

Abstract
Despite compelling evidence that the accumulation of amyloid-beta (Aβ) promotes cortical MAPT (tau) aggregation in familial and idiopathic Alzheimer’s disease (AD), murine models of cerebral amyloidosis are not considered to develop tau-associated pathology. The absence of neurofibrillary lesions in amyloidosis mice remains a challenge for the amyloidocentric paradigm of AD pathogenesis. It has resulted in the generation of transgenic mice harboring mutations in their tau gene, which may be inappropriate for studying a disease with no known TAU mutations, such as AD. Here, we have used APPswe/PS1ΔE9 mice to show that tau pathology can develop spontaneously in murine models of familial AD. Tauopathy was abundant around Aβ deposits, with Gallyas- and thioflavin-S-positive perinuclear inclusions accumulating in the APPswe/PS1ΔE9 cortex by 18 months of age. Age-dependent increases in Gallyas signal correlated positively with binding levels of the paired helical filament (PHF) ligand [18F]Flortaucipir, in all brain areas examined. Sarkosyl-insoluble PHFs were visualized by electron microscopy. Tandem mass tag proteomics identified sequences of hyperphosphorylated tau in transgenic mice, along with signs of RNA missplicing, ribosomal dysregulation and disturbed energy metabolism. Human frontal gyrus tissue was used to validate these findings, revealing primarily quantitative differences between the tauopathy observed in AD patient vs. transgenic mouse tissue. Levels of tau mRNA were not different between APPswe/PS1ΔE9 and littermate control animals. As physiological levels of endogenous, ‘wild-type’ tau aggregate secondarily to Aβ in transgenic mice, this study demonstrates that amyloidosis is both necessary and sufficient to drive tauopathy in experimental models of familial AD.
Introduction
Genetically-inherited and sporadic forms of Alzheimer’s disease (AD) are characterized by a common set of hallmark brain lesions, which include the accumulation of amyloid-β (Aβ) peptides into plaques, neuroinflammation, aggregation of hyperphosphorylated MAPT (tau) into neurofibrillary tangles (NFTs), and neurodegeneration. Transgenic mouse models that reproduce aspects of the aforementioned lesions have been generated based on mutations in the amyloid precursor protein (APP) and presenilin 1 (PSEN1) and 2 (PSEN2) genes, which are known to cause familial AD (1). Despite playing important roles in evaluating APP processing, Aβ toxicity, and amyloid-targeting therapeutic strategies, transgenic mice are not being regarded as models that can replicate the full spectrum of AD histopathology (2). In particular, while the overexpression of mutant APP and APP/PSEN1 has been shown to yield amyloidosis (3), neuroinflammation (4) and neurodegeneration (5) in mice, it is generally not considered to promote the conversion of endogenous tau into neurofibrillary structures (6).
To address the in vivo role of tau hyperphosphorylation and NFT formation in AD pathogenesis, human MAPT (TAU) has been introduced into the mouse genome, either mutated or non-mutated, on a Tau-knockout background (7, 8). TAU overexpressing mice demonstrate progressive neurofibrillary pathology, albeit in the marked absence of cerebral amyloidosis, which is required for a neuropathological diagnosis of AD. Moreover, mutations in TAU have been linked to non-AD tauopathies, most commonly frontotemporal lobar degeneration [FTLD; (9)], a condition with neuropathological hallmarks distinct from AD. Thus, murine models of amyloidosis and combined amyloidosis-tauopathy models have been widely criticized for their translational relevance to the human condition. It has been argued that virtually all existing murine models would be considered as ‘not’ AD (10) according to the ABC scoring system of neuropathology (11). The inability of amyloidosis mice to develop neurofibrillary lesions is thought to contribute to the poor translation of preclinical research into clinical benefits (12), and has raised concern about the amyloidocentric model of AD pathogenesis (13).
Two principal explanations have been put forward for the lack of tau-associated pathology in amyloidosis mice (14). First, adult mice express fewer isoforms of the tau protein than humans (three vs. six), which might render them less liable to the post-translational modifications (PTMs) that are associated with the accumulation of tau into NFTs, such as phosphorylation (15). However, murine tau has been shown to readily fibrillize in vitro upon treatment with polyanionic factors, including RNA (16), and there is ample evidence of tau hyperphosphorylation in the transgenic mouse brain [(17), Table S1], indicating that no differences exist in the propensity of murine and human tau for aggregation and PTMs. A second reason that is often cited for the absence of tauopathy in amyloidosis models is that the murine lifespan may be too short for the complete sequence of neurofibrillary pathology to unfold in transgenic mice. Although age scaling studies suggest otherwise (18), the aging factor has been neglected in the design of preclinical studies.
Transgenic Fischer rats (TgF344-AD), expressing human APP harboring the Swedish double mutations (KM670/671NL) and PSEN1 lacking exon 9 (APPswe/PS1ΔE9), both under control of the mouse prion protein promoter, develop progressive neurofibrillary pathology (19). In this study, transgenic APPswe/PS1ΔE9 mice that were constructed in an identical manner as TgF344-AD rats were used to demonstrate neurofibrillary pathology in aging amyloidosis mice.
