

Abstract
DNA replication origin licensing, the process of MCM helicase loading, is an early essential step in replication and should be restricted to G1 phase to avoid re-replication and genome instability. Cdt1 is a critical MCM loading factor whose licensing activity must be restrained after G1. We discovered that Cdt1 hyperphosphorylation during G2 and M phase is essential to prevent re-replication and DNA damage, the first example of direct Cdt1 licensing activity control by post-translational modification. This hyperphosphorylation specifically requires Cyclin A/CDK1 and occurs at a cluster of phosphorylation sites in a disordered Cdt1 linker region. Hyperphosphorylation interferes with Cdt1-MCM binding independently of protein degradation or inhibition by the Cdt1 inhibitor, Geminin. At the M-G1 transition, Cdt1 is re-activated by protein phosphatase 1-dependent dephosphorylation. We propose that distinct, non-redundant re-replication inhibition mechanisms act in a sequential relay from early S through mitosis to ensure once, and only once, chromosome duplication.
Introduction
Accurate DNA replication in S phase must be completed precisely once per cell cycle. A prerequisite for DNA replication in eukaryotic cells is the DNA loading of the central core of the replicative helicase, the minichromosome maintenance complex (MCM). The process of MCM loading is called DNA replication origin licensing, and it should be tightly restricted to the G1 cell cycle phase1,2. In proliferating mammalian cells, many hundreds of thousands of replication origins are licensed in G1, then a subset of these origins initiate replication in S phase, but no origin should initiate more than once per cell cycle3-6. Improper re-licensing in S, G2, or M phases can lead to re-initiation and re-replication, a source of DNA damage and genome instability that can promote cell death or oncogenesis7,8. For example, de-regulated licensing factors in adult mice caused replication stress, DNA re-replication, and oncogenesis9-11. Licensing factors are also deregulated in a wide variety of spontaneous human cancers12-14.
Re-replication is normally avoided by inhibiting essential MCM loading proteins throughout S, G2, and M phase. These essential proteins include the origin recognition complex (ORC), a direct origin DNA binding complex, which recruits and cooperates with the Cdc6 (cell division cycle 6) protein. ORC and Cdc6 then recruit Cdt1 (Cdc10-dependent transcript 1) bound to the MCM complex, and altogether these factors load MCM onto DNA during G1. Each of the loading factors is tightly regulated through combinations of transcription, protein phosphorylation, and ubiquitin-mediated degradation to restrict licensing activity to G1 phase15-17. Although many mechanisms to avoid re-licensing and re-replication have been described, we postulate that the set of known mechanisms is not comprehensive enough to fully explain how hundreds of thousands of mammalian origins are tightly controlled, nor how re-replication control mechanisms might vary in different cell cycle phases.
One of the known mechanisms to avoid re-replication is degradation of mammalian Cdt1 in S phase. Beginning in late S phase however, Cdt1 re-accumulates and reaches levels in G2 phase similar to its levels in G117,18,19. Cdt1 licensing activity is blocked in late S, G2, and M phases by binding to a dedicated inhibitor protein, Geminin, which interferes with Cdt1-MCM binding17,20,21. Cdt1 is also hyperphosphorylated in G2 phase18,19, but the molecular and physiological consequences of those modifications are largely unknown. In general, mechanisms of Cdt1 activity control are poorly understood because the role of Cdt1 in licensing itself is also incompletely understood. Unlike the other licensing proteins that are AAA+ ATPases22-24, Cdt1 is not an enzyme. Moreover, Cdt1 is not as highly conserved among eukaryotic species as ORC, Cdc6, and MCM are, and there is no full-length structure of any metazoan Cdt117. Determining mechanisms of Cdt1 activity control can shed light on Cdt1 function. Here, we elucidated a novel phosphorylation-dependent mechanism of Cdt1 inhibition that complements other re-replication control mechanisms to ensure precise genome duplication.
We analyzed the re-replication consequences of mutationally altered Cdt1 variants that cannot be hyperphosphorylated in G2. We discovered that Cdt1 phosphorylation in a disordered linker region previously implicated in stress-induced licensing inhibition prevents re-replication in human cells. We demonstrated that Cyclin A/CDK1 is the major Cdt1 kinase in G2 and M phases, and defined a phosphorylation-mediated mechanism to block Cdt1-MCM binding that is independent of contributions from Geminin. This inhibition then creates a requirement for PP1-dependent Cdt1 dephosphorylation to reactivate licensing in the subsequent G1. We propose that multiple re-licensing inhibition mechanisms are not simply redundant, but rather act in a sequential relay from early S phase (replication-coupled destruction) through mid-S phase (degradation plus Geminin) to G2 and M phase (Geminin plus Cdt1 hyperphosphorylation) to achieve stringent re-replication control for large metazoan genomes.
Results
Cdt1 phosphorylation inhibits DNA re-replication
