational analyses, see Fig. S3). We conclude that Sp SPAC1093.04 possesses a distinctive C addition activity.

(a) SPAC1093.04 adds tails enriched for polycytosine. Left, tail-o-gram depicting nucleotide composition in each added tail length and number of tails normalized to unique heptamer sequences. Right, most abundant tail sequences added to tRNA reporter containing a 3’ CC, or 3’ CCA end. (b) S. cerevisiae Cca1 adds cytosines and adenosines. Left, tail-o-gram depicting nucleotide composition in each added tail length and number of tails normalized to unique heptamer sequences. Right, most abundant tail sequences added to tRNA reporter containing a 3’ CCA end.
(c) Sequence motif effect analysis of tails added by Sp SPAC1093.04 (red) and Sc Cca1 (black). Each adjusted p-value quantifies the significance of contribution of the indicated oligonucleotide to the variation in tail sequence read counts. Significances for dinucleotide (CC) and trinucleotides (CCA) after multiplicity correction with the Bonferroni procedure are shown. A dashed line indicates significance level 0.05. The −log10 p-values from left to right in the figure are 300, 148, 0.87, and 313.
(d) Expression of both SPAC1093.04 and SPCC645.10, but of neither protein alone, rescues cca1-1 temperature sensitivity. cca1-1 mutant strains containing CEN plasmids expressing indicated plasmids were serially diluted, spotted on SD-Ura-Leu media and grown at 37°C for 3 days or 23°C for 4 days.
The S. pombe genome encodes a second enzyme (SPCC645.10) with sequence similarity to CCA-adding enzymes. This enzyme yielded tails of almost entirely adenosines (Fig. 2c, 96.3%, s.d. 0.7%). Thus, we wondered whether Sp SPAC1093.04 and Sp SPCC645.04 might act sequentially to add CCA to tRNAs, with Sp SPAC1093.04 first adding two C’s and then Sp SPCC645.10 adding the terminal A. The use of two enzymes to add CCA has not previously been demonstrated in eukaryotes, though it occurs in certain bacteria55,56.
To test our hypothesis, we determined whether the two S. pombe genes, SPAC1093.04 and SPCC645.04, could rescue lethality due to loss of CCA-adding activity in S. cerevisiae. We used a cca1-1 mutant strain of S. cerevisiae strain bearing a temperature-sensitive (ts) allele of the essential CCA1 gene. CCA1 encodes the single protein that adds CCA to tRNAs in S. cerevisiae57,58. SPAC1093.04 and SPCC645.10 were expressed in the cca1-1 strain using the CCA1 promoter and terminator sequences on single-copy plasmids. Effects on temperature sensitivity were assessed in strains expressing the S. pombe proteins either together or with an empty vector (Fig. 3d, Fig. S4).
Coexpression of both S. pombe enzymes rescued loss of endogenous CCA addition activity in S cerevisiae. cca1-1 temperature sensitivity at 37°C was fully rescued by co-expression of SPAC1093.04 and SPCC645.10, and by the wild-type CCA1 positive control. Expression of SPAC1093.04 alone only partially suppressed the cca1-1 ts phenotype. Expression of SPCC645.10 alone or catalytic-inactive versions of SPAC1093.04 and SPCC645.10 failed to rescue the temperature sensitivity. Thus, our data suggest that SPAC1093.04 and SPCC645.10 collaborate to add CCA to tRNAs to rescue the cca1-1 ts phenotype. We propose that this collaboration is also necessary for CCA addition to tRNAs in S. pombe because both enzymes are essential59,60. To our knowledge, this would be the first dual enzyme system that adds CCA to tRNAs in a eukaryote.
An enzyme with broad specificity
C. elegans F31C3.2 displayed a uniquely broad nucleotide specificity (Fig. 2b, Fig. 4a). The majority of nucleotides added by the enzyme were adenosines and uridines, but guanosines and cytosines were also prominent (Fig. 4b). Analysis of enrichment of short oligonucleotide sequences within the added tails yielded no discernible pattern or sequence motif (of p-value less than 0.05). This is further emphasized by computational analysis of all 16 possible dinucleotide sequences, none of which displays statistically significant enrichment among the added tails (Fig. S5). The base composition of the added tails paralleled intracellular ribonucleotide concentrations in S. cerevisiae61 (Fig. 4b). Taken together with the random nature of the sequences added, we suggest that Ce F31C3.2 may be relatively indiscriminate in its nucleotide preference. Hereafter, we refer to Ce F31C3.2 as nucleotide polymerase-1 (NPOL-1).

