

Abstract
Malaria parasites (Plasmodium spp.) contain a nonphotosynthetic plastid organelle called the apicoplast, which houses essential metabolic pathways and is required throughout the parasite life cycle. Hundreds of proteins are imported across 4 membranes into the apicoplast to support its function and biogenesis. The machinery that mediates this import process is distinct from proteins in the human host and may serve as ideal drug targets. However, a significant concern is whether inhibition of apicoplast protein import will result in a “delayed-death” phenotype that limits clinical use, as observed for inhibitors of apicoplast housekeeping pathways. To assess the growth inhibition kinetics of disrupting apicoplast protein import, we targeted a murine dihydrofolate reductase (mDHFR) domain, which is stabilized by the compound WR99210, to the apicoplast to enable inducible blocking of apicoplast-localized protein translocons. We show that stabilization of this apicoplast-targeted mDHFR disrupts parasite growth within a single lytic cycle in an apicoplast-specific manner. Consistent with inhibition of apicoplast protein import, stabilization of this fusion protein disrupted transit peptide processing of endogenous apicoplast proteins and caused defects in apicoplast biogenesis. These results indicate that disruption of apicoplast protein import avoids delayed-death growth inhibition and that target-based approaches to develop inhibitors of import machinery may yield viable next-generation antimalarials.
Importance Malaria is a major cause of global childhood mortality. To sustain progress in disease control made in the last decade, new antimalarial therapies are needed to combat emerging drug resistance. Malaria parasites contain a relict chloroplast called the apicoplast, which harbors new targets for drug discovery, including import machinery that transports hundreds of critical proteins into the apicoplast. Unfortunately, some drugs targeting apicoplast pathways show delayed growth inhibition, which results in a slow onset-of-action that precludes their use as fast-acting, frontline therapies. We used a chemical biology approach to disrupt apicoplast protein import and showed that chemical disruption of this pathway avoids delayed growth inhibition. Our finding indicates that prioritization of proteins involved in apicoplast protein import for target-based drug discovery efforts may aid in the development of novel fast-acting antimalarials.
Introduction
Plasmodium spp. parasites cause malaria and are responsible for over 200 million human infections and over 400,000 deaths annually (1). Despite a reduction in malaria-related mortality in the past 15 years, emerging resistance to frontline antimalarials necessitates the continued development of new chemotherapies (2, 3). One key source of drug targets is the apicoplast, a nonphotosynthetic plastid organelle found in many apicomplexan pathogens (4, 5). The apicoplast produces essential metabolites required for parasite replication (6). Apicoplast function requires the import of over 300 nuclear-encoded gene products into the organelle, including biosynthetic enzymes and pathways that support organelle biogenesis and maintenance (7). Most apicoplast proteins are 1) synthesized on cytosolic ribosomes, 2) trafficked to the apicoplast via the endoplasmic reticulum (ER), and 3) translocated across the 4 apicoplast membranes. This multistep import pathway involves more than a dozen proteins, including homologs of the translocation and ubiquitylation machinery typically involved in ER-associated degradation (ERAD) (8-12) as well as homologs of the TOC and TIC machinery found in plant plastids (9, 13-15).
Apicoplast protein import machinery are potential drug targets, but a key unresolved question is whether inhibition of apicoplast protein import causes a “delayed-death” phenotype that has been observed for inhibitors of apicoplast housekeeping functions (16, 17). During delayed death, parasite growth is unaffected during the first lytic cycle of inhibitor treatment but is severely inhibited in the second lytic cycle even after drug removal. This in vitro phenotype manifests as a slow onset-of-action that limits clinical use of these drugs. Unfortunately, there are no inhibitors known to act directly on apicoplast protein import, precluding direct assessment of growth inhibition kinetics. Furthermore, most genetic tools available in Plasmodium parasites act at the DNA or RNA levels (18), which can result in different growth inhibition kinetics than direct chemical inhibition of that same target (19-21). Destabilization domains that conditionally target proteins for degradation by the cytosolic ubiquitin-proteasome enable protein-level disruption (22, 23), but these systems are not suitable to study apicoplast-localized proteins, which are inaccessible to the cytosolic proteasome.