Results
Neurofibrillary pathology in aging APPswe/PS1ΔE9 mice
Fresh-frozen brain sections from 3-, 6-, 12-, 18- and 24-month-old APPswe/PS1ΔE9 transgenic (TG) mice and their wild-type (WT) counterparts were processed for the detection of neurofibrillary alterations with the Gallyas silver stain (n=6/group). Thioflavin-S and DAPI (4′,6-diamidino-2-phenylindole) were used to detect perinuclear β-pleated structures. Co-staining for amyloid and Gallyas was used to probe the relationship between amyloidosis and tau-associated pathology in aging TG animals. Fresh-frozen sections of the middle frontal gyrus from a patient with definite AD were processed in parallel with sections from APPswe/PS1ΔE9 mice, to compare Gallyas-positive structures in mouse vs. human tissue.
Aβ deposition was the predominant lesion in the 6-month-old APPswe/PS1ΔE9 brain (Fig. 1A&B), with age-dependent increases in argyrophilic density observed exclusively in TG mice (Fig. 1C-F). Only mild and diffuse silver staining was observed in the neocortex of 6-month-old animals (Fig. 1G). Densely-labeled, round structures, surrounded by a halo of argyrophilic staining, constituted the majority of Gallyas-positive signal in the neuropil of the neocortex and hippocampus at 12-24 months of age (Fig. 1H&I). In addition, diffuse and compact argyrophilic staining was observed surrounding red-stained nuclei in the neocortex of 18- and 24-month-old APPswe/PS1ΔE9 mice (Fig. 1J&K). The perinuclear structures were positive for thioflavin-S (Fig. 1L-N), which colocalized with nuclear DAPI (Fig. 1O) and was further detected in cell-sized structures lacking a stainable nucleus (Fig. 1P). There were no apparent differences in morphology between the argyrophilic structures in brain tissue from 24-month-old TG mice (Fig 1Q-U) and AD-confirmed patient material (Fig. 1V-Z), although neuropil threads were detected exclusively in AD tissue (Fig. 1Q-Z). Coronal brain sections of 20-month-old Tg2576 mice, harboring the Swedish double mutations, were used to examine 6E10- and Gallyas-positive pathology in a second mouse model of amyloidosis (Fig. 1AA-AD). Amorphous argyrophilic signal (AC) and perinuclear lesions (AD) were also present in the Tg2576 mouse brain, albeit at lower levels compared to 18-month-old APPswe/PS1ΔE9 mice.

Sagittal brain sections of 6-month-old APPswe/PS1ΔE9 mice, processed for 6E10 immunohistochemistry (A) and the Gallyas silver stain (B). Silver-labeled sections were counterstained with nuclear fast red. β-amyloidosis dominates over argyrophilic pathology in 6-month-old APPswe/PS1ΔE9 mice. (C-F) Progressive increase in Gallyas-positive signal in 12- (C), 18- (D), and 24-month-old transgenic mice (E). Wild-type animals showed no silver deposition up to 24 months of age (F). (G-P) All photomicrographs are from the neocortex of APPswe/PS1ΔE9 miceArgyrophilic signal was scarce in 6-month-old TG animals (G). Gallyas-positive structures in 18- (H) and 24-month-old animals (I), likely of neuritic nature. Gallyas silver (J & K) and thioflavin-S stainings (L-P), showing perinuclear and intranuclear signal in 18- and 24- month-old transgenic mice. The insert in J shows compact Gallyas staining in the absence of nuclear fast red. Note potential fragmented nuclei in (M) and (N), intranuclear signal in (O), and absence of DAPI signal in (P). (Q-Z) Gallyas/6E10 doubly-labeled sections from a 24-month-old transgenic mouse (Q-U) and an AD patient (V-Z), showing dense-core plaques (Q & V), teardrop-shaped structures (R & W, arrows), tuft-shaped filaments (S & X, arrows), and globose structures in close proximity (T & Y) and over 200 µm afar from Aβ deposits (U & Z). (AA- AD) 6E10/Gallyas- AA) and Gallyas-labeled (AB-AD) sections of 20-month-old Tg2576 mice. Scale bar is 2 mm for A&B, 1 mm for C-F, 10 µm for G-I, 5 µm for J-K, L-P, 10 µm for Q & V, 20 µm for R-U & W-Z, 200 µm for AA&AB, 20 µm for AC, and 5 µm for AD.
The fraction of brain tissue occupied by Gallyas-positive staining in aging APPswe/PS1ΔE9 mice is shown in Fig. S1A. Conformationally-altered tau was detected with the MC-1 monoclonal antibody (Fig. S1B). Vascular and meningeal lesions were present in 18- and 24-month-old animals (Fig. S1C).
[18F]Flortaucipir autoradiography
The paired helical filament (PHF) ligand [18F]Flortaucipir ([18F]AV-1451, [18F]T807) was used to quantify tau pathology in aging APPswe/PS1ΔE9 TG mice by autoradiography (Table 1). Increased binding was observed in the neocortex, hippocampus, amygdala and the cerebellum of 12-month-old APPswe/PS1ΔE9 mice, compared to age-matched WT, 3- and 6-month-old TG animals (P<0.001 for all regions; Bonferroni post-hoc tests). [18F]Flortaucipir binding was further elevated in the visual (P<0.001), somatosensory (P<0.001), motor cortex (P<0.001), and the amygdala (P<0.05) of 18- vs. 12-month-old APPswe/PS1ΔE9 TG mice. Increased binding over age-matched WT mice was first observed in the thalamus of TG animals at 18 months of age (P<0.001). In 24-month-old APPswe/PS1ΔE9 mice, [18F]Flortaucipir signal had increased in all brain regions examined compared to age-matched controls. Three-way ANOVA confirmed genotype-[F(1,476)=2603.1, P<0.001], age-[F(4,476)=457.3, P<0.001] and brain region-specific increases in the binding levels of [18F]Flortaucipir [F(9,476)=42.9, P<0.001], as well as significant age x genotype x region interaction effects [F(36,476)=5.5, P<0.001].