(a) Top, tail-o-gram of Ce F31C3.2 activity. Bottom, example tail sequences added by Ce F31C3.2. (b) Ce F31C3.2 addition vs. measured rNTP concentrations in yeast61.
A poly(UG) polymerase required for RNA silencing
C. elegans MUT-2 protein yielded tails with a 1:1 ratio of uridines and guanosines (Fig. 2b, Fig. 5a). Surprisingly, we found that Ce MUT-2 added alternating U and G nucleotides, yielding striking, polymeric sequences of alternating U and G (Fig. 5b). Computational analysis confirmed repetitive UG addition, and revealed that tails began with either uridine or guanosine. Of the two predicted splicing isoforms of Ce MUT-2 (mut-2a, mut-2b, https://wormbase.org/species/c_elegans/gene/WBGene00003499#0-1-3), only MUT-2a protein exhibited polymerase activity (Fig. 6b). We refer to this enzyme as a poly(UG) polymerase.

(a) Tail-o-gram depicting Ce MUT-2 nucleotide addition activity in yeast. (b) The most abundant tail sequences identified in two biological replicates of Ce MUT-2 TRAID-Seq assays. (c) Schematic of experiment to test Ce MUT-2 activity on RNase P RNA reporter in yeast. (d) Ce MUT-2 adds UG repeats to RNase P RNA in three biological replicates, regardless of the 3’ end sequence of the reporter RNA. (e) Schematic of experiment to test Ce MUT-2 activity in Xenopus laevis oocytes. (f) UG tail sequences from two biological replicates of Ce MUT-2 activity in Xenopus laevis oocytes. From replicate 1, we cloned 43 independent reporter sequences, 16 had added tails, and all contained UG. From replicate 2, we cloned 31 independent reporter sequences, 11 had added tails, and all contained UG. (g) Statistical analysis of all possible dinucleotides in the tails added by MUT-2. A heatmap of p-values for individual dinucleotides with minus logarithm (base 10) is shown. Each p-value quantifies the significance of adjusted contribution of each dinucleotide to the variation in tail sequence read counts. Dinucleotides with a significant effect after multiplicity correction at significance level 0.05 are marked with an asterisk (*).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(a) Schematic of Ce MUT-2 isoforms and tested mutations, known catalytic mutants (pink), mutants identified in forward genetic screen35 (blue). NTD, Nucleotidyl transferase domain; PAPd, Poly(A) polymerase-associated domain. (b) Percent of nucleotides added by each Ce MUT-2 enzyme variant. Percent of tails containing UG repeats, standard deviation, and number of biological replicates are indicated. (c) Model depicting the potential roles of poly(UG) tails in small RNA amplification in C. elegans. Poly(UG) tails could directly recruit RNA-dependent RNA polymerase (RdRP) for small RNA amplification (left). Alternatively, poly(UG) tails could be identified by a poly(UG) binding protein (UG-BP), which then recruits an RdRP (right).
To test whether this unusual specificity was independent of the RNA substrate, we used a different RNA, derived from RNase P RNA (Fig. 5c, Fig. S1). This RNA neither had a CCA 3’ end nor resembled a tRNA. Ce MUT-2 again added tandem UG repeats, as demonstrated by representative sequences from three biological replicates (Fig. 5d). Indeed, it added alternating UG to any of the multiple termini formed on the RNase P reporter RNA.
To further examine whether addition of UG repeats was intrinsic to the protein, we tested Ce MUT-2 in a different organism and cell type – Xenopus laevis oocytes. Ce MUT-2 was expressed in X. laevis oocytes by microinjecting an mRNA encoding Ce MUT-2a fused to MS2 coat protein. After allowing the protein to accumulate, an RNA containing a polylinker and three MS2 loops was injected (plgMS2-luc, Fig. 5e). Untemplated nucleotides were detected on 35-37% of the reporter RNA molecules, and all of these tails contained UG repeats, most commonly tandem UG repeats (Fig. 5f). We also observed a few instances of short uridine stretches, perhaps due to endogenous Xenopus TUT4 or TUT7 poly(U) polymerase activity on reporter RNAs containing UG repeats.