Given these limitations, we developed a protein-level tool to dissect growth inhibition kinetics following disruption of apicoplast protein import. A murine dihydrofolate reductase (mDHFR) domain which can be conditionally stabilized by a small molecule has been used to characterize protein translocation steps during yeast mitochondrial protein import in vitro (24) and, more recently, during export of malarial proteins across the parasitophorous vacuole (PV) membrane into the host red blood cell (25, 26). Strikingly, in the context of malarial protein export, mDHFR stabilized with the compound WR99210 can block the translocon to prevent export of other Plasmodium proteins and disrupt parasite growth (27). Given that there are at least three putative translocation steps during apicoplast protein import (7), we reasoned that we could use a similar strategy to block apicoplast translocons and disrupt protein import in a manner that resembles inhibition with a small molecule (Fig. 1A).

(A) Model for protein-level disruption of apicoplast protein import by a conditional stabilization domain. In the absence of WR99210, an apicoplast-targeted mDHFR domain successfully targets to the apicoplast lumen along with endogenous apicoplast proteins. Addition of WR99210 stabilizes the mDHFR domain to prevent unfolding, which causes the domain to stall during translocation across one or multiple apicoplast membranes and block import of essential endogenous apicoplast cargo. (B) Schematic (not to scale) of constructs targeting GFP-mDHFR to the apicoplast via the ACP leader sequence or to the PV via a mutant ACP leader sequence. (C) Live-cell imaging of GFP-mDHFR fusions. Nuclei were stained with Hoechst. Brightness/contrast adjustments were not held constant between the two cell lines due to differences in GFP fluorescence intensity in the apicoplast versus the PV. Scale bars, 5 µm.
Results and Discussion
To generate a protein-level conditional tool for disruption of apicoplast protein import, we targeted a GFP-mDHFR fusion to the apicoplast via the N-terminal leader sequence of acyl carrier protein (ACP) (Fig. 1B). As a negative control, we generated a cell line expressing the same fusion with a lysine 18 to glutamate (K18E) mutation in the ACP leader sequence that renders this transit peptide nonfunctional and causes mistargeting to the PV (28). Both GFP-mDHFR fusions were expressed in P. falciparum Dd2attB parasites (29) and localized to the expected compartments (Fig. 1C).
To test whether stabilization of these GFP-mDHFR fusions disrupted parasite growth, we treated ring-stage parasites with increasing doses of WR99210 and assessed parasitemia after 3 days as a read-out for parasite growth inhibition during the first lytic cycle. Parental Dd2attB parasites, which express a WR99210-resistant human DHFR allele, were unaffected at the WR99210 concentrations tested (Fig. 2A). Parasites expressing the PV-localized ACPL(K18E)-GFP-mDHFR fusion from the attB site were also insensitive to WR99210. In contrast, parasites expressing the apicoplast-targeted ACPL-GFP-mDHFR fusion showed dose-dependent growth inhibition in response to WR99210 (Fig. 2A).

(A) Growth of parental Dd2attB, ACPL-GFP-mDHFR, and ACPL(K18E)-GFP-mDHFR parasites after 3 days in response to increasing doses of WR99210. ACPL-GFP-mDHFR parasites were assayed in both the presence and absence of 200 µM IPP. (B) Growth of ACPL-GFP-mDHFR parasites in the presence of 10 nM WR99210 over a 5-day time course. Parasites were grown in the presence or absence of 200 µM IPP, and parasitemia was normalized to an untreated control at each time point. Error bars in both panels represent standard deviation of the mean of 3 biological replicates. Biological replicates in (A) were performed in technical triplicate.