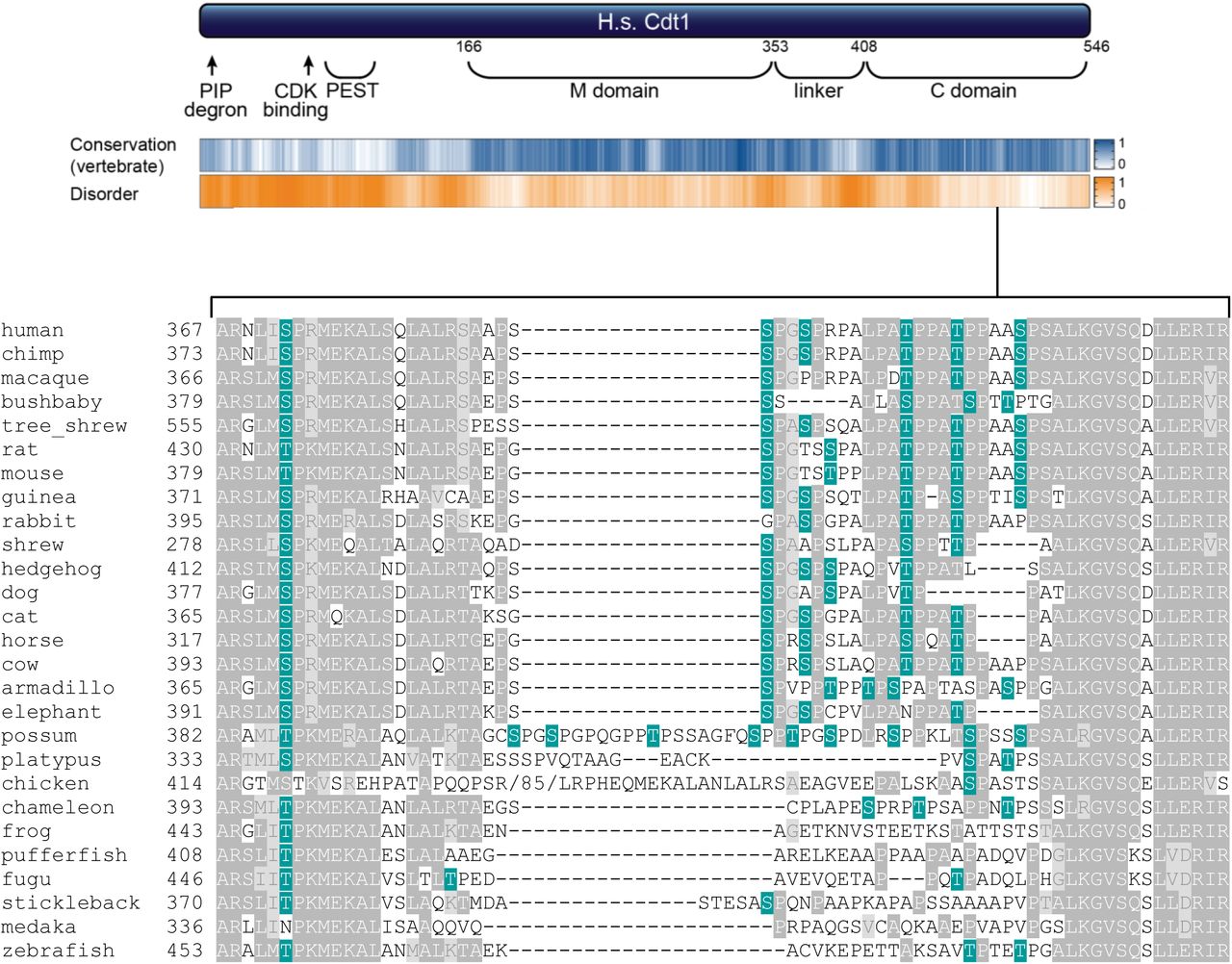
Human Cdt1 is phosphorylated in G2 phase and mitosis, and we hypothesized that this phosphorylation inhibits re-replication by directly inhibiting Cdt1 activity. To test that hypothesis, we compared the activity of normal Cdt1 (wild-type, WT) or an unphosphorylatable Cdt1 variant, “Cdt1-5A”. We had previously shown that this variant, “Cdt1-5A” (S391A, T402A, T406A, S411A, and S491A) is both virtually unphosphorylatable in vitro by stress-induced MAP kinases, and is compromised for G2 hyperphosphorylation detected by gel mobility shift19. Cdt1-5A bears alanine substitutions at five sites, and four are in a region of low sequence conservation and high-predicted intrinsic disorder (Fig. 1a and Supplementary Fig. S1). This “linker” region connects the two domains of Cdt1 that have been structurally characterized for Geminin binding (a middle “M domain”) and for MCM binding (C-terminal “C domain”)25,26. Because both cyclin dependent kinases (CDK) and MAP kinases share similar substrate sequence requirements (serine/threonine-proline), and because both are active in G2, we postulated that during normal G2 and M phases these Cdt1 sites are phosphorylated by CDK or MAPK27. We inserted cDNAs encoding either wild-type Cdt1 (Cdt1-WT) or Cdt1-5A into a single chromosomal FRT recombination site under doxycycline-inducible expression control in U2OS osteosarcoma cells. Both Cdt1 constructs bear C-terminal HA epitope and polyhistidine tags to distinguish ectopic Cdt1 from endogenous Cdt1.

A representative selection of 28 vertebrate sequences for comparison was taken from Miller et al.81 and Cdt1 protein sequences retrieved from https://www.uniprot.org/. For the alignment, Xenopus tropicalis Cdt1 was replaced with Xenopus laevis Cdt1, Tupaia belangeri was replaced with Tupaia chinensis, and no Cdt1 sequence for Echinops telfairi (tenrec) was available. These 27 full-length sequences were aligned with ClustalW at https://www.genome.jp/tools-bin/clustalw using the default settings, and the resulting alignment was visualized with BoxShade, 50% identity or similarity were shaded medium and light grey (https://embnet.vital-it.ch/software/BOX_form.html). The portion corresponding to the Cdt1 linker domain is shown using common names. All potential CDK/MAPK phosphorylation sites are shaded green, and an 85 residue insertion in chicken Cdt1 (lacking any potential CDK/MAPK phosphorylation sites) was deleted for clarity. The 27 sequences are from the following species: Homo sapiens, Pan troglodytes, Macaca mulatta, Otolemur garnettii, Tupaia chinensis, Rattus norvegicus, Mus musculus, Cavia porcellus, Oryctolagus cuniculus, Sorex araneus, Erinaceus europaeus, Canis familiaris, Felis catus, Equus caballus, Bos Taurus, Dasypus novemcinctus, Loxodonta Africana, Monodelphis domestica, Ornithorhynchus anatinus, Gallus gallus, Anolis carolinensis, Xenopus laevis, Tetraodon nigroviridis, Takifugu rubripes, Gasterosteus aculeatus, Oryzias latipes, and Danio rerio.

(a) Schematic of the human CDT1 protein. CDT1 contains two structurally-characterized domains, the Geminin and MCM binding domain (M) and a C-terminal MCM binding domain (C). The Ser/Thr-Pro sites that were altered for this study are marked with green ovals, and the cyclin binding motif is marked with a green triangle. Positions are T29, S31, S372, S391, S394, T402, T406, S411, and S491; the cyclin binding motif (Cy) is 68-70. Human CDT1 was aligned with 26 other vertebrate Cdt1 sequences using ClustalW, and a relative conservation score was derived (see also Methods and Supplementary Fig. S1). The blue heatmap indicates relative conservation at each amino acid position of human Cdt1. An intrinsic disorder score was also derived for human Cdt1 and shown as the corresponding orange heatmap.
(b) Asynchronously-growing U2OS cells with integrated inducible Cdt1 constructs or the parent line (control) were cultured in 1 μg/ml doxycycline for 48 hours. Whole cell lysates were subjected to immunoblotting for ectopic (HA) or endogenous and ectopic Cdt1; Ponceau S staining of the blot serves as a loading control.
(c) Asynchronously-growing U2OS cells were treated with 1 μg/mL doxycycline for 48 hours and labeled with EdU for 1 hour before harvesting. Cells were analyzed by flow cytometry for DNA content with DAPI and for DNA synthesis by EdU detection; the gating scheme is illustrated. One representative of more than four independent biological replicates is shown. The bar graph plots the percentages of re-replicating cells across all experiments. Asterisks indicate statistical significance (*** p<0.001) determined by Mann–Whitney U-test. n.s. = not significant. Error bars indicate standard deviation.
(d) Asynchronously-growing U2OS cells were treated with 1 μg/mL doxycycline for 48 hours, and whole cell lysates were probed for phospho-Chk1 (S345), total Chk1, HA-Cdt1, and total protein (Ponceau S); at least two independent experiments were probed and one example is shown.
(e) The same cell lines in (b) were synchronized in prometaphase (“G2/M”) by sequential thymidine and nocodazole treatment. Whole cell lysates were separated by standard SDS-PAGE (top) or Phos-tag SDS-PAGE followed by immunoblotting for ectopic Cdt1 (HA); Ponceau S staining serves as a loading control.