To evaluate the data statistically, we compiled the sequences of Ce MUT-2-catalyzed tails from all TRAID-Seq experiments and examined the statistical significance of the occurrence of each of the possible 16 dinucleotide pairs (Fig. 5g). 5’ -GU-3’ and 5’ -UG-3’ were highly enriched, with −log10 (p-values) of 7.3 and 6.2. We conclude that Ce MUT-2 catalyzes the addition of alternating UG. To our knowledge, Ce MUT-2 is the first example of a poly(UG) polymerase. The UG repeats are essentially perfectly repeated throughout the tails added, a remarkable pattern not observed in sequences added by other known nucleotidyl transferases.
The UG-adding activity of Ce MUT-2 likely is critical for RNAi. Ce MUT-2 was first identified in a screen for mutants with increased frequency of transposon Tc1 excision in the C. elegans germline27. The same gene was later identified in a forward genetic screen designed to detect genes involved in the efficacy of “feeding RNAi” in C. elegans, and so was referred to as RDE-3 (“RNAi-defective”)35. Ce MUT-2 has since been implicated in the production of secondary small RNAs (22G RNAs)35,37. The original RNAi-defective screen yielded six independent alleles, each of which alters a region predicted to be important for catalytic activity (Fig. 6a). We tested the nucleotide addition activities of MUT-2 fusion proteins that correspond to each of these mutants, as well as a negative control D105A/D107A (DADA) in which two predicted catalytic aspartates were mutated to alanine (Fig. 6b). All of the Ce MUT-2 mutations identified previously35 lacked UG addition activity, and the nucleotide compositions of any tails added resembled the negative control and the catalytically inactive enzyme (DADA mutant). Because C. elegans mutant strains harboring these same alleles are defective for multiple forms of RNA silencing and for secondary small RNA production in RNAi interference, we propose that poly(UG) polymerase activity is important in those events.
DISCUSSION
TRAID-Seq is facile, sensitive, and enables parallel analyses of many rNTases. Thousands of independently captured tail sequences are identified, permitting sensitive determinations of their sequences and abundances, and thus the identity and patterns of nucleotides added. Since neither the protein nor its RNA product need to be purified, multiple proteins can be analyzed in parallel and thousands of tail sequences determined precisely. While we tested proteins identified through their sequence similarity to rNTases, the approach could be applied to a much broader range of ORFs, and identify enzymes that catalyze any RNA modifications detectable through sequencing, including certain base modifications.
Our analyses reveal the activities of 19 previously uncharacterized members of the rNTase protein family, from six species. The active site regions of the PAPs and PUPs we identified bear on how U and A are distinguished by different enzymes in the same family. Prior work demonstrated that a histidine in the active-site regions of Human Gld2 and S. pombe Cid1 dictates their apparent preferences for A and U, respectively62-67. Similarly, U-adding enzymes appear to have arisen repeatedly in evolution by the insertion of histidine into ancestral A-adding enzymes62. However, among the U-adding enzymes we uncovered here, several (Sp Cid16, Ce PUP-3 and Ce F43E2.1) lack that histidine, and one that possesses a histidine (Hs TUT1/Star-PAP) adds adenosines21 (Table 1). Both Ce NPOL-1, the broad specificity NTase, and Ce MUT-2, which adds alternating U and G, possess a histidine, further emphasizing that purines can be accommodated. These findings illustrate that the basis of nucleotide discrimination is more complex than previously thought. Analysis of the structures of these enzymes bound to their nucleotide substrates should be illuminating.
Our findings suggest that protein partners or small molecules may contribute to the specificities of certain nucleotidyl transferases. In vivo, Hs TUT1 (also known as Star-PAP) adds U’s to U6-snRNA51, but adds A’s to a variety of mRNAs21,68,69. In TRAID-Seq, we detected a strong preference for A (adenosine= 89.5%, s.d. 1.4%), and only low levels of incorporation of other nucleotides (uridine=3.2%, s.d. 0.7%; cytosine= 2.5%, s.d. 0.4%; guanosine=4.8%, s.d. 0.6%). A specific phosphoinositide enhances A addition activity of this enzyme in vitro21, and may underlie these differences. Aspergillus (An) CutA adds CU-rich 3’ terminal extensions to RNAs in vivo and prefers CTP in vitro70,71. In TRAID-Seq, An CutA added predominantly adenosine (91.8%, s.d. 0.4%) vs C (5.9%, s.d. 0.2%) or U (1.9%, s.d. 0.3%). In vivo in Aspergillus, An CutB collaborates with An CutA to form CU-rich tails72 but added virtually all A’s in TRAID-Seq [98.7%, s.d. 0.2% vs. C (0.4%, s.d. 0.05%) or U (0.3%, s.d. 0.07%)]. These findings suggest that additional cofactors or the nature of the RNA substrates can influence specificity in vivo.