During in vitro culture of blood-stage P. falciparum, biosynthesis of the isoprenoid precursor isopentenyl pyrophosphate (IPP) is the only essential function of the apicoplast. As such, supplementation with exogenous IPP can rescue apicoplast defects and can be used to identify apicoplast-specific phenotypes (30). IPP supplementation reversed the WR99210 sensitivity of parasites expressing ACPL-GFP-mDHFR in both a 3-day dose-response experiment and over a 5-day time course (Fig. 2A and B). These results indicate that the WR99210 sensitivity conferred by ACPL-GFP-mDHFR is due to disruption of an apicoplast-specific pathway.
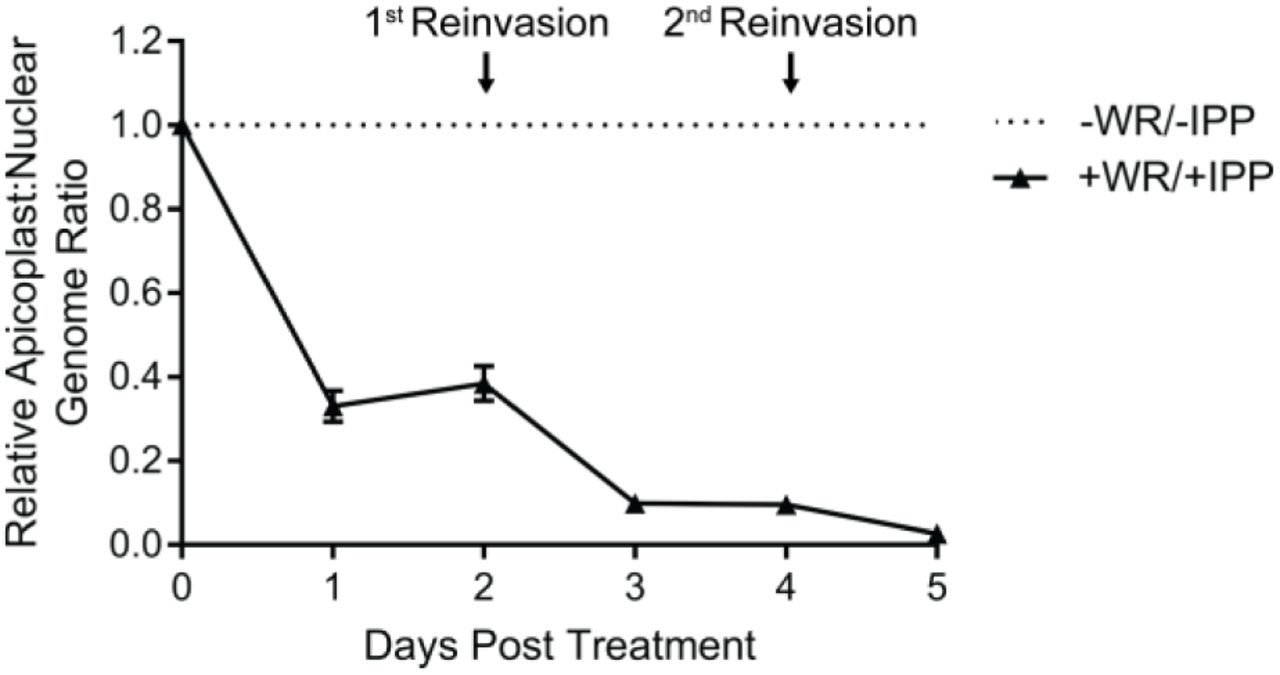
Given our hypothesized mechanism-of-action inhibiting apicoplast protein import (Fig. 1A), we expected that ACPL-GFP-mDHFR stabilization would cause global disruption of the apicoplast, including organelle maintenance and biogenesis, leading to apicoplast loss. To assess the status of the apicoplast, we assayed the presence of the apicoplast genome in WR99210-treated, IPP-rescued parasites by quantitative PCR (qPCR). WR99210 treatment caused a decrease in the apicoplast:nuclear genome ratio beginning at 1 day post-treatment and near-complete loss of the apicoplast genome after 2 lytic cycles (Fig. 3). The loss of the apicoplast genome, a key marker of the apicoplast, indicates that chemical stabilization of ACPL-GFP-mDHFR-expressing parasites caused apicoplast loss.