Within each brain area analyzed, there was a positive correlation between the age-dependent increase in the binding levels of [18F]Flortaucipir and the progressive increases in the density of Gallyas-positive lesions (Pearson r for all brain regions: 0.92, P<0.001; Fig. S2).
Fresh-frozen brain sections from APPswe/PS1ΔE9 and age-matched wild-type (WT) animals were incubated with 38.4±9.6 MBq [18F]Flortaucipir for a period of 60 min (specific activity: 145±68 GBq/µmol). Autoradiography data are presented as the mean specific binding of [18F]Flortaucipir (kBq/mL) ± standard error of the mean in brain regions of 5-6 animals/group. By 24 months of age, [18F]Flortaucipir binding in APPswe/PS1ΔE9 mice had increased across all brain areas examined vs. age-matched WT animals. The age-dependent increase in [18F]Flortaucipir binding levels was positively correlated with the progressive increase in Gallyas-positive argyrophilic signal, in all TG brain areas examined. **P<0.01, ***P<0.001 vs. age-matched littermate control mice, Bonferroni post-hoc tests. Symbols of significant differences between groups of 24 & 18 vs. 3-, 6- and 12-month-old-mice were omitted from the table for clarity of presentation.
Representative autoradiograms of [18F]Flortaucipir binding sites are shown in Fig. 2. Binding was decreased in the presence of 50 µM unlabeled flortaucipir but was not reversed by 1 µM of the amyloid-preferring Pittsburgh compound B (PIB).

(A) Sagittal brain sections of aging transgenic (top panel) and wild-type mice (WT, lower panel), taken at the level of the entorhinal cortex [lateral 2.88±0.12 mm of the Paxinos and Franklin mouse atlas (74)]. Images were analyzed on a black & white display mode, and presented as a pseudocolor interpretation of black & white pixel intensity, calibrated in kBq/mL of [18F]Flortaucipir solution. Age-dependent increases in binding levels were observed exclusively in APPswe/PS1ΔE9 mice. (B) [18F]Flortaucipir binding in sections from the middle frontal gyrus of AD-confirmed patients, 18-month-old APPswe/PS1ΔE9 mice and 20-month-old Tg2576 animals, showing the magnitude of tau pathology in patient vs. transgenic mouse tissue. Non-specific binding (NSB) was assessed in the presence of 50 µM ‘cold’ flortaucipir. (C) Binding was not blocked by co-incubating sections with [18F]Flortaucipir and 1 µM of the amyloid-targeting agent Pittsburgh Compound B (PIB).
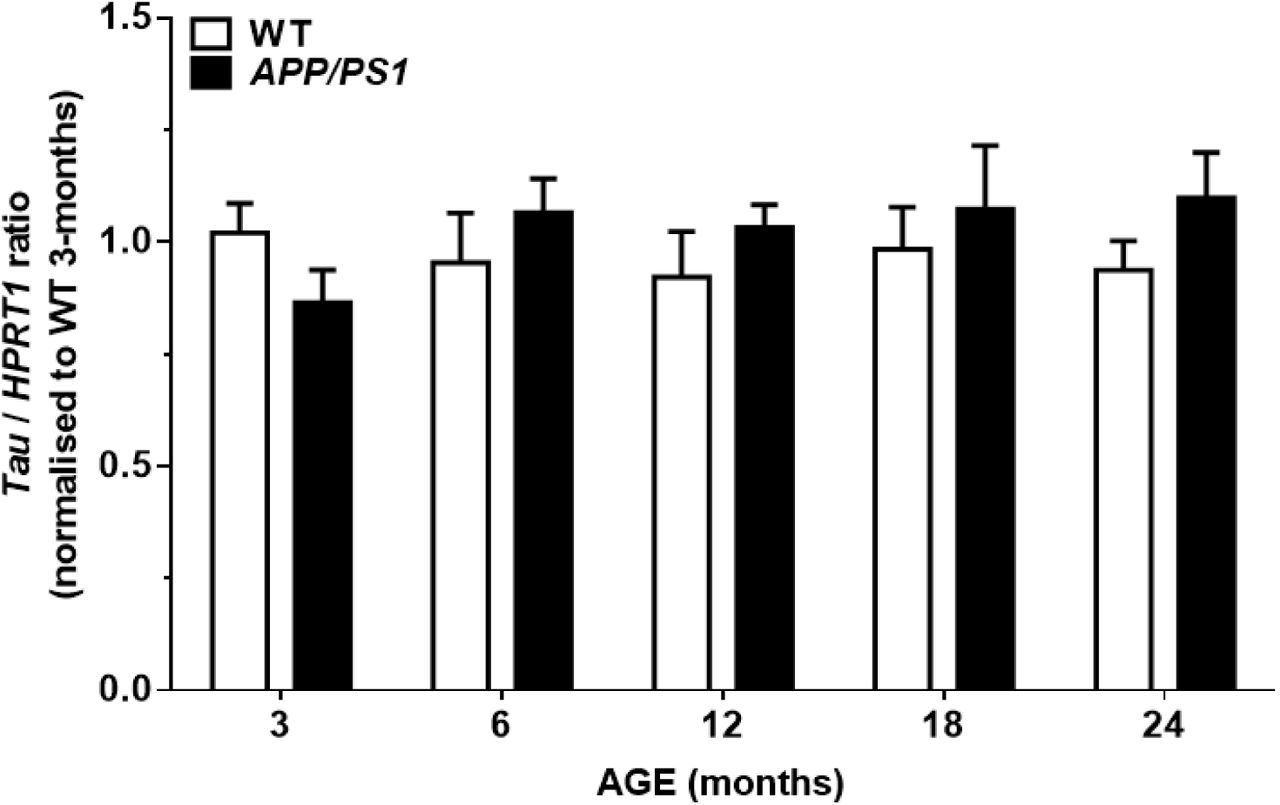
Mapt expression
Relative expression of total Mapt mRNA was determined by RT-qPCR (Fig. 3). There were no age [F(4,50)=0.29, P>0.05], genotype [F(1,50)=0.93, P>0.05], or age x genotype interaction effects on the expression levels of Mapt [F(4,50)=1.21, P>0.05].