The sensitivity of TRAID-Seq revealed previously undetected nucleotide addition capabilities that may underlie the addition of in vivo tails that have been enigmatic. For example, three human PAPs (TENT2, TENT4b, and TUT1) are capable of G addition, albeit at a low level in our system (Fig. 2a), and could explain the observation of G addition on mRNAs in human cells24. Indeed, TENT4a and TENT4b were recently implicated in G addition to mRNAs, which then are protected from deadenylation73. Perhaps the ability of several human PAPs to add G’s might indicate that other classes of RNAs are subject to such regulation. The abilities of Sp Cid13 and Sp Cid14 to add C and G, respectively, in addition to A, and suggests an analogous mechanism of RNA regulation in S. pombe.
C. elegans NPOL-1 added tails composed of all four nucleotides without a discernible sequence pattern, and is distinct in specificity from the other enzymes tested. The levels of incorporation mirror intracellular concentrations of ribonucleoside triphosphate concentrations, which may determine the proportions of nucleotides added. The broad specificity of Ce NPOL-1 echoes the activities of terminal deoxynucleotidyl transferase (TdT) and E. coli poly(A) polymerase (EcPAP), which also can add all four nucleotides without a template74-76. However, NPOL-1 diverges in sequence from TdT and EcPAP, which belong to a different subfamily of nucleotidyl transferases19. Indeed, the closest ortholog of NPOL-1 is human TENT2 (aka GLD2/PAPD4; 37% sequence homology; https://wormbase.org/species/c_elegans/gene/WBGene00001596#0-1-3). The addition of random nucleotides within, or at the end of, homopolymeric tails could interfere with their function26,73,77. It will be of interest to test whether binding partners, RNA substrates, or cofactors alter the nucleotide preferences of NPOL-1, as has been observed with Hs TUT1/Star-PAP69.
We propose that SPAC1093.04 and SPCC645.10 constitute a two-enzyme system that catalyzes CCA addition to tRNAs in S. pombe. This is strongly suggested by their specificities and ability to jointly complement a S. cerevisiae strain lacking a functional CCA-adding enzyme. This would be the first report of a two-enzyme CCA addition system in a eukaryote. Studies in S. pombe will test this proposal in its natural context, and determine whether either these enzymes act on other RNAs as well.
MUT-2, the poly(UG) polymerase, is remarkable both in its enzyme activity and roles in RNA biology. Its capacity to polymerize tails composed of as many as 18 perfect UG repeats is striking. Even longer UG tails likely were present, but were undetected due to sequencing read limitations. Alternating U and G addition bears comparison to that of CCA-adding enzymes, which switch nucleotide specificities as they sequentially add C, C and then A to a tRNA. They do so through a single active site, repositioning the growing 3’ end relative to the enzyme78-82. Redesign of the polarity of hydrogen bonds in a CCA-adding enzyme enable it to add UUG to a tRNA substrate in vitro76 and two CCAs can be added by shifting the 3’ end relative to the protein83,84. Repetitive UG addition by Ce MUT-2 may be promoted by repositioning the 3’ -most UG relative to the Ce MUT-2 active site.
The functions of Ce MUT-2 in vivo are diverse. mut-2 was first isolated in a genetic screen for elevated transposition frequency in C. elegans27, and later in a screen for mutants with impaired RNAi in response to exogenous double-stranded RNA35. mut-2 mutants possess reduced levels of secondary small RNAs35,37,39 (22G and 26G RNAs), suggesting that the protein stabilizes or helps to generate them. Ce MUT-2 function in vivo likely hinges on its poly(UG) polymerase activity, since the mutations identified in RNAi-defective mut-2 mutants abrogate poly(UG) polymerase activity in our assays (Fig. 6a,b).