As a measure of relative Cdt1 activity, we induced Cdt1 overproduction to approximately 5-10 times more than endogenous Cdt1 (Fig. 1b, compare lanes 1 and 2). The amount of re-replication induced by Cdt1 overproduction is directly related to Cdt1 licensing activity28. As previously reported29,30, Cdt1-WT overproduction in human cells induced re-replication, which we detected by analytical flow cytometry as a population of cells with DNA content greater than the normal G2 amount (>4C, Fig. 1c, and Supplementary Figure S2b). Strikingly however, overproducing Cdt1-5A to the same level as Cdt1-WT induced substantially more re-replication suggesting that it is intrinsically more active. DNA re-replication can also induce the formation of giant nuclei31,32, and we noted that the average nuclear area of cells overproducing Cdt1-WT was somewhat larger than control nuclei, whereas nuclei of cells overproducing Cdt1-5A were even larger (Supplementary Fig. S3a). Thus, Cdt1-5A expression not only induces more cells to re-replicate, but it also induces a higher degree of re-replication in those cells than Cdt1-WT.

(a) U2OS cells ectopically expressing HA-tagged Cdt1-WT were arrested with nocodazole and treated with 20 μM MG132 to prevent premature anaphase. Alternatively, cells were released for 3 hours to obtain G1 cells. The cells were mock treated (lanes 3 and 4) or treated with 10 μM RO3306 (CDK1i, lane 5). Cell lysates were also mock treated (lane 3) or with lambda and CIP phosphatase (lane 4) for 30 minutes. The samples were then subjected to Phos-tag SDS-PAGE followed by immunoblotting with HA antibody.
(b) Quantification of the experiments in (Fig. 1c) showing all cell cycle phase distributions (G1, S, G2/M, and re-replication). n >4.

(a) U2OS cells were treated with 1 μg/mL doxycycline for 48 hours before fixation and staining with DAPI. Nuclear sizes were analyzed by measuring DAPI area using Photoshop software. The average nuclear area of cells overproducing Cdt1-WT was 1.2 fold larger than control cells, whereas cells expressing Cdt1-5A had even larger average nuclear area (~1.7 fold higher than control cells). Representative results of two independent experiments are shown; total numbers of cells analyzed is listed under the histograms. Asterisks indicate statistical significance (*** p<0.001, ** p<0.01) determined by Mann–Whitney U -test. Mean +/-standard deviation is indicated.
(b) U2OS cells were treated as indicated in (a) and stained with an anti-γH2AX antibody (green). Nuclei were stained with DAPI (blue). Representative results of two independent experiments are shown. Quantification of the percentage of γH2AX positive cells is shown with the total number of cells analyzed listed under the histogram.
Re-replication is an aberrant genotoxic phenomenon characterized by molecular markers of DNA damage6,33,34. As an independent measure of re-replication, we analyzed lysates of Cdt1-overproducing cells for Chk1phosphorylation, a marker of the cellular DNA damage response. Cdt1-5A consistently induced more Chk1 phosphorylation than WT Cdt1 (Fig. 1d, compare lanes 2 and 3). We also noted that the accumulation of re-replicated cells came at the expense of G1 cells, consistent with a checkpoint arrest (Supplementary Fig. S2b). Moreover, cells overproducing Cdt1-5A were also ~3 times more likely to generate γ-H2AX foci, another marker of re-replication-associated DNA damage20,35,36 (Supplementary Fig. S3b). We thus conclude that phosphorylation at these sites negatively regulates Cdt1 activity.
In nocodazole-arrested (early mitotic) cells, phosphorylated Cdt1-WT migrated more slowly by SDS-PAGE than Cdt1-5A (Fig. 1e, lanes 2 and 5). As a better measure of Cdt1 phosphorylation, we analyzed Cdt1 gel migration in the presence of Phos-tag reagent which retards protein mobility proportional to the extent of phosphorylation37. Endogenous Cdt1 from nocodazole-arrested cells migrates much more slowly on Phos-tag gels than endogenous Cdt1 from G1 cells, and this slow migration was reversed by phosphatase treatment (Supplementary Fig S2a). We detected similar slow Cdt1 migration in G2 cells synchronized by release from a thymidine arrest without the nocodazole block (data not shown), so we presume that the phosphorylation in prometaphase reflects phosphorylation from late S phase through mid-mitosis (i.e. “G2/M”). The distribution of ectopic Cdt1-5A bands was lower than Cdt1-WT bands on Phos-tag gels, demonstrating that these sites are indeed phosphorylated late in the cell cycle.
Phosphorylation at two additional candidate CDK/MAPK target sites in the linker region has been reported from global phosphoproteomics studies38. To test the potential additional contribution of these sites to Cdt1 regulation, we included the mutations S372A and S394A to Cdt1-5A to create Cdt1-7A (Fig. 1a). This variant had slightly increased mobility on Phos-tag gels relative to Cdt1-5A (Fig. 1e, compare lanes 5 and 6). On the other hand, Cdt1-7A overproduction did not consistently induce more re-replication or DNA damage than Cdt1-5A (Fig. 1c, bar graph and Fig 1d, lanes 3 and 4). From this observation, we infer that Cdt1-5A is already at the lowest activity that can be achieved from phosphorylation in the linker region, and that additional phosphorylations do not cause further activity decreases.
To assess the importance of the four sites in the linker relative to the single site in the C domain, we generated Cdt1-4A and Cdt1-S491A (Fig. 1a). Cdt1-4A migrated on Phos-tag gels with a pattern very similar to Cdt1-5A whereas Cdt1-S491A migration was undistinguishable from Cdt1-WT (Fig. 1e, lanes 2-5). Furthermore, Cdt1-4A was as active as Cdt1-5A for inducing re-replication, whereas Cdt1-S491A only induced as much re-replication as Cdt1-WT (Fig. 1c). Like Cdt1-5A, Cdt1-4A induced substantially more DNA damage (phospho-Chk1) than Cdt1-WT (Fig. 1d, lanes 7-9). Thus, linker region phosphorylation is responsible for Cdt1 inactivation during G2 and M phases.
Cdt1 is also known to be phosphorylated at both T29 and S3138,39. Phosphorylation at T29 generates a binding site for the SCFSkp2 E3 ubiquitin ligase, which contributes to Cdt1 degradation during S phase40,41. Robust Cdt1 degradation in S phase is important for avoiding re-replication30,42,43. The stress MAPK JNK (c-Jun N-terminal kinase) has also been reported to inhibit Cdt1 by phosphorylating T2939. To determine if these N-terminal phosphorylations add to the effects of linker region phosphorylations, we added the two mutations, T29A and S31A, to Cdt1-7A to generate Cdt1-9A. Cdt1-9A from nocodazole-arrested cells migrated even faster than Cdt1-7A on Phos-tag gels (similar to phosphatase-treated WT Cdt1, not shown), demonstrating that one or both T29 and S31 are phosphorylated after S phase, although Cdt1 is not as unstable in G2 as it is in S phase. Cdt1-9A overproduction induced even more re-replication than Cdt1 bearing only linker region mutations, Cdt1-4A, 5A, and 7A (Fig. 1c), and similar amounts of DNA damage checkpoint activation (pChk1, Fig 1d, lanes 5 and 9). We presume that compromised S phase degradation from loss of SCFSkp2 targeting contributes to this enhanced re-replication30,41,44.