The relative apicoplast:nuclear genome ratio of ACPL-GFP-mDHFR parasites grown in 10 nM WR99210 and 200 µM IPP was measured by qPCR over 5 days of treatment. Values are normalized to an untreated control at each time point. Error bars represent standard deviation of the mean of 3 biological replicates, each analyzed in technical triplicate.
Sorting of apicoplast cargo occurs in the parasite endomembrane system, after which proteins are thought to traffic to the apicoplast by vesicle transport. While we expected that stabilization of ACPL-GFP-mDHFR would disrupt transit of apicoplast cargo across the apicoplast-localized ERAD-and TOC/TIC translocons, there was a possibility that this stabilized fusion protein could disrupt the upstream sorting process and prevent apicoplast proteins from ever reaching the organelle. Previous data suggest that defects in sorting apicoplast cargo manifest as mistargeting of apicoplast proteins to the PV (28). To assess whether stabilization of ACPL-GFP-mDHFR blocked protein import early during sorting or later after arrival at the apicoplast, we used live-and fixed-cell imaging to assess whether this fusion protein and the endogenous apicoplast protein ACP were successfully sorted in WR99210-treated parasites. As expected, in untreated parasites ACPL-GFP-mDHFR localized to an elongated structure characteristic of the apicoplast and co-localized with ACP (Fig. 4A and B). After 1 day of WR99210 treatment (i.e., within the same lytic cycle as treatment), the majority of cells exhibited ACPL-GFP-mDHFR and ACP signal in a single punctum or elongated pattern that likely indicates an intact apicoplast (Fig. 4A and B). After 3 days of WR99210 treatment (i.e., after 1 full lytic cycle of treatment), both ACPL-GFP-mDHFR and ACP were present almost exclusively in diffuse puncta that are thought to represent vesicles post-sorting in parasites that have lost their apicoplast (30) (Fig. 4A and B). Altogether, the localization of ACPL-GFP-mDHFR and ACP to either organelle-like structures or diffuse puncta following mDHFR stabilization indicates that sorting in the parasite endomembrane system remained intact. This suggests that the defect in WR99210-treated ACPL-GFP-mDHFR parasites occurs after this initial protein sorting step.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACPL-GFP-mDHFR parasites were grown with 10 nM WR99210 and 200 µM IPP for either 1 or 3 days (corresponding to the same lytic cycle or the first lytic cycle post-treatment, respectively). (A) Live imaging of Hoechst-stained parasites. Scale bars, 5 µm. (B) Fixed imaging of parasites stained with an antibody against the endogenous apicoplast marker ACP (recognizing an epitope not present on the ACPL fused to GFP-mDHFR). Scale bars, 5 µm. (C) Western blot to assess transit peptide processing of ACPL-GFP-mDHFR and the endogenous apicoplast protein ClpP. pre-, precursor (unprocessed) protein; m-, mature (processed) protein. (D) Quantification of transit peptide processing for ACPL-GFP-mDHFR and ClpP. Data are expressed as the percentage of total GFP or ClpP signal that is mature (processed). Error bars represent standard deviation of the mean of 3 biological replicates. **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA with Tukey’s multiple comparisons test. ns, not significant.