The multiple roles of Ce MUT-2 – preserving genome integrity27-29, silencing transgenes30-34 and promoting RNAi due to exogenous dsRNA35-39 – all likely reflect the same underlying molecular mechanisms. MUT-2 increases the abundance of secondary RNAs during RNAi, suggesting that UG tails are important in RdRP-based secondary siRNA synthesis or stabilization35,37. In one simple model, MUT-2 adds poly(UG) to the 3’ end of sliced RNAs generated in an Ago-dependent process. The poly(UG) tails would then provide a distinctive mark on sliced RNAs and bind RdRP directly, or via a separate UG-binding protein (Fig 6c). In either case, the tail could be single-stranded, or, as we favor, form a more complex structure involving U-G, U-U, or G-G pairing interactions (depicted as UG pairing in Fig. 6c, left). By recruiting RdRP enzymes to amplify siRNA pools, and perhaps by directly stabilizing sliced RNAs, poly(UG) tails could promote long-term gene silencing known to occur in C. elegans85-88. Regardless, identification of the natural RNA targets of MUT-2 should provide a powerful entree into the breadth and biological roles of poly(UG) polymerases and poly(UG) tails.
MATERIALS AND METHODS
Plasmid Construction
To enable overexpression of rNTases as MS2 coat protein fusions in S. cerevisiae, the MAP72 MS2 cassette vector was constructed. YEplac 181 (LEU2 2μ)89 was digested with HindIII and XhoI. Then each portion of the MS2 cassette was subcloned with unique restrictions sites, resulting in the following insert: S. cerevisiae TEF1 promoter, MS2 coat protein, a multiple cloning site to insert the rNTase to test (consisting of BamHI, XmaI/SmaI, NotI, XbaI, PstI, and KpnI sites), SV40 nuclear localization signal, an RGS(H6) sequence to verify rNTase expression by Western blotting, and S. cerevisiae ADH1 terminator sequence.
Each rNTase tested was cloned into MAP72 by amplifying the genes indicated in Table S1 using the primers listed. All inserts were sequenced to confirm identity and lack of mutations. Site-directed mutations were made using standard methods with oligomers corresponding to the mutated sequences.
The tRNA reporter was constructed using a tRNAHis expression cassette, MAB812A90. tRNAHis sequence was removed by digestion with XhoI and BglII. Then DNA corresponding to the tRNA reporter sequence was inserted by annealing overlapping oligomers to construct both strands of the DNA sequence. The tRNA reporter is an S. cerevisiae tRNASer(AGA) altered to contain an MS2 stem loop sequence (underlined) in place of the endogenous tRNASer(AGA) variable arm (5’ - GGCAACTTGGCCGAGTGGTTAAGGCGAAAGATTAGAAATCTTTACATGAGGATCACCCATGTCGC AGGTTCGAGTCCTGCAGTTGTCG-3’).
A CCA1 cassette vector was constructed using YCplac 111 (LEU2 CEN)89 in order to express CCA1, SPAC1093.04, or SPCC645.10 with the same promoter and C-terminal epitope tag [RGS(H)6]. BY4741 yeast genomic DNA was used as a template to generate an amplicon consisting of LEU2 CEN vector sequence at the 5’ end, the CCA1 promoter sequence, and a 3’ terminal sequence corresponding to the multiple cloning site of MAP72 using 5’ -GAAACAGCTATGACCATGATTACGCCAAGCTTACTAGTAGCTACTTCAGGGACAAGCAAC-3’, and 5’ - ACCCTGCAGTCTAGAAGGCGGCCGCGTGGATCCACACAAAAAAAGCCCTTATAACCCACG-3’. MAP72 was used as a template to generate an amplicon consisting of the multiple cloning site, RGS(H6) sequence, ADH1 terminator sequence of MAP72, and LEU2 CEN vector sequence at the 3’ end using 5’ -GGATCCACGCGGCCGCCTTCTAGACTGCAGGGTACCAGAGGTTCTCACCACCACCACCAC-3’ and 5’ - CCAGTCACGACGTTGTAAAACGACGGCCAGTGAATTCCTCGAGCGGTAGAGGTGTGGTCA-3’. These two amplicons were combined with LEU2 CEN vector (YCplac111) linearized with PstI/SacI and assembled by Gibson cloning91. The CCA1 cassette sequence was confirmed by Sanger sequencing. CCA1, SPAC1093.04, or SPCC645.10 sequences were subcloned from their respective MAP72-based constructs into the CCA1 cassette for expression in cca1-1 yeast.