Levels of endogenous murine tau mRNA were not altered by age or genotype. PCR products of x4 diluted tau cDNA were determined after 24 cycles. A single peak was obtained by melt-curve analysis, and no signal was detected in the genomic DNA and buffer controls. The efficiency of amplification was 99.2±0.2% for Hprt1 and 100.3±2.1% for Mapt.
Isolation and Transmission Electron Microscopy (TEM) of sarkosyl-insoluble tau
The general methods of Sahara et al. (20) and Greenberg and Davies (21) were evaluated for the extraction of PHFs from the 24-month-old APPswe/PS1ΔE9 TG brain (Fig. S3). Although longer filaments were isolated by the procedure of Sahara et al., the Greenberg and Davies method was chosen for the isolation of sarkosyl-insoluble tau from 3- and 24-month-old mice, based on immunoblotting experiments, solubility considerations, and to allow for comparisons with literature data (22). Soluble and insoluble tau levels were measured in mouse brain homogenates by using the mouse Total Tau Meso Scale kit (Meso Scale Diagnostics LLC). TEM was used to visualize filaments in the sarkosyl-insoluble extracts from the TG mouse and AD patient brains by negative staining.
Tau protein levels increased with age in the pellet obtained by centrifuging WT and APPswe/PS1ΔE9 homogenates at 27,000 × g [Fig. 4A; age effect: F(1,18)=50.0, P<0.001; genotype effect: F(1,18)=2.4, P>0.05]. Levels of tau in the supernatant fraction were not different between 3- and 24-month old, WT and APPswe/PS1ΔE9 TG animals [age: F(1,16)=0.6, P>0.05; genotype: F(1,16)=0.0, P>0.05]. Treatment of the supernatant with 1% sarkosyl for 2 h at 37°C increased the concentration of tau in the detergent-soluble fraction by >3-fold. Sarkosyl-soluble tau levels were lower in the 24- vs. 3-month-old mouse brain [F(1,16)=12.5, P<0.01], irrespective of genotype [F(1,16)=0.5, P>0.05]. Sarkosyl-insoluble tau was not detected in 3-month-old animals, and its levels were not different between 24-month-old APPswe/PS1ΔE9 and WT mice [t(8)=0.7, P>0.05; independent two-tailed Student’s t-test].

(A) Levels of soluble and insoluble tau were determined with the mouse Total Tau Meso Scale kit. Tau levels increased with age in the pellet obtained by centrifuging brain homogenates at 27,000 × g. The resulting supernatant was treated with 1% sarkosyl and centrifuged at 200,000 × g. The solubility of tau in sarkosyl was decreased with age, irrespective of genotype. (B) Overview of negatively-stained filament types in the sarkosyl-insoluble fraction from 24-month-old APPswe/PS1ΔE9 and AD brain tissue. Fibrils of ∼20 nm in width, appearing as straight filaments (a) or as two intertwined fibrils (e), each with a diameter of ∼10 nm. PHFs with axial periodicities of ∼80 nm (b & f; arrows) were present in APPswe/PS1ΔE9 mice, and more frequently observed in AD patient material. The inserts show ‘stacked’ PHFs, which were denser in the AD preparation. Structures commonly identified in the detergent-insoluble fractions of the mouse and human brain included bent fibrils of ∼7 nm in width (c & g), and rod-shaped particles (d & h; arrows). Scale bars: 200 nm (a, b, e, f), 100 nm (c, d, g, h).
Fig. 4B shows negatively-stained filaments in the sarkosyl-insoluble extract from the 24-month-old APPswe/PS1ΔE9 mouse and AD patient brain. Fibrils of mean length 104.9±8.3 nm and width 10.1±0.5 nm were isolated from TG mice. Wider fibrils (∼20 nm), with or without a pronounced twist, were readily detected (a & e). Longer filaments (271.7±11.3 nm), with axial periodicities of 78.7±9.8 nm, constituted ∼8% and ∼34% of the fibril population analyzed in the APPswe/PS1ΔE9 and AD brains, respectively (b & f). Clusters of long filaments, which were denser in AD patient material, were present in the insoluble preparation from APPswe/PS1ΔE9 mice (b & f, inserts). Thin, bent fibrils (c & g) and rod-shaped particles (d & h) were observed in both 24-month-old APPswe/PS1ΔE9 and AD brains. There were no between-species differences in the dimensions of the isolated filaments [short filaments, length: t(82)=0.1, P>0.05, width: t(82)=1.2, P>0.05; long filaments, length: t(16)=0.3, P>0.05, width: t(16)=0.8, P>0.05; independent two-tailed t-tests].