Most apicoplast-targeted proteins contain an N-terminal transit peptide that is proteolytically processed upon successful import into the organelle lumen, and an accumulation of unprocessed protein can be used as a marker for a protein import defect. We expected that stabilized ACPL-GFP-mDHFR would block translocation of apicoplast cargo into the organelle lumen and cause a transit peptide processing defect in WR99210-treated parasites. We therefore assessed the processing of the ACPL-GFP-mDHFR fusion and the endogenous apicoplast protein ClpP by western blotting. Consistent with a defect in protein translocation, WR99210-treated parasites exhibited a modest but reproducible accumulation of unprocessed ACPL-GFP-mDHFR after 1 day of WR99210 treatment (Fig. 4C and D). ClpP showed a comparable transit peptide processing defect (Fig. 4C and D), suggesting that stabilization of ACPL-GFP-mDHFR affected import not only of the fusion protein itself but also of endogenous apicoplast cargo. Processing of both the GFP-mDHFR fusion and ClpP was almost completely ablated after 3 days of WR99210 treatment (Fig. 4C and D). Because apicoplast proteins are still sorted to the apicoplast (Fig. 4A and B) but show a transit peptide processing defect (Fig. 4C and D), these data are consistent with a disruption in the translocation of apicoplast proteins across one or more apicoplast membranes.
In lieu of direct inhibitors of apicoplast protein import, we used a chemical biology approach to conditionally block apicoplast protein import by addition of a small molecule. Altogether, our results suggest a model in which chemically stabilized ACPL-GFP-mDHFR traffics to the apicoplast and stalls within putative membrane translocons, preventing import of endogenous apicoplast cargo and disrupting apicoplast biogenesis and function. Given that apicoplast protein import is likely required for biogenesis pathways such as apicoplast genome replication, the emergence of both a transit peptide processing defect (a readout for protein import; Fig. 4C and D) and a genome replication defect (Fig. 3) after 1 day of ACPL-GFP-mDHFR stabilization are consistent with this model. Unfortunately, because we could not detect direct biochemical interaction of stabilized ACPL-GFP-mDHFR with apicoplast translocons, we cannot rule out translocation-independent mechanisms-of-action, such as toxicity due to accumulation of stably folded ACPL-GFP-mDHFR in the apicoplast lumen. However, given the previous uses of mDHFR fusions to block protein translocation (24-27) and the defect in apicoplast protein import observed after just 1 day of WR99210 treatment (Fig. 4C and D), our model seems the most parsimonious.
These findings suggest that the proteins required for apicoplast protein import can serve as antimalarial targets that avoid delayed-death growth inhibition. Over a dozen proteins have been implicated in import of nuclear-encoded apicoplast proteins, but of particular interest are proteins related to established drug targets in other systems. For example, the apicoplast-localized AAA ATPase CDC48 is related to mammalian p97 and is likely involved in translocation of apicoplast cargo across the periplastid membrane (12). Notably, mammalian p97, which plays an important role in ERAD and other cellular processes, is of interest as an anti-cancer drug target. Specific inhibitors of mammalian p97 have been developed (31-34), suggesting that the same could be accomplished for the apicoplast-localized CDC48 in apicomplexans. Similarly, components of an apicoplast-localized ubiquitylation system are essential for apicoplast protein import and may also represent potential drug targets (11). A small molecule inhibitor of UAE, a ubiquitin-activating enzyme in humans, has recently been reported to have activity in multiple tumor models (35), indicating that the apicoplast-localized ubiquitin activating enzyme may also be a valuable drug target. The druggability of other members of apicoplast protein import complexes has not been explored and may yield additional targets.
Overall, our data suggest that chemical inhibition of apicoplast protein import machinery may be a viable strategy for development of next-generation antimalarials. Target-based drug discovery efforts against known import machinery may therefore yield specific inhibitors of apicoplast biogenesis with mechanisms-of-action orthogonal to those of current antimalarials.
Materials and Methods
Ethics statement
Human erythrocytes were purchased from the Stanford Blood Center (Stanford, California) to support in vitro P. falciparum cultures. Because erythrocytes were collected from anonymized donors with no access to identifying information, IRB approval was not required. All consent to participate in research was collected by the Stanford Blood Center.