To construct the MAP136 MUT-2 oocyte expression vector (pCS2 3HA MS2-MUT-2 WT), MUT-2a was PCR-amplified from its MAP72-based vector using 5’ -CTACCATGGATGGCTTCTAACTTTACTCAGTTCGTTCTCGTCGAC-3’ and 5’ -ACTCTCGAGTTAGTGGTGGTGGTGGTGGTGAGAACCTCTGGTACCCTGCAGTACAAATGA-3’ and then cloned into the NcoI/XhoI site of pCS2 3HA MS2. MUT-2 DNA sequence was verified prior to oocyte injections.
Yeast Growth
BY4741 yeast were co-transformed using standard methods92 with a plasmid expressing the reporter RNA and a plasmid expressing the rNTase of interest, or vector controls, and selected on synthetic yeast medium lacking uracil and leucine (SD-Ura-Leu). Cultures were inoculated with single colonies, grown to saturation, and then diluted to 0.1 OD600/mL and grown to log phase (0.8-1 OD600/mL). Cells were spun down in pellets of 25 OD600 (approximately 5 x 108 cells) and stored at −80°C until RNA extraction or protein expression analysis. We performed Western blotting with mouse anti-RGS-His Antibody (1:2500 dilution, 5PRIME/Qiagen). Only those samples with clear expression of the rNTase fusion protein were analyzed by high-throughput sequencing.
cca1-1 yeast were co-transformed with vectors as listed in Figs. 3 and S3 using standard methods92, and selected on SD-Ura-Leu plates at room temperature. Colonies were selected and grown to saturation in SD-Ura-Leu liquid media. Cultures were diluted to 0.5 OD/mL followed by three 10-fold serial dilutions, spotted on SD-Ura-Leu plates, and incubated at room temperature (23°C) for 4 days or 37°C for 3 days.
RNA Extraction
RNA was extracted from 25 OD of yeast corresponding to each sample by modification of a previously described method93. To each sample, 0.5 g of 0.5 mm acid washed beads (Sigma-Aldrich), 0.5 mL of RNA ISO buffer (500 mM NaCl, 200 mM Tris-Cl pH 7.5, 10 mM EDTA, 1 % SDS) and 0.5 mL of phenol-chloroform-isoamyl alcohol pH 6.7 (PCA, Fisher Scientific) was added. Samples were lysed with 10 cycles that each consisted of vortexing for 20 seconds and incubation on ice for 30 seconds. 1.5 volumes (relative to starting amount of ISO Buffer) of RNA ISO Buffer and of PCA were added, and samples were centrifuged at 4°C to separate phases. The aqueous layer was transferred to a pre-spun phase-lock gel (heavy) tube (5PRIME/Quantabio); an equal volume of PCA was added and mixed prior to centrifugation at room temperature to separate phases. The aqueous layer was transferred to 2 new tubes for ethanol precipitation with 2 volumes of 100% ethanol followed incubation at −80°C for 1 hour to overnight. Precipitated RNA was pelleted by centrifugation at 4°C. Each pellet was dissolved in 25 μL nuclease-free water and combined into 1 tube per sample. Co-purifying DNA was digested with 20 U of Turbo DNase (Invitrogen) at 37°C for 4 hours, and RNA was cleaned up with the GeneJET RNA Purification Kit (Thermo Scientific), and eluted with 50 μL of DEPC-treated water.
RT-PCR Experiments
RT-PCR experiments to detect A tails or U tails on an RNase P RNA reporter (see Fig. S1) were performed by using a tail-specific reverse transcription step with 5 pmol of a T33 or A33 DNA primer and 100 ng of total RNA using ImProm-II Reverse Transcriptase (Promega Corporation). Then the resulting reactions were PCR-amplified using reporter-specific primers (5’ - TCGAGCCCGGGCAGCTTGCATGC-3’ and 5’ - GGGAATTCCGATCCTCTAGAGTC-3’). If a tail was added to the RNase P RNA reporter, then the RT reaction would produce cDNA, and the PCR would result in an amplicon.
RT-PCR experiments to detect tails added to the tRNA reporter were performed as described with the RNase P RNA reporter but with the following modifications. PCR amplification was performed with a forward primer specific to the 5’ end of the tRNA (5’ -GGCAACTTGGCCGAGTGGTTAAGG-3’) and a reverse primer specific to the 3’ end of the tRNA with an A tail or U tail, respectively: 5’ - AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAATGGCGACAACTGC-3’ or 5’ - TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTGGCGACAACTGC-3’. If a tail was added to the tRNA reporter, then the RT reaction would produce cDNA, and the tail-specific PCR would result in an amplicon.