Cyclin A/CDK1 is the primary Cdt1 kinase during G2 and M phases
To determine which kinase(s) is responsible for Cdt1 inactivation, we assessed the effects of kinase inhibitors. All nine of the sites in Cdt1-9A can be targeted by both CDKs and MAPKs since all nine are serine or threonine followed by proline (Supplementary Fig. S1). We synchronized cells in nocodazole when Cdt1 is maximally phosphorylated and then tested the effects of pharmacological MAPK and CDK inhibitors on the migration of endogenous Cdt1 by Phos-tag gel analysis. We first treated nocodazole-arrested cells with pharmacological inhibitors of p38 or JNK, two stress-activated MAP kinases which we previously showed can phosphorylate the linker region and inactivate origin licensing during a stress response19 (p38 inhibitor SB203580 and c-Jun N-terminal kinase JNK inhibitor VIII). These MAPK inhibitors, either alone or in combination, had no effect on mitotic Cdt1 migration on Phos-tag gels (Fig. 2a, lanes 8-10, compared to lane 4). We confirmed that the inhibitors were active in these cells at these concentrations by analyzing known downstream substrates (Supplementary Fig. S4a-c)19,45-48. We also tested inhibitors of CDK1 and CDK2 singly or in combination. The slow migration of phospho-Cdt1 was largely reversed by treatment with CDK1 inhibitor RO3306 for just 15 minutes (Fig. 2a, compare lanes 5, 7, and 11 to lane 4), but not when treated with the CDK2 inhibitor CVT313, (Fig. 2a, lane 6). This effect occurred even in the presence of the proteasome inhibitor MG132, which we included in all kinase inhibitor experiments to prevent premature anaphase onset.

(a) U2OS cells were treated as indicated in Fig. 3a. Mitotic phosphoproteins were analyzed by immunoblotting with an anti-Mpm-2 antibody. Mitotic phosphoprotein mAb2 (MPM-2) is a mitotic marker that recognizes a large subset of mitotic phosphoproteins and reflects CDK1 activity in M phase47.
(b) U2OS cells were mock treated (lane 1) or treated with 6 μM CVT313 (lane 2), then probed for endogenous Cdc6. Cdc6 is stabilized by CDK2/Cyclin E activity during late G1 phase48.
(c) U2OS cells were mock treated (lane 2), treated with UV light (lane 1), or arrested in G2/M phase (lane 3) followed by 30 μM SB203580 treatment (lane 4). The mitogen-activated protein kinase-activated protein kinase 2 (MK2) is a direct substrate of p3845. The phosphorylation and total protein levels of MK2 were analyzed by immunoblotting. Equal loading was confirmed by Ponceau S staining in all experiments, and representative results of two independent experiments are shown.

(a) U2OS cells were synchronized with nocodazole then mock treated or treated with 10 μΜ RO3306 (lane 5), 6 μΜ CVT313 (lane 6), 30 μΜ SB203580 (lane 8), 10 μΜ JNK inhibitor VIII (lane 9), or combinations of inhibitors (lane 7,10,11) as indicated for 1 hour except the RO3306 was treated for only the final 15 minutes. All cells were simultaneously treated with 20 μΜ MG132 to prevent premature mitotic exit. Alternatively, cells were released for 3 hours to obtain G1 cells or subjected to 20 J/m2 to induce Cdt1 degradation. Endogenous Cdt1 phosphorylation was assessed by standard or Phos-tag SDS-PAGE followed by immunoblotting; Ponceau S staining serves as a loading control. The example shown is representative of more than three independent experiments.
(b) HEK 293T cells expressing GFP, His-tagged Cdt1-WT or a Cdt1-variant that cannot bind CDKs (Cdt1-Cy) were synchronized with nocodazole and harvested by mitotic shake off. Cdt1 was retrieved on nickel-agarose, then both the whole cell lysates (lanes 1-3) and bound fractions (lanes 4-6) were probed for the indicated proteins by immunoblotting. The result is representative of at least two independent experiments.
CDK1 is normally activated by either Cyclin A or Cyclin B, and we sought to determine which cyclin is responsible for directing CDK1 to phosphorylate Cdt1. We therefore took advantage of the polyhistidine tag at the C-terminus of the Cdt1-WT construct to retrieve Cdt1 from lysates of transiently transfected, nocodazole-arrested 293T cells. As a control, we included a Cdt1 variant with a previously-characterized mutation in the cyclin binding motif, Cdt1-Cy41 (RRL to AAA at positions 66-68, Fig. 1a). We analyzed His-Cdt1-bound proteins from these lysates for the presence of endogenous cyclin and CDK subunits. Cdt1-WT interacted with both CDK1 and CDK2, and strongly interacted with Cyclin A, but not at all with either Cyclin B or Cyclin E (Fig. 2b). Cdt1-Cy retrieved no cyclins or CDKs, indicating that the only CDK binding site in Cdt1 is the RRL at positions 66-68. Since Cdt1 binds Cyclin A, CDK1 and CDK2, but only inhibition of CDK1 activity affected Cdt1 phosphorylation, we conclude that Cyclin A/CDK1 is responsible for the inactivating Cdt1 phosphorylations during G2 and M phases.
Cdt1 phosphorylation blocks MCM binding
We next sought to determine by what mechanism Cyclin A/CDK1-mediated phosphorylation inhibits Cdt1 licensing activity. The inhibitory phosphorylation sites are in the linker region between the middle and C-terminal domains (Fig. 1a), and these positions are not visible in any currently available Cdt1 atomic structures. Nonetheless, our recently-generated homology model of the human Cdt1-MCM complex49 led us to speculate that phosphorylation-induced changes at this linker could inhibit MCM binding, either because the linker contacts MCM directly or because phosphorylation alters Cdt1 conformation or relative positions of the two MCM binding domains (Fig. 3a). We thus set out to test if MCM interacts with hypophosphorylated G1 Cdt1 more effectively than with hyperphosphorylated G2 Cdt1 (i.e. if phosphorylation impairs Cdt1-MCM binding). We noted however that simply comparing co-immunoprecipitations from lysates of G1 and G2 phase cells is complicated by the presence of the Cdt1 inhibitor, Geminin, which interferes with the Cdt1-MCM interaction50,51 and is only present in the G2 cells. Because Geminin is differentially expressed in G1 and G2 cells, the comparison would not be fair. To account for the effects of Geminin, we prepared a lysate of asynchronously-proliferating cells which contain mostly G1 hypophosphorylated Cdt1; Cdt1 is degraded in S phase, and cells spend a relatively small fraction of total cell cycle time in G2 (e.g. Supplementary Fig. S2b). We mixed this lysate with lysate from nocodazole-arrested cells that contains both Geminin and hyperphosphorylated Cdt1. In this way, we created a similar opportunity for MCM to bind either hyper- or hypo-phosphorylated Cdt1. We then immunoprecipitated endogenous MCM2 and probed for MCM6 as a marker of the MCM complex and for tagged Cdt1. As a control, we immunoprecipitated MCM2 from an unmixed lysate of nocodazole-arrested cells. As expected, Geminin did not co-precipitate with MCM since the Cdt1-Geminin and Cdt1-MCM interactions are mutually exclusive50,51 (Fig. 3b). We found that the MCM complex retrieved from the mixed lysates was enriched for the faster-migrating hypophosphorylated Cdt1 relative to hyperphosphorylated Cdt1 and that the total amount of Cdt1 bound to MCM was much higher when hypophosphorylated Cdt1 was available (Fig. 3b, compare lanes 5 and 6).

(a) Two views of a homology model of the human MCM2-7-Cdt1 complex as described in Pozo et al.49; numbers refer to individual MCM subunits. The disordered linker containing phosphorylation sites is hand-drawn connecting the two structured Cdt1 domains (MD and CD) in the model.