Parasite growth
P. falciparum Dd2attB parasites (MRA-843) were obtained from MR4 and were grown in human erythrocytes (2% hematocrit) obtained from the Stanford Blood Center in RPMI 1640 media (Gibco) supplemented with 0.25% AlbuMAX II (Gibco), 2 g/L sodium bicarbonate, 0.1 mM hypoxanthine (Sigma), 25 mM HEPES, pH 7.4 (Sigma), and 50 μg/L gentamicin (Gold Biotechnology) at 37°C, 5% O2, and 5% CO2.
Vector construction
Oligonucleotides (Table 1) were purchased from the Stanford Protein and Nucleic Acid facility and molecular cloning was performed using In-Fusion cloning (Clontech). GFP and mDHFR were PCR amplified from pARL2-SBP1-mDHFR-GFP (27) using primers MB119/MB120 and MB121/MB122, respectively. These products were simultaneously cloned into the BsiWi/AflII sites of the plasmid pRL2-ACPL-GFP (36) or a similar plasmid containing the ACPL K18E mutant (AAA to GAA codon change) to generate pRL2-ACPL-GFP-mDHFR and pRL2-ACPL(K18E)-GFP-mDHFR for expression of GFP-mDHFR fusions from the mitochondrial ribosomal protein L2 promoter (37).
Primers used in this study.
Parasite transfection
Transfections into Dd2attB parasites were performed using a variation of the spontaneous uptake method (38, 39). Briefly, 50 μg each of pINT (29) and the desired pRL2 plasmid were ethanol precipitated and resuspended in a 0.2 cm electroporation cuvette in 100 μL TE buffer, 100 μL RPMI 1640 containing 10 mM HEPES-NaOH, pH 7.4, and 200 μL packed uninfected erythrocytes. Erythrocytes were pulsed with 8 square wave pulses of 365 V x 1 ms separated by 0.1 s and were allowed to reseal for 1 hour in a 37°C water bath before allowing parasites to invade. Drug selection with 2.5 μg/mL Blasticidin S (Research Products International) was initiated 4 days after transfection.
Microscopy
For live imaging, parasites were settled onto glass-bottomed microwell dishes (MatTek P35G-1.5-14-C) in PBS containing 0.4% glucose and 2 μg/mL Hoechst 33342 stain (ThermoFisher H3570).
For fixed imaging, parasites were processed as previously described (40) with modifications. Briefly, parasites were washed in PBS and fixed in 4% paraformaldehyde (Electron Microscopy Sciences 15710) and 0.0075% glutaraldehyde (Electron Microscopy Sciences 16019) in PBS for 20 minutes. Cells were washed once in PBS, resuspended in PBS, and allowed to settle onto poly-L-lysine-coated coverslips (Corning) for 1 hour. Coverslips were washed once with PBS, permeabilized in 0.1% Triton X-100 in PBS for 10 minutes, and washed twice more in PBS. Coverslips were treated with 0.1 mg/mL sodium borohydride in PBS for 10 minutes, washed once in PBS, and blocked in 5% BSA in PBS. Following blocking, parasites were stained with 1:500 rabbit-α-PfACP (41) diluted in 5% BSA in PBS overnight at 4°C. Coverslips were washed three times in PBS, incubated for 1 hour in 1:3000 donkey-α-rabbit 568 secondary antibody (ThermoFisher A10042) in 5% BSA in PBS, washed three times in PBS, mounted onto slides with ProLong Gold antifade reagent with DAPI (ThermoFisher P36935), and sealed with nail polish prior to imaging.
Cells were imaged with a 100X, 1.35 NA objective on an Olympus IX70 microscope with a DeltaVision system (Applied Precision) controlled with SoftWorx version 4.1.0 and equipped with a CoolSnap-HQ CCD camera (Photometrics). Brightness and contrast were adjusted in Fiji (ImageJ) for display purposes.