TRAID-Seq Library Preparation
Total RNA (100 ng) was ligated with 20 pmol of a 5’ adenylated primer containing a 7-nucleotide random DNA sequence (random heptamer), Illumina TruSeq adapter sequence and a 3’ dideoxycytidine (5’ - A(pp) NNNNNNN TGGAATTCTCGGGTGCCAAGG ddC-3’) using 200 U of T4 RNA ligase 2, truncated KQ (New England BioLabs) in a 20μL reaction with 16°C overnight incubation. This ligation added the random heptamer and Illumina TruSeq adapter sequence to the 3’ end of the RNAs in the sample.
Half of the ligation reaction (10 μL) was reverse transcribed using 5 pmol of Illumina RNA RT primer (5’ -GCCTTGGCACCCGAGAATTCCA-3’) and ImProm-II Reverse Transcriptase (Promega Corporation) with 1.5 mM MgCl2 and 0.5 mM dNTPs, according to manufacturer’s instructions.
Samples were then PCR-amplified with a forward primer consisting of Illumina-specific sequences and sequence (underlined), specific to the tRNA reporter (5’ - AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGACGATCGAGGATCACCCATGTCGCAG-3’) and a reverse Illumina RNA PCR Primer with various indices used for multiplexing, using GoTaq Green PCR Master Mix (Promega Corporation). PCR products were run on an 8% polyacrylamide 8M urea gel and gel extracted. Resulting samples for each sequencing run were combined in equimolar amounts and run on an Illumina HiSeq2000 or HiSeq2500 (2×50 bp or 2×100 bp), to produce approximately 1 x 106 reads per sample.
Experiments with the RNase P RNA reporter were performed essentially as described above but with a few modifications. For TRAID-Seq, the 5’ primer used for PCR amplification was specific for the RNase P RNA reporter (5’ - AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGACGATCGTCTGCAGGT CGACTCTAGAAA-3’).
TRAID-Seq Data Analysis
Reads resulting from sequencing of TRAID-Seq samples were analyzed using a group of Python scripts that we call the “puppyTails” program. Briefly, puppyTails identifies sequences corresponding to the tRNA reporter, CCA end of the tRNA, and added tail in read 1. In read 2, the program identifies the random heptamer sequence, added tail sequence, and, if read length allows, the CCA end and tRNA reporter sequence. Reads were collapsed into unique ligation events using the random heptamer and then compared to identify and remove sequences resulting from PCR amplification (PCR duplicates). The number of unique times that each tail sequence is observed is counted. Tail sequences are sorted by length to calculate the nucleotide composition at each tail length and the number of tails per million heptamers (TPMH) measured for each tail length; these data are plotted as tail-o-grams (for example, Fig. 1e-g). A subsequent Perl script was used to calculate the overall nucleotide composition of tails added by a given rNTase, accounting for the number of times that a tail sequence was observed (for example, Fig. 2a-c).
Computational Analyses of Sequence Motifs
To analyze tail sequences, a general feature screening with a random forest application94 was performed at the replicate level. We first quantified the number occurrences of all oligonucleotides (k=1, 2, 3, 4) within each tail sequence and utilized the resulting set of 340 features, as well as the length of the tail. The variable importance, defined as the percent mean decrease in accuracy (with 500 trees, 113 candidate variables at each split, minimum node size of 5), were estimated for all features. We define the selected features those whose importance measures are greater than 4% across replicates. We fitted a Poisson regression model in which the response variable was tail sequence counts.
Tails added by S. cerevisiae Cca1, S. pombe SPAC1093.04, and predicted CCA-adding enzymes
The above selected features were used as covariates. P-values from individual replicates, calculated from one-sided Wald’s test, were aggregated using Fisher’s (n<4) or Wilkinson’s (n>=4) method, followed by multiplicity correction with the Bonferroni procedure. This process identified oligonucleotides that differ between S. cerevisiae Cca1 and S. pombe SPAC1093.04 at level 0.05.
Tails added by C. elegans MUT-2
We evaluated the impacts of 16 dinucleotides by formally testing for their effects by a comparison of a null model without each dinucleotide and the alternative model deduced from random forest filtered set of features plus other dinucleotides. This procedure identified UG and GU as the most significant dinucleotides.