Proteomics of sarkosyl-insoluble tau
The sarkosyl-insoluble fractions extracted from 3- and 24-month-old mouse brain, AD and non-AD individuals, were pooled and digested with trypsin & Lys-C. The peptides were labeled with Tandem Mass Tags (TMT), fractionated, and analyzed by nanoLiquid Chromatography-Electrospray Ionization Mass Spectrometry (LC-ESI MS/MS).
A list of tau-associated proteins quantified in the sarkosyl-insoluble proteome is shown in Table 2. Lists of between-group abundance ratios for all regulated proteins are shown in Data File S1. There were 583 proteins identified in the sarkosyl-insoluble mouse proteome, of which 456 were also present in the human samples. Isoforms of tau with three (3R) and four (4R) microtubule-binding repeats were extracted from both human and the murine brain. In mice, all isoforms collapsed under the term microtubule associated protein (MAP; UniProt accession number: B1AQW2). Mouse MAP was regulated by age, rather than genotype. The protein was enriched 2.1-fold in 24- vs. 3-month-old TG mice, and 1.8-fold in 24- vs. 3-month-old WT mice. Genotype-specific enrichment was observed for mouse tau isoform-B (UniProt accession number: P10637-3), a 3R isoform of tau with an extended C-terminal domain, which was identified by the sequence 205KVQIVYKPVDLSKV218. Tau isoform-B increased 3.2-fold in 24-month-old TG vs. WT mice, and 4.5-fold in 24- vs. 3-month-old TG animals. Human MAP (UniProt accession number: A0A0G2JMX7), containing tau isoforms P10637-2, -4, -6 & -8, was 37-fold enriched in the sarkosyl-insoluble fraction of AD compared to non-AD brain.
Tau-associated proteins quantified in the detergent-insoluble fractions of the mouse and human brain. The presented proteins have been selected for their documented roles in the regulation and binding of tau.
The mouse MAP sequence 174KVAVVRTPPKSPSASKS190, phosphorylated at Threonine (T) 180 and Serine (S) 188, was more than 20-fold enriched in 24-month-old TG, compared to age-matched WT and 3-month-old APPswe/PS1ΔE9 mice. The peptide sequence was not regulated in aging WT animals. An orthologous sequence of the human MAP was phosphorylated at Threonine (T) 566 and Serine (S) 573 (560KVAVVRTPPKSPSSAKS576). The reported phosphorylation sites correspond to amino acids (aa) T231 and S238 of the human tau isoform with 441 aa. Indications of additional phosphorylation sites were obtained by searching modified peptides against tau isoform- and species-specific databases. Phosphorylated S396, S400 and S404 on the conserved sequence 396SPVVSGDTSPR406 of the human 441 aa isoform were identified in the sarkosyl-insoluble mouse proteome. Phosphorylation at S404 in 24-month-old TG mice was confirmed by immunoblotting (Fig. S4). In addition to phosphorylation, murine MAP was deamidated at Asparagine (N) 44, a site on the N-terminal domain of tau that is not conserved in humans (34AEEAGIGDTPNQEDQAAGHVTQAR57). Human MAP was sdeamidated at position N484, corresponding to N167 of the 441 aa tau isoform (473GAAPPGQKGQANATRIPAK491).
The database for annotation, visualization and integrated discovery (DAVID, v6.8) was used for gene ontology (GO) enrichment analysis of the sarkosyl-insoluble proteome (23, 24). RNA splicing, mRNA processing and translation were among the 10 most enriched biological processes associated with protein upregulation in 24-month-old APPswe/PS1ΔE9 vs. WT mice and AD vs. non-AD subjects. Ribonucleoprotein complexes, ribosomes, and exosomes were among the 10 most enriched cellular components in the insoluble extracts from the mouse and human brain (Fig. 5A). The top 10 molecular functions of the enriched proteins were associated with poly(A) RNA binding, as well as binding of molecules contributing to the structural integrity of ribosomes and the cytoskeleton (Fig. 5B). Pathway-based enrichment analysis of upregulated proteins in 24-month-old APPswe/PS1ΔE9 vs. WT mice involved GO terms such as Alzheimer’s and Huntington’s disease, long-term depression, cholinergic, serotonergic and glutamatergic synapse (Fig. 5C). Glycolysis/gluconeogenesis and the Krebs cycle were among the top 10 pathways for downregulated proteins (Fig. 5D).

(A) Enriched cellular components; (B) Enriched molecular functions; (C) Top 10 enriched pathways based on protein upregulation in 24-month-old TG vs. WT mice, according to the Kyoto Encyclopedia of Genes and Genomes (KEGG); (D) Top 10 enriched KEGG pathways based on protein downregulation in 24-month-old TG vs. WT mice. Functional annotation clustering was generated by using DAVID software. Maximum enrichment probability (P value) was based on an EASE score threshold value of 0.05.
Discussion
The present study describes tauopathy in murine models of familial AD. Neurofibrillary alterations in APPswe/PS1ΔE9 and Tg2576 mice were demonstrated by a set of tools that are currently used for the evaluation of pathological tau clinically, such as the Gallyas silver stain and [18F]Flortaucipir. The presence of PHF tau was confirmed by TEM of sarkosyl-insoluble preparations from the APPswe/PS1ΔE9 mouse brain. As murine tau possesses a remarkably high number of 76 potential serine/threonine and 4 tyrosine phosphorylation sites, an antibody-free proteomics approach was used for the detection of tauopathy-related epitopes. Of the five hyperphosphorylated sites identified, S404 has been associated with the intraneuronal and extracellular deposition of NFTs in AD (25). The pathology observed in the present study occurred at physiological levels of endogenous tau, as there was no difference in total tau mRNA and protein between APPswe/PS1ΔE9 and WT mice. Hence, in addition to progressive amyloidosis (3), neuroinflammation (4) and neurodegeneration (5), APPswe/PS1ΔE9 mice develop progressive neurofibrillary pathology of the AD type, mimicking a range of AD pathologies, in a translationally-relevant manner. The observation that endogenous tau accumulates secondarily to Aβ in models of cerebral amyloidosis is entirely consistent with post-mortem (26) and in vivo imaging data (27), showing that the development of cortical tau pathology in AD patients is associated with, and may depend on, pre-existing amyloid pathology.