(b) A lysate of nocodazole-arrested (Cdt1 hyperphosphorylated, Geminin-expressing) U2OS cells ectopically expressing HA-tagged Cdt1-WT was subjected to immunoprecipitation with anti-MCM2 antibody either alone or mixed with lysate from the same cells growing asynchronously. Asynchronous cells contain mostly hypophosphorylated Cdt1. Input lysates (lanes 1-3) and bound proteins (lanes 4-6) were probed for HA, MCM6 (as a marker of the MCM complex), and Geminin; Ponceau S staining serves as a loading control. An asterisk indicates light chain IgG from the immunoprecipitation. The results are representative of three independent experiments.
(c) Asynchronously growing or nocodazole-arrested HEK293T cells ectopically expressing HA-tagged Cdt1-WT or the Cdt1-Cy variant were lysed and subjected to immunoprecipitation with anti-MCM2 antibody. Whole cell lysates (lanes 1 - 4) and bound proteins (lanes 5 - 10) were probed for HA, MCM6 and Geminin, respectively; Ponceau S staining serves as a loading control. The results are representative of two independent experiments.
Furthermore, we compared the MCM binding ability of Cdt1-WT to the Cdt1-Cy variant that cannot bind Cyclin A/CDK1 (Fig. 2b). We transiently transfected 293T cells and then immunoprecipitated MCM2 from asynchronously growing cells or from cells arrested in nocodazole. As expected in asynchronously growing cells (mostly hypophosphorylated Cdt1), there was little difference in MCM binding ability between Cdt1-WT and Cdt1-Cy (Fig. 3c, lanes 6 and 7). In contrast, in nocodazole-arrested cells, Cdt1-Cy bound MCM significantly better than did Cdt1-WT (Fig. 3c, lanes 9 and 10), even in the presence of Geminin (lanes 3 and 4). In summary, these results suggest that Cdt1 phosphorylation disrupts its interaction with MCM complex, and that this disruption contributes to re-replication inhibition in G2 and M phases. We note that this is the first example of direct regulation of the Cdt1-MCM interaction by post-translational modification.
Cdt1 dephosphorylation at the M-G1 transition requires PP1 phosphatase activity
Our finding that Cdt1 phosphorylation in G2 and M phase inhibits its ability to bind MCM suggests that Cdt1 must be dephosphorylated in the subsequent G1 phase to restore its normal function. To explore this notion, we first monitored Cdt1 expression and phosphorylation in cells progressing from M phase into G1. We released nocodazole-arrested cells and collected time points for analysis by immunoblotting (Fig. 4a). Geminin is a substrate of the Anaphase Promoting Complex/Cyclosome (APC/C)52, and as expected for an APC/C substrate, Geminin was rapidly degraded within 60 minutes of mitotic release. In contrast, Cdt1 was not degraded during the M-G1 transition but rather, was rapidly dephosphorylated coincident with Geminin degradation (Fig. 4a, compare lanes 3 and 4). We next explored which phosphatase is required for Cdt1 dephosphorylation. We first tested phosphatase inhibitors for the ability to prevent Cdt1 dephosphorylation after CDK1 inhibition. We tested inhibitors of protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A), which are responsible for the majority of protein dephosphorylation in cells53. We treated nocodazole-arrested cells with the CDK1 inhibitor to induce Cdt1 dephosphorylation in the presence or absence of calyculin A (Cal A) or okadaic acid (OA)54. Both compounds are potent inhibitors of both PP1 and PP2A, but calyculin A is more effective than okadaic acid for inhibiting PP1, particularly at the concentrations we tested55,56. We found that calyculin A maintained Cdt1 hyperphosphorylation (Fig. 4b, compare lanes 2 and 3) whereas concentrations of okadaic acid that inhibit PP2A but not PP1 did not affect Cdt1 dephosphorylation (Supplementary Fig. S5). In addition, we released nocodazole-arrested cells into G1 phase for 30 minutes (to initiate mitotic progression) and then treated the cells with calyculin A. As a control, we probed for MCM4, a known PP1 substrate that is normally dephosphorylated in G1 phase57; calyculin A largely prevented MCM4 dephosphorylation (Fig. 4c). PP1 inhibition also largely prevented Cdt1 dephosphorylation during the mitosis-G1 phase transition without blocking mitotic progression as evidenced by Geminin degradation (Fig. 4c, lanes 2 and 3). These results suggest that the PP1 family phosphatase is required for Cdt1 dephosphorylation. By extension, we suggest that PP1 activity is required to re-activate Cdt1-MCM binding and origin licensing in G1 phase.
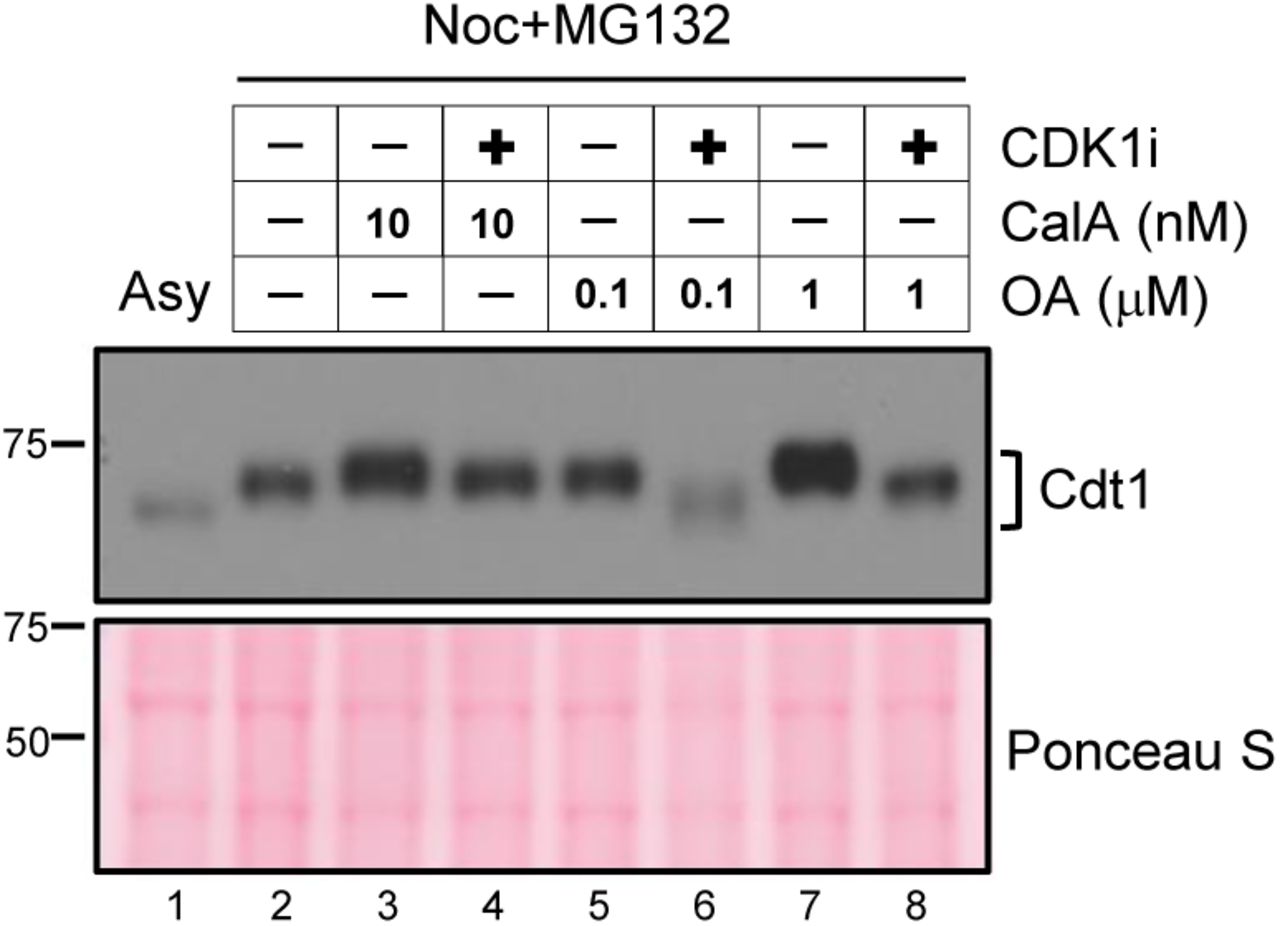
U2OS cells arrested with nocodazole were treated with MG132 followed by mock treatment (lanes 2, 3, 5, and 7) or a CDK1 inhibitor treatment (lanes 4, 6, and 8) to induce dephosphorylation. Cells were also treated with okadaic acid (OA, lanes 5-8) or with calyculin A (CalA lanes 3-4) at the indicated concentrations. Okadaic acid is expected to inhibit PP2A at low concentrations and can only inhibit PP1 at high concentrations86. Cells were harvested by mitotic shake off, and whole cell lysates were subjected to standard SDS-PAGE followed by immunoblotting with HA antibody. A representative of two independent experiments is shown.