In vitro Transcription
pCS2 3HA MS2-MUT-2 (MAP136) was linearized with SacII, and 3 μg of linearized plasmid was transcribed with Ampliscribe SP6 High Yield Transcription Kit (Epicentre), according to manufacturer’s instructions. pLGMS2-luc (RNA with three MS2-binding sites)16,95 was linearized with BglII, and 1 μg of linearized plasmid was transcribed with T7 Flash In Vitro Transcription Kit (Epicentre), according to manufacturer’s instructions. Transcription reactions included m7G(5’)ppp(5’)G RNA Cap Structure Analog (New England Biolabs).
Tethered Function Assays and Oocyte RNA Extraction
Xenopus laevis oocyte manipulations and injections were performed as in previous studies16,95,96.
Tethered function assays were conducted essentially as previously described22. Briefly, Stage VI oocytes were injected with 50 nL of 600 ng/µL capped mRNA encoding MS2-HA-MUT-2 protein. After 6 hours, the same oocytes were injected with 50 nL of 3 ng/µL pLGMS2-luc reporter mRNA. After 16 hours, oocytes were collected, lysed, and assayed. Three oocytes were used to confirm protein expression. Total RNA was extracted from oocytes using TRI reagent (Sigma-Aldrich), as described previously22, then treated with 8 U of Turbo DNase (Invitrogen) at 37°C for 1 hour, and cleaned up with the GeneJET RNA Purification Kit (Thermo Scientific).
Oocyte RNA Analysis and Tail Sequencing
Oocyte total RNA (100 ng) was ligated with 20 pmol of the 5’ adenylated primer as described above. This ligation added the random heptamer sequence and a known sequence to the 3’ ends of RNAs in the sample for tail sequence-independent analyses. Half of the ligation reaction (10 μL) was reverse transcribed as described above.
Samples were PCR-amplified with a forward primer specific to the RNA reporter (5’ - CTCTGCAGTCGATAAAGAAAACATGAG-3’) and a reverse primer specific to the known sequence added to the 3’ end of the RNA (5’ - GCCTTGGCACCCGAGAATTCCA-3’), using GoTaq Green PCR Master Mix (Promega Corporation). PCR products were run on a 1.5% agarose gel, and purified with the GeneJET Gel Extraction Kit (Thermo Scientific). Non-templated A overhangs were added by treating the purified PCR products with 10 U of TaqPlus Precision Polymerase Mixture (Agilent Genomics) in TaqPlus Precision buffer supplemented with 0.2 mM dATP at 70°C for 30 minutes. The PCR products were then subjected to cloning with the TOPO TA Cloning Kit for Subcloning (ThermoFisher Scientific) as follows: 6% of the A addition reaction volume (2.4 μL) was combined with 0.6 μL of Salt Solution and 0.7 μL of TOPO Vector and incubated at room temperature for 30 minutes. Reactions were diluted 1 in 4 with water, transformed into DH5α competent cells, and selected on LB agar with 100 μg/mL ampicillin and 75 μg/mL X-Gal for blue/white screening. White colonies were selected, plasmids were extracted, and inserts were sequenced to identify tails added to the reporter. All reporter sequences with added tails are reported in Fig. 5f.
AUTHOR CONTRIBUTIONS
M.A.P. and M.W. designed experiments; M.A.P. performed the experiments and analyzed data unless otherwise noted. D.F.P. wrote the PuppyTails program used to analyze TRAID-Seq data, including “tailograms.” F.C. performed statistical analyses of tail sequence motifs. N.B. prepared N. crassa and C. albicans TRAID-Seq samples. C.P.L wrote the Perl script used to determine total nucleotide incorporation. M.A.P. and M.W. wrote the paper, with contributions from all authors.
ACKNOWLEDGEMENTS
We are very grateful to the Wickens and Kimble labs for advice throughout the work. We are particularly grateful to Scott Kennedy for discussions concerning MUT-2 and the manuscript, and Anita Hopper and Eric Phizicky for advice on S. pombe CCA-adding enzymes. We thank the Kennedy and Anderson labs for discussions. We thank the University of Wisconsin Biotechnology Center DNA Sequencing Facility, especially Marie Adams and Michael Sussmann, for providing Illumina sequencing facilities and services. We acknowledge Michael Harte for assistance with cloning S. pombe rNTases. We are grateful to Laura Vanderploeg of the UW Biochemistry Media Laboratory for help with the figures. This work was supported by a Ruth Kirschstein National Research Service Award (1F32GM103130-01A1) to M.A.P. and an NIH Grant to M.W (GM50942).
REFERENCES