Current approaches to induce tauopathy in mice have been criticized for generating models that poorly recapitulate the situation in the AD brain, as TAU in AD is neither overexpressed, nor mutated (28). FLTD-linked mutations, in particular, induce tauopathy that is not only morphologically different than that of AD (e.g. Pick bodies), but further characterized by distinct neurodegenerative processes. For example, cholinergic neurons are extensively lost in AD, but not in FTLD (29). Acetylcholinesterase inhibitors, which are prescribed for the symptomatic relief of cognitive impairment in AD, are largely ineffective in FTLD and may even worsen its symptoms (30). Thus, the pathophysiology that differentiates AD from primary tauopathies is unlikely to be modeled in mutant TAU models. Moreover, neurofibrillary alterations in TAU overexpressing mice occur in the absence of Aβ deposition, which is a defining feature of AD histopathology. The present results indicate that amyloidosis models may overcome these limitations, by reproducing both the neurofibrillary pathology of familial AD and the molecular heterogeneity that is associated with it. In addition to the spontaneous aggregation of tau in APPswe/PS1ΔE9 and Tg2576 mice, analysis of the sarkosyl-insoluble APPswe/PS1ΔE9 proteome identified proteins that have been strongly linked to AD pathogenesis, in general, and tau pathology in particular. Among them, APOE and BIN1 are encoded by genes whose variants are known to increase the risk of late-onset AD, through pathways involving interactions with both APP (31, 32) and tau (33-35). Core components of the spliceosome, on the other hand, particularly Sm-D1 and Sm-D2, are closely related to the deposition of NFTs, but not plaques in familial AD (36). This literature implicates multiple mechanisms in AD tauopathy, which occur downstream of Aβ processing in cases of autosomal dominant AD (ADAD) and, as shown here, APPswe/PS1ΔE9 mice.
Although the sporadic and familial forms of AD share common clinical and histopathological features, it is becoming increasingly recognized that they are not precisely equivalent (37). Positron emission tomography (PET) with [11C]PIB demonstrates accumulation of Aβ in the cerebellum of familial AD cases, which is not typical of sporadic AD (38). Cerebellar deposition of hyperphosphorylated tau has been observed in ADAD cases harboring the PSEN1 E280A mutation, but not in sporadic AD (39). Thus, the pronounced cerebellar involvement in APPswe/PS1ΔE9 mice, which are known to accumulate Aβ in this region (40), suggests that the model mimics familial, rather than the sporadic forms of AD. Reports of cerebellar pathology in ADAD cases and APPswe/PS1ΔE9 mice warrant caution in using the cerebellum as a reference region for the quantification of [11C]PIB and [18F]Flortaucipir PET (41, 42), as this is likely to underestimate cortical Aβ and tau pathology, respectively.
Unlike the imbalance in Aβ homeostasis, which is thought to be central in the pathogenesis of AD, gross changes in tau production and clearance were not observed in this study. On the one hand, Gallyas-, [18]Flortaucipir- and thioflavin-S-positive signal was observed in the vasculature of 18-24-month-old APPswe/PS1ΔE9 mice. Moreover, there were age- and genotype-specific changes in multiple components of the phagosome and proteasome in TG vs. WT animals (Data File S1). On the other hand, total tau mRNA and protein levels were not different between 24-month-old WT and TG mice, as evidenced by tau mesoscale, proteomics and PCR. Moreover, age-dependent decreases in the solubility of tau were equally observed in APPswe/PS1ΔE9 and control animals. Notwithstanding that the contribution of individual pathways to tau degradation was not assessed in this study, these findings suggest that the neurofibrillary alterations observed in APPswe/PS1ΔE9 mice are not mediated by an imbalance between the production and clearance of tau. It is important to note that, unlike in TG mice, sarkosyl-insoluble tau was increased in AD vs. non-AD tissue, a finding that is consistent with literature data on the regulation of human tau in AD (43). It might be that the increased concentration of brain tau in late-stage AD is associated with heavily impaired clearance pathways or pronounced neuronal damage, processes that may not be modeled in 24-month-old APPswe/PS1ΔE9 mice. Alternatively, the present data may highlight the involvement of transcriptional and translational mechanisms, rather than production and clearance pathways, in the assembly of PHF tau.