(a) Nocodazole-arrested U2OS cells were released into fresh medium and collected at the indicated time points. Endogenous Cdt1 phosphorylation (top) and Geminin (middle) degradation were analyzed by immunoblotting; Ponceau S staining and a non-specific band (*) serve as loading controls. The results are representative of two independent experiments.
(b) Nocodazole-arrested U2OS cells were mock treated (lane 1) or treated with 10 μM RO3306 (CDK1i, lane 2), or treated with both 10 μM RO3306 and with 20 nM calyculin A as indicated (CalA, lane 3). Endogenous Cdt1 phosphorylation was analyzed by standard or Phos-tag SDS-PAGE followed by immunoblotting; Ponceau S staining serves as a loading control. The results are representative of three independent experiments.
(c) Nocodazole-arrested U2OS cells (lane 2) were released into fresh medium for 3 hours and mock treated (lane 1) or treated with 20 nM calyculin A 30 minutes after release (lane 3). Endogenous Cdt1 or MCM4 phosphorylation and total Geminin were detected by immunoblotting; Ponceau S staining serves as a loading control. The results are representative of three independent experiments.
Discussion
Cell cycle-dependent Cdt1 phosphorylation
Mammalian Cdt1 is degraded during S phase, and this degradation is essential to prevent re-replication18,30,42. Perhaps counter-intuitively, Cdt1 then accumulates beginning in late S phase58, and by mitosis reaches a level similar to Cdt1 in G1 phase59,60. Despite the potential danger from re-licensing and re-replicating G2 DNA, these high Cdt1 levels serve two purposes: 1) Cdt1 is essential for stable kinetochore-microtubule attachments61, and 2) high levels of Cdt1 in mitosis can improve licensing efficiency in the next G1 phase60. In this study, we discovered that Cdt1 phosphorylation by Cyclin A/CDK1, a kinase active during G2 phase, inhibits Cdt1 licensing activity and contributes to preventing DNA re-replication while Cdt1 levels are high in G2 and M phase.
We analyzed a cluster of inhibitory Cyclin A/CDK1 phosphorylation sites that are distinct from the only previously-characterized CDK sites at T29 and S31. T29 phosphorylation contributes to Cdt1 degradation during S phase by creating a binding site for the SCFSkp2 E3 ubiquitin ligase. The sites we identified here, S391, T402, T406, S411, and S491, do not induce Cdt1 degradation however. In fact, we previously demonstrated that phosphorylation at these sites stabilizes, rather than destabilizes Cdt119. Aside from separating the four linker sites from the most C-terminal S491 site in the 5A allele, we did not attempt to systematically dissect these “linker” phosphorylation sites, largely because this region of Cdt1 is not strongly conserved among vertebrates (Fig. 1a and Supplementary Fig. S1). Most vertebrate Cdt1 linker sequences are nonetheless predicted to be similarly disordered, and most have at least one candidate CDK phosphorylation site (Supplementary Fig. S1). Interestingly, altering two additional sites in this region (converting Cdt1-5A to Cdt1-7A) did not exacerbate the re-replication phenotype suggesting that four phosphorylations are sufficient to achieve maximal Cdt1 inhibition. It may be that the four linker sites vary in their relative importance for inhibiting human Cdt1 activity, or it may simply be the need for a total amount of phosphorylation in this region regardless of specific position. In that regard, multisite Cdt1 linker phosphorylation may resemble other examples of cell cycle-dependent multisite phosphorylation in which the total negative charge is more important than the specific phosphorylated position62. If so, then all four sites in Cdt1 may work additively to achieve maximal inhibition.
Although we had previously established that stress-activated MAP kinases (p38 and JNK) can phosphorylate these inhibitory sites in Cdt1 during a stress response, and both p38 and JNK are active during a G2 arrest, we could detect no contribution of stress MAPK activity to endogenous Cdt1 phosphorylation during G2 and M phases. (The ability of a JNK inhibitor to reverse Cdt1 phosphorylation in nocodazole-arrested cells may be attributed to off-target indirect effects of the drug on CDK1 activity63.) On the other hand, our findings here do shed light on the molecular mechanism of stress-induced origin licensing inhibition. We postulate that MAPK-mediated Cdt1 hyperphosphorylation at the linker region blocks Cdt1-MCM binding just as it does in a normal G2 or M phase, even in cells that do not express Geminin.
The nine phosphorylation sites we tested in this study are by no means the only phosphorylation sites in human Cdt1. Unbiased phosphoproteomics studies have detected phosphorylation at a total of 22 sites, 13 of which are also serine/threonine-proline sites17. In addition to the nine sites included here, Agarwal et al. recently reported that Cdt1 is a substrate for Aurora B kinase, and that phosphorylation at eight sites distributed across the N-terminal two-thirds of the Cdt1 primary sequence regulates its direct microtubule binding activity64. In addition, an incompletely-characterized PEST domain that restrains Cdt1 licensing activity by influencing chromatin association (but not stability) includes other untested candidate CDK sites59. Clearly the spectrum of Cdt1 biological activities can be determined by combinations of phosphorylations and dephosphorylations, and continued in-depth analyses will yield additional insight into Cdt1 regulation and function.