Parasite growth assays
For dose-response assays, sorbitol-synchronized ring-stage parasites were grown in 96-well plates containing 2-fold serial dilutions of WR99210 (Jacobus Pharmaceutical Company). After 3 days of growth, parasites were fixed in 1% paraformaldehyde in PBS and were stained with 50 nM YOYO-1 Iodide (ThermoFisher Y3601). Parasitemia was analyzed on a BD Accuri C6 flow cytometer. Each biological replicate of dose-response assays was performed in technical triplicate.
For time course growth experiments, sorbitol-synchronized parasites were untreated or were grown with 10 nM WR99210 with or without 200 μM IPP (Isoprenoids, LLC) for 5 days. Cultures were treated identically in terms of media changes and splitting into fresh erythrocytes. Samples to assess growth were collected daily, fixed in 1% paraformaldehyde in PBS, and stored at 4°C until completion of the experiment. Samples were then stained with YOYO-1 and analyzed as above.
qPCR
Samples for DNA isolation were harvested daily during growth time course experiments. Parasites were released from erythrocytes by treatment with 0.1% saponin, washed in PBS, and stored at −80°C until analysis. Total parasite DNA was isolated using the DNeasy Blood & Tissue kit (Qiagen). qPCR was performed using Power SYBR Green PCR Master Mix (Thermo Fisher) with 0.15 µM each CHT1 F and CHT1 R primers targeting the nuclear gene chitinase or TufA F and TufA R primers targeting the apicoplast gene elongation factor Tu (30). qPCR was performed on Applied Biosystems 7900HT or ViiA 7 Real-Time PCR systems with the following thermocycling conditions: initial denaturation 95°C/10 minutes; 35 cycles of 95°C/1 minute, 56°C/1 minute, 65°C/1 minute; final extension 65°C/10 minutes. Relative quantification was performed using the ΔΔCt method.
Western blotting
Sorbitol-synchronized parasites were untreated or were treated with 10 nM WR99210 and 200 μM IPP for 1 or 3 days. Parasites were separated from RBCs by lysis in 0.1% saponin, washed in PBS, and were stored at −80°C until analysis. Parasite pellets were resuspended in PBS containing 1X NuPAGE LDS sample buffer with 50 mM DTT and were heated to 95°C for 10 minutes before separation on NuPAGE Bis-Tris gels and transfer to nitrocellulose. Membranes were blocked in 0.1% Hammarsten casein (Affymetrix) in 0.2X PBS with 0.01% sodium azide. Antibody incubations were performed in a 1:1 mixture of blocking buffer and TBST (Tris-buffered saline with Tween 20: 10 mM Tris, pH 8.0, 150 mM NaCl, 0.25 mM EDTA, 0.05% Tween 20. Blots were incubated with primary antibody at 4°C overnight at the following dilutions: 1:20,000 mouse-α-GFP JL-8 (Clontech 632381); 1:4000 rabbit-α-PfClpP (42); 1:20,000 rabbit-α-PfAldolase (Abcam ab207494). Blots were washed once in TBST and were incubated for 1 hour at room temperature in 1:10,000 dilutions of IRDye 800CW donkey-α-rabbit or IRDye 680LT goat-α-mouse secondary antibodies (LI-COR Biosciences). Blots were washed three times in TBST and once in PBS before imaging on a LI-COR Odyssey imager. Band intensities of precursor and mature protein were quantified using Image Studio Lite version 5.2 (LI-COR).
Statistics
One-way ANOVAs with Tukey’s multiple comparisons tests were performed in GraphPad Prism version 7.04.
Acknowledgements
Funding for this work was provided by National Institutes of Health grants K08 AI097239 and DP5 OD012119 (E.Y.), a Burroughs Wellcome Fund Career Award for Medical Scientists (E.Y.), the Chan Zuckerberg Biohub Investigator Program (E.Y.), and a William R. and Sara Hart Kimball Stanford Graduate Fellowship (M.J.B.).
We thank Tobias Spielmann for providing plasmids encoding mDHFR, Sean Prigge for α-PfACP antibody, Walid Houry for α-PfClpP antibody, and Jacobus Pharmaceutical Company for WR99210.
References