A prevalence of 3R isoforms in the composition of NFTs has been observed in the AD hippocampus by immunohistochemical and biochemical methods (44). Moreover, a shift from 4R to 3R isoforms has been associated with the morphological evolution of tau-positive neurons from a pre-tangle to the NFT state (45). Although the literature on the regulation of tau isoforms in AD remains scarce, the present results support the notion that an imbalance in tau isoform ratio is involved in the neurofibrillary alterations of AD, with 3R isoforms being preferentially sequestered into the insoluble tau fraction. The identification of tau isoform-B, a 3R isoform that is predominantly expressed in the fetal mouse brain, supports the suggestion that immature tau isoforms participate in AD tauopathy (46), and implicates aberrant transcription and translation mechanisms in the disease process. A re-induction of fetal tau may be attributed to the deregulation of core splicing machinery, which was marked in this study and considered to occur early and selectively in AD (47). Moreover, as the selection of splice sites is determined by canonical sequences encoded into the genome, the re-expression of fetal isoforms might be a consequence of aberrant DNA replication during cell cycle re-entry (48). Cell cycle proteins that were deregulated in an age- and genotype-specific manner in this study include Sub1, cdc42, CEND1, Histone H3 and nucleolin (Data File S1). Clearly, the exact mechanisms underlying tauopathy in AD cannot be resolved by the present set of experiments. The data demonstrate, however, that the formation of PHF tau is associated with loss of regulatory control over tau splicing in vivo, which may have important implications for the origins and management of tauopathy in AD. It is tempting to speculate that tau hyperphosphorylation may partly be due to the re-emergence of fetal isoforms, which are known to be over-phosphorylated compared to adult tau (49). Moreover, it is plausible that an imbalance in tau isoform ratio mediates protein mislocalization from the axonal to the somatodendritic compartment, as distinct tau isoforms are differentially sorted across the cell (50). Of note, cofilin-dependent, ‘classical’ pathways of tau missorting (51) may also be involved in the pathology observed in this study, as cofilin was reduced in the sarkosyl-insoluble proteome of 24-month-old APPswe/PS1ΔE9 mice. Collectively, these data highlight the relevance of amyloidosis models for studying the diverse macroscopic and molecular aspects of AD tauopathy.
The limitations associated with models overexpressing APP and PSEN mutations have been discussed previously (2). To exclude the possibility that tauopathy is an artefact of APP or PSEN overexpression, it would be important to determine whether it develops in second-generation amyloidosis models, carrying AD-related mutations in endogenous genes. Moreover, as there is evidence of [18F]Flortaucipir binding to monoamine oxidases (MAO; 52), signal quantification in the presence of MAO inhibitors is warranted to determine the extent of off-target binding, if any (53). Practical considerations in using amyloidosis mice to study tauopathy include long waiting times for the accumulation of endogenous murine tau, mouse-on-mouse antibody issues, and the low abundance of pathology as compared to human AD. While ∼30% of all Nissl-positive cells in the prefrontal cortex of Braak stage V-VI brains may contain NFTs (54), Gallyas-positive signal in this study occupied ∼1% of the frontal cortex of 24-month-old APPswe/PS1ΔE9 mice, neuritic structures included. Nevertheless, cognitive impairment in AD is known to correlate with the spread of tau pathology, and the number of brain areas containing at least one NFT has been shown to be the best explanatory variable of intellectual status in AD (55). In this context, it is worth evaluating whether measurements of tauopathy in aging APPswe/PS1ΔE9 mice correlate with the progressive cognitive impairment that these animals exhibit in the Barnes maze assay (56).
Materials and Methods
Study design
Mice were grouped according to age and genotype. Sample numbers were based on preliminary studies, showing absence of Gallyas and [18F]Flortaucipir signal in 18-month-old WT vs. TG animals. (Immuno)histochemistry, autoradiography, the isolation of sarkosyl-insoluble tau, and electron microscopy studies were repeated at least three independent times. Tau Meso Scale was performed two independent times. Remaining samples were pooled and subjected to proteomics. To avoid cross-contamination during the isolation of sarkosyl-insoluble tau, glassware was washed in ultrapure de-ionized H2O (dH2O, Ultra Clear™, Siemens), followed by rinses in formic acid (FA, 98-100%; Merck Millipore), dH2O, ethanol (99%; VWR International) and dH2O. No samples were excluded from data analysis, which was performed in an unblinded manner. To compare tau pathology in transgenic mouse vs. human brain, tissue from the middle frontal gyrus of an AD-confirmed patient [BB08-002, Female, 80 years old, post-mortem interval (PMI): 9 h] and a non-AD subject (BB16-023, Female, 83 years old, PMI unknown) were processed along with the murine samples for the Gallyas silver stain, autoradiography, electron microscopy and proteomics experiments. The AD and non-AD samples were chosen for their abundance and complete lack of tau pathology respectively, as assessed by Gallyas silver staining and [18F]Flortaucipir autoradiography. To avoid confounding effects of anesthesia on tau phosphorylation, mice were euthanized by cervical dislocation.
Ethical statement
Mouse tissue: All procedures complied with Danish law (Dyreværnsloven-Protection of Animals Act, nr 344/2005) and European Union directive 2010/63/EU, regulating animal research. Ethical permission was granted by the Animal Ethics Inspectorate of Denmark (nr 2011/561-1950).
Human tissue: Fresh-frozen samples from the middle frontal gyrus were obtained from the Maritime Brain Tissue Bank, Department of Medical Neuroscience, Faculty of Medicine, Dalhousie University, Sir Charles Tupper Building, 5850 College Street, Halifax Nova Scotia B3H 4R2. Ethical approval was obtained from the Nova Scotia Health Authority Research Board in Halifax, Canada, and the Danish Biomedical Research Ethical Committee of the region of Southern Denmark (Project ID: S-20070047). Informed, written consent forms were obtained for all subjects.