Only Cyclin A/CDK1 phosphorylates Cdt1 in G2/M phase
Cdt1 only binds endogenous Cyclin A and neither Cyclin E nor Cyclin B. As a result, we presume that SCFSkp2 targeting for degradation can only happen once Cyclin A accumulates in mid-S phase. The fact that endogenous Cyclin E cannot bind Cdt1 means that Cdt1 is both stable and active throughout G1 phase even after Cyclin E/CDK2 becomes active in late G1. Cdt1 is thus not a substrate for SCFSkp2 until after Cyclin A has accumulated during S phase, but by then, most of Cdt1 has already been destroyed at the onset of S phase by CRL4Cdt2-mediated replication-coupled destruction43,65-67. The basis for cyclin specificity is still not fully understood however. A hydrophobic patch on cyclins is critical for substrate recognition68, and subtle differences in this sequence may explain why Cdt1 is not a substrate for either Cyclin E or Cyclin B. Alternatively, the catalytic CDK1 subunit itself may contribute to substrate specificity69.
Our binding assays indicate that the Cy docking motif is the only CDK binding site in Cdt1. Efficient phosphorylation requires simultaneous interaction of the CDK with both the substrate phospho-acceptor site(s) and the CDK docking motif 70,71. We demonstrate here that the Cdt1 docking motif at positions 68-70 is required for phosphorylation not only at the previously-investigated T29 position, but also at sites more than 300 residues towards the C-terminus. This finding prompts us to speculate that in free Cdt1 (i.e. not bound to MCM, ORC, or Cdc6), the linker region is relatively close to the N-terminal regulatory domain containing the Cy motif. The structure of the yeast Cdt1-MCM complex indicates that when bound to MCM, Cdt1 is in a relatively extended conformation with the linker distant from the N-terminal domain72,73,74. This extended binding surface with multiple MCM contacts supports a role for Cdt1 in maintaining the MCM ring in an open state compatible with DNA loading72.
Phosphorylation inhibits Cdt1 binding to MCM
We found that hyperphosphorylated Cdt1 binds MCM poorly relative to hypophosphorylated Cdt1. This observation provides a simple mechanism for Cyclin A/CDK1-mediated phosphorylation to inhibit Cdt1 licensing activity. Both the Cdt1 N-terminal domain and the linker region are predicted to be intrinsically disordered, and the fact that these regions were excluded from mammalian Cdt1 fragments subjected to structure determination supports that prediction25,26. The only structure of full-length Cdt1 available to date is part of the budding yeast Cdt1-MCM or ORC/Cdc6/Cdt1/MCM complexes73,74, and budding yeast Cdt1 lacks candidate phosphorylation sites in the linker region. For this reason, we cannot determine precisely how phosphorylation in the linker inhibits MCM binding. We suggest however, that the introduction of multiple phosphorylations either induces a large conformational change in Cdt1 that prevents it from extending around the side of the MCM ring or alternatively, these phosphorylations may directly repel Cdt1 from the MCM surface (Fig. 3a). Of additional note the linker phosphorylations are not in the Cdt1 domain that is both necessary and sufficient for Geminin binding25,75. Thus as expected and consistent with previous findings40, this mutation does not affect the binding of Cdt1 to Geminin (data not shown), indicating that the contribution of the phosphorylation site mutations to induced re-replication is Geminin-independent.
PP1-dependent Cdt1 dephosphorylation
Approximately one-third of all eukaryotic proteins may be dephosphorylated by PP153. PP1 binds some of its substrates directly via a short motif, RVxF, KGILK or RKLHY53,76. Human Cdt1 contains several such predicted PP1 binding motifs and thus may be a direct target of PP1. Alternatively, Cdt1 dephosphorylation may require an adapter to bind PP1 similar to the role of the Rif1 adapter for MCM dephosphorylation77. In either case, the fact that hyperphosphorylated Cdt1 binds MCM poorly, plus the fact that the levels of Cdt1 do not change from M phase to G1 (i.e. Cdt1 is not degraded and resynthesized at the M-G1 transition), means that PP1-dependent Cdt1 dephosphorylation activates origin licensing. In that regard, dephosphorylation is the first example of direct Cdt1 activation, and it complements indirect activation by Geminin degradation in M phase.
A sequential relay of re-replication inhibition mechanisms
We propose that Cdt1 activity is restricted to G1 through multiple regulatory mechanisms during a single cell cycle, but that the relative importance of individual mechanisms changes at different times after G1 (Fig. 5). At the onset of S phase Cdt1 is first subjected to rapid replication-coupled destruction via CRL4Cdt2 which targets Cdt1 bound to DNA-loaded PCNA78. This degradation alone is not sufficient to prevent re-replication however, and a contribution from Cyclin A/CDK2 to create a binding site for the SCFSkp2 E3 ubiquitin ligase is also essential30. We suggest that SCFSkp2-targeting occurs primarily in mid and late S phase based on the dynamics of Cyclin A accumulation41,79. A reinforcing mechanism for Cdt1 degradation is more important in mid and late S phase than in early S phase because the amount of DNA that has already been copied increases throughout S phase. Licensing DNA that hasn’t been copied yet is presumably benign, but as S phase proceeds, the amount of DNA that has been copied already (i.e. the substrate for re-replication) also increases. The Cdt1 inhibitor, Geminin, begins to accumulate in early S phase, and its levels increase along with the amount of replicated DNA until Geminin is targeted for degradation by the APC/C during mitosis21,52. Geminin binding to Cdt1 interferes with Cdt1-MCM binding, and since Cdt1-MCM binding is essential for MCM loading, Geminin prevents re-licensing31,32. This inhibition is particularly important once Cdt1 re-accumulates after S phase is complete; in late S phase the responsibility for restraining Cdt1 is passed from the ubiquitin ligases to Geminin and Cyclin A/CDK1. Just as CRL4Cdt2-mediated degradation in S phase is not sufficient to fully prevent re-replication, we demonstrated that the presence of Geminin alone is not sufficient to inhibit Cdt1 during G2. Cyclin A/CDK1-mediated Cdt1 phosphorylation in a linker domain between two MCM binding sites49,72,73,74 also prevents Cdt1-MCM binding. These (and potentially more) mechanisms to restrain Cdt1 activity are also reinforced by regulation to inhibit ORC, Cdc6, PR-Set7, and other licensing activators3,16,29,80. Given that there are many thousands of potential origins in mammalian genomes, and the consequences of even a small amount of re-replication are potentially dire, precise once-and-only-once replication requires that Cdt1 be inhibited by at least two mechanisms at all times from G1 through mitosis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cdt1 levels are indicated by the dashed blue line whereas Cdt1 licensing activity is indicated by the solid blue line. Geminin protein levels are indicated by the dashed pink line. G1 phase: Cdt1 plays an essential role in MCM helicase loading. S phase: Cdt1 is first targeted by the CRL4Cdt2 E3 ubiquitin ligase at the onset of S phase and then additionally by SCFSkp2 after phosphorylation by Cyclin A/CDK2. Geminin accumulates beginning in early S phase. The amount of duplicated DNA at risk of re-replication is lowest in early S and highest in G2. Late S and G2 phase: Cdt1 re-accumulates when Geminin is at peak levels. Cyclin A/CDK1 phosphorylates Cdt1, and both Geminin and hyperphosphorylation independently interfere with Cdt1-MCM binding. M-G1 transition: Protein Phosphatase 1 is required for Cdt1 dephosphorylation to reactivate MCM loading.