Animals and tissue sectioning
APPswe/PS1ΔE9 mice (57), originally purchased from the Jackson Laboratories (MMRRC Stock No: 34832-JAX), were bred and maintained on a C57BL/6J background. The animals were group-housed (4-8/cage) in a temperature (21±1°C) and humidity controlled environment (45-65%), under a 12:12 h light:dark cycle (lights on: 7 am). Food and water were available ad libitum. Female APPswe/PS1ΔE9 mice were used at 3, 6, 12, and 18 months of age. Sex- and age-matched WT littermates were used as control. Both male and female mice were used in the 24-month-old groups (n=6/genotype & age-group, total animal number: 60). The animals were euthanized by cervical dislocation, and brains immediately removed and bisected along the midline. Right hemispheres were frozen in isopentane on dry-ice (−30°C). The olfactory bulb, striatum, cortex, hippocampus, diencephalon, brainstem and cerebellum from the left hemisphere were dissected on a petri dish on ice, collected in Eppendorf® tubes, and frozen on dry-ice. The tissue was stored at −80°C until use.
Sectioning was carried out at −17°C using a Leica CM3050S cryostat (Leica Biosystems GmbH). Series of 20 µm-thick sagittal sections were collected at 300 µm intervals. The sections were mounted onto ice-cold Superfrost™. Plus slides (Thermo Fisher Scientific), dried at 4°C in a box containing silica gel for at least 2 h, and stored at −80°C for future experiments. Every 13th and 14th section was collected in Eppendorf® tubes for RNA extraction with Trizol™.
Fresh-frozen coronal brain sections of male and female, 20-month-old Tg2576 and WT mice were provided by the Centre for Biological Sciences, University of Southampton, U.K.
(Immuno)histochemistry, autoradiography and proteomics
The Gallyas silver stain was performed according to Kuninaka et al. (58), thioflavin-S according to Sun et al. (59), [18F]Flortaucipir autoradiography according to Marquié et al. (60), proteomics according to Kempf et al. (61). Protocol details are provided in Supplementary Materials and Methods.
Statistical analysis
Parametric testing was employed following inspection of the data for normality with the Kolmogorov-Smirnov test in Prism (v6.01; GraphPad Software). Data sets were analyzed by Statistica ™ v10 (TIBCO Software Inc., USA). The effects of age, genotype and brain region on the binding levels of [18F]Flortaucipir were analyzed by three-way ANOVA. Gallyas-positive area fraction and tau gene/protein levels were analyzed by two-way ANOVA for the independent factors age and brain region or genotype, respectively. Where ANOVA yielded significant effects, Bonferroni post-hoc comparisons were used to detect between-group regional and age-dependent differences. Levels of sarkosyl-insoluble tau between 24-month-old TG and WT mice, and PHF dimensions extracted from TG vs. AD brain were compared by two-tailed independent Student’s t-tests. Significance was set at α=0.05. A 1.3-fold change cut-off value for all TMT ratios was used to rank proteins as up- or down-regulated in the proteomics study (62).
Funding
This work was supported by SDU2020 (CoPING AD: Collaborative Project on the Interaction between Neurons and Glia in AD) and the A.P. Møller og Hustru Chastine Mc-Kinney Møllers Fond.
Author contributions
AM designed the project and wrote the manuscript, performed (immuno)histochemistry, autoradiography, tau isolation studies, and assisted with TEM. CT and SJK performed proteomics and immunoblotting. MA and RV performed tissue sectioning, (immuno)histochemistry, autoradiography and tau filament analysis. SP performed PCR and tau Meso Scale. AML organized and performed autoradiography. HA synthesized [18F]Flortaucipir. JLT performed (immuno)histochemistry and provided Tg2576 tissue, SD provided human tissues and critically reviewed the manuscript. DJB, MRL and BF supervised the project and contributed to its design. All authors assisted with data interpretation, participated in drafting the manuscript and approved its final version.
Competing interests
None related to the design and completion of this study.
Data and materials availability
The proteomics data have been deposited to the ProteomeXchange Consortium (63) via the PRIDE partner repository with the dataset identifier PXD009306 [username: reviewer39090@ebi.ac.uk; password: HdJxxVxU (64)]. The MC-1 antibody, and material for the synthesis of [18F]Flortaucipir were obtained through an MTA.
DATA FILE S1
REGULATED PROTEINS 3 MONTHS: TG vs. WT
DATA FILE S1
REGULATED PROTEINS 24 MONTHS: TG vs. WT
DATA FILE S1
REGULATED PROTEINS TG: 24 vs. 3 months
DATA FILE S1
REGULATED PROTEINS WT: 24 vs. 3 months
DATA FILE S1
REGULATED PROTEINS AD vs. non-AD
Acknowledgments
We thank Andrew Reid, senior technician and manager of the Maritime Brain Tissue Bank, for organizing the transportation of human tissue. We acknowledge the Core Facility for Integrated Microscopy, Faculty of Health and Medical Sciences, University of Copenhagen, for assisting with TEM. The Villum Center for Bioanalytical Sciences at SDU is acknowledged for supporting the proteomics part of the study. Precursor material for the synthesis of [18F]Flortaucipir was generously supplied by AVID Radiopharmaceuticals, Philadelphia, PA, USA.
Footnotes
↵† Shared first authorship
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.
- 73.↵
- 74.↵
- 75.
- 76.
- 77.
- 78.
- 79.
- 80.
- 81.
- 82.
- 83.
- 84.
- 85.
- 86.
- 87.
- 88.
- 89.
- 90.
- 91.
- 92.
- 93.
- 94.
- 95.
- 96.
- 97.
- 98.
- 99.
- 100.
- 101.
- 102.
- 103.
- 104.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}