Methods
Sequence analysis
A representative selection of vertebrate sequences for comparison was taken from Miller et al.81 and Cdt1 protein sequences retrieved from https://www.uniprot.org/. For the alignment, Xenopus tropicalis Cdt1 was replaced with Xenopus laevis Cdt1, Tupaia belangeri was replaced with Tupaia chinensis, and no Cdt1 sequence for Echinops telfairi (tenrec) was available. Full-length sequences were aligned with ClustalW at https://www.genome.jp/tools-bin/clustalw using the default settings. Conservation scores were determined according to Capra and Singh82, and intrinsic disorder predicted at https://iupred2a.elte.hu/ 83. Heat maps were generated in GraphPad Prism.
Cell Culture and Manipulations
U2OS Flp-in Trex cells84 bearing a single FRT site (gift of J. Aster) and HEK 293T cells were cultured at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum, 2 mM L-glutamine, 10 U/mL of penicillin and 10 μg/mL of streptomycin. For kinase inhibitor treatments, cells arrested by thymidine-nocodazole synchronization were incubated with each inhibitor combination for 1 hour or 15 minutes and harvested by mitotic shake-off as described19. Cells were treated with 10 μM, RO-3306 (Sigma), 6 μM CVT313 (Sigma), 10 μM JNK inhibitor VIII (Sigma), 30 μM SB203580 (Sigma) or 20 nM, calyculin A (LC Laboratories) as indicated. For the transient expression of Cdt1 variants, HEK 293T cells were transfected with Cdt1 expression plasmids using PEI Max (Sigma). Transfected cells were harvested after 16 hours and processed for subsequent co-immunoprecipitation assays. All cell lines were validated by STR profiling and mycoplasma test.
Antibodies
Antibodies were purchased from the following sources: Cdt1 (Cat# 8064), Chk1 (Cat# 2345), phospho-Chk1 S345 (Cat# 2341), Cyclin E1 (Cat#4129), MAPKAPK-2 (Cat#), Phospho-MAPKAPK-2 T334 (Cat#3007), phospho-Histone H2A.X Ser139 (Cat#9718) from Cell Signaling Technologies; hemagglutinin (HA) (Cat#11867423001) from Roche; Geminin (Cat#sc-13015), Cdc6 (Cat#sc-9964), MCM6 (Cat#sc-9843), Cyclin A (Cat#sc-596), Cyclin B1 (Cat#sc-245) and CDK2 (Cat#sc-163) from Santa Cruz Biotechnology; MCM4 (Cat#3728) from Abcam. MCM2 antibody (Cat#A300-191A) used for co-immunoprecipitation experiment was purchased from Bethyl Laboratories. Serum to detect CDK1 was a gift from Y. Xiong (University of North Carolina), and MPM2 antibody was a gift from R. Duronio85 (University of North Carolina). Alexa 647-azide used in flow cytometry analyses was purchased from Life Technologies, and secondary antibodies for immunoblotting were purchased from Jackson ImmunoResearch.
Flow cytometry
U2OS cells were cultured in complete medium with 1 μg/mL doxycycline for 48 hours to induce expression of wild type or mutant Cdt1. Cells were labeled with EdU (Sigma) using a final concentration of 10 μM in the medium for 1 hour followed by trypsinization. The cells were fixed with 4% formaldehyde (PFA) for 15 minutes, extracted with 0.5% Triton X-100 for 15 minutes, then processed for EdU detection by conjugation to Alexa Fluor 647 azide in 1 mM CuSO4 and 100 mM ascorbic acid; DNA was stained with 1 μg/mL DAPI (Life Technologies) in 100 μg/mL RNAse A (Sigma). Samples were analyzed using a Beckman Coulter CyAn ADP cytometer and Summit software 4.3.
Protein-protein interaction assays
For polyhistidine pulldown assays, cells were lysed in lysis buffer (50 mM HEPES pH 8.0, 33 mM KAc, 117 mM NaCl, 20 mM imidazole, 0.5% triton X-100, 10% glycerol) plus protease inhibitors (0.1 mM AEBSF, 10 μg/mL pepstatin A, 10 μg/mL leupeptin, 10 μg/mL aprotinin), phosphatase inhibitors (5 μg/Ml phosvitin, 1 mM β-glycerol phosphate, 1 mM Na-orthovanadate), 1 mM ATP, 1 mM MgCl2, 5 mM CaCl2 and 15 units of S7 micrococcal nuclease (Roche). Lysates were sonicated for 10 seconds at low power followed by incubation on ice for 30 minutes and clarification by centrifugation at 13,000 x g for 15 minutes at 4°C. The supernatants were incubated with nickel NTA agarose beads (Qiagen) for 2 hours at 4°C with rotation. Beads were rinsed 4 times rapidly with ice-cold lysis buffer followed by boiling in SDS sample buffer for 5 minutes prior to immunoblot.
For co-immunoprecipitation assays, cells were lysed in Co-IP buffer (50 mM HEPES pH 7.2, 33 mM KAc, 1 mM MgCl2, 0.5% triton X-100, and 10% glycerol) containing protease inhibitors (0.1 mM AEBSF, 10 μg/mL pepstatin A, 10 μg/mL leupeptin, 10 μg/mL aprotinin), phosphatase inhibitors (5 μg/mL phosvitin, 1 mM β-glycerol phosphate, 1 mM Na-orthovanadate), 1 mM ATP, and supplemented with 5 mM CaCl2 and 15 units of S7 micrococcal nuclease (Roche). Lysates were sonicated for 10 seconds at low power followed by incubation on ice for 30 minutes and clarification by centrifugation at 13,000 x g for 15 minutes at 4°C. The supernatants were incubated and rotated with Protein A beads (Roche) with an anti-Mcm2 antibody (Bethyl, 1:1000) at 4°C with rotation for 4 hours. Beads were rinsed three times with ice-cold co-IP buffer then eluted by boiling in sample buffer for subsequent immunoblot analysis.
Immunofluorescence microscopy
U2OS cells cultured on cover glass were fixed with 4% PFA for 15 minutes and permeabilized with 0.5% Triton in PBS for 5 minutes. Cells were blocked in 1% BSA for 30 minutes followed by incubation with primary antibody overnight at 4°C and secondary antibody for 1 hour at room temperature. Cells were stained with 1 μg/ml DAPI for 5 minutes before mounting with the ProLong® Gold Antifade mounting medium (life technologies). Fluorescent images were captured on a Nikon 2000E microscope. The areas of nuclei were measured by using the Adobe Photoshop software.
Statistical analysis
The differences were considered significant with a p-value less than 0.05 in an unpaired Student t-test or a Mann–Whitney U -test using Sigmaplot software (Systat Software).
Acknowledgements
We thank Y. Xiong, R. Duronio, L. Graves, and J. Aster for the generous gifts of antibodies and reagents, and all members of the Cook lab for comments on the manuscript. We thank Jeffrey Jones for research support assistance. The UNC Hooker Imaging Core and the UNC Flow Cytometry Core Facility are supported in part by a National Institutes of Health Cancer Core Support Grant to the UNC Lineberger Comprehensive Cancer Center (CA016086). This work was also supported by National Institutes of Health grants F31GM121073 to P.N.P. and R01GM102413 to J.G.C.
References




