

Abstract
RNAseq technology provides an unprecedented power in the assesment of the transcription abundance and can be used to perform a variety of downstream tasks such as inference of gene-correlation network and eQTL discovery. However, raw gene expression values have to be normalized for nuisance biological variation and technical covariates, and different normalization strategies can lead to dramatically different results in the downstream study. Here we present a simple three-parameter transformation, DataRemix, which can greatly improve the biological utility of gene expression datasets without any specific knowledge on the dataset. As we optimize the transformation with respect to the downstream biological objective, this parametric framework reweighs the contribution of each hidden factor and makes the biological signals visible. We demonstrate that DataRemix can outperform normalization methods which make explicit use of dataset specific technical factors. Also we show that DataRemix can be efficiently optimized via Thompson Sampling approach, which makes it feasible for computationally expensive objectives such as eQTL analysis. Finally we reanalyze the Depression Gene Networks (DGN) dataset, and we highlight new trans-eQTL networks which were not reported in the initial study.
Genome-wide gene expression studies have become a staple of large scale systems biology and clinical projects. However, while gene expression is the most mature high-throughput technology, technical challenges remain. Raw gene expression values must be normalized for any technical and nuisance biological variation and the normalization strategy can have dramatic effects on the results of downstream analysis. This is especially true in cases where the sought-after gene expression effects are likely to be small in magnitude, such as expression quantitative trail loci (eQTLs). Increasingly sophisticated normalization methods have been proposed and many are computational intensive and/or can have multiple free parameters that must be optimized (Leek & Storey 2007; Stegle et al.. 2010; Listgarten et al.. 2010; Kang et al.. 2008; Mostafavi et al.. 2013). Moreover, it is not uncommon for one dataset to yield multiple normalized versions that maximize performance in a particular setting (such as the discovery of cis-and trans-eQTLs Battle et al.. 2014), highlighting the complexity of the normalization problem.
Singular value decomposition (SVD) is one of the most widely used gene expression analysis tools (Alter et al.. 2000, 2003) that can also be used for data normalization. Using the SVD we can simply remove the first few principle components that are presumed to represent technical factors such as batch-effects or other nuisance variation. In some cases this dramatically improves downstream performance, for example in the case of eQTL analysis (Mostafavi et al.. 2013). The drawback of this method is that the exact number of components to remove must be determined empirically and some meaningful biological signals may be lost in the process.
More sophisticated approaches attempt to partition data structure into true biological and nuisance variation and remove only the latter (Leek & Storey 2007; Stegle et al.. 2010; Listgarten et al.. 2010; Kang et al.. 2008; Mostafavi et al.. 2013). These can improve on the naive SVD-based normalization but require additional input such as technical covariates, or the study design. The success of these methods ultimately depends on the availability and quality of such meta data and some methods still rely on parameter optimization to maximize performance. These widely used normalization approaches all have a common theme that the rely in part on the intrinsic data structure. One key property that contributes to the success of these approaches is that for many biological questions of interest nuisance variation (of technical or biological origin) is larger in magnitude than true biological variation. Our proposed method, DataRemix, explicitly formalizes this view of the data normalization problem.
In this work we demonstrate that biological util-ity of gene expression datasets can be dramatically improved with a simple three-parameter transformation, DataRemix. Our method does not require any dataset specific knowledge but rather optimizes the transformation with respect to some independent objective of data quality, such as the quality of the genecorrelation network or the number of trans-eQTL discoveries. Because our method requires only the gene expression data and biological validity objective, it can be applied to any publicly available dataset. We focus our study on gene expression data for which methods for quantifying biological validity are well established, but our approach can be readily applied to any high-throughput molecular data for which similar quality metrics can be defined. We show that this strategy can outperform methods that make explicit use of dataset specific factors, and can further improve datasets that have been extensively normalized via an optimized, parameter rich model. We also show how the optimal parameters of DataRemix can be found efficiently by Thompson Sampling with a dual learning setup, making the approach feasible for computationally expensive objectives such as eQTL analysis.
Result
The DataRemix framework
We formulate DataRemix as a simple parametrized version of SVD which can be directly optimized to improve the biological utility of gene expression data. Given a gene-by-sample matrix X, SVD decomposition can be thought of as a solution to the low-rank matrix approximations problem defined as:
 where U and V are unitary matrices. With the SVD decomposition U ΣV T, the product of k-truncated matricies
where U and V are unitary matrices. With the SVD decomposition U ΣV T, the product of k-truncated matricies  gives the rank-k approximation of X. We introduce two additional parameters p and µ to define a new reconstruction:
gives the rank-k approximation of X. We introduce two additional parameters p and µ to define a new reconstruction:

Here, k is the number of principle components of SVD and p ∈ [-1, 1] is a real number which alters the scaling of each eigenvalue. For p = 1, this approach reduces to the original SVD-based reconstruction. For p = 0 the transformation gives the frequently used whitening operation (Friedman 1987). As depicted in Figure 1, generally, different choices of p reweigh the contribution of each variance component, possibly making some low-variance biological signals visible while down-weighting technical and other systematic noise. The parameter µ is a non-negative weight that adds the residual back to the reconstruction in order to make the transformation lossless.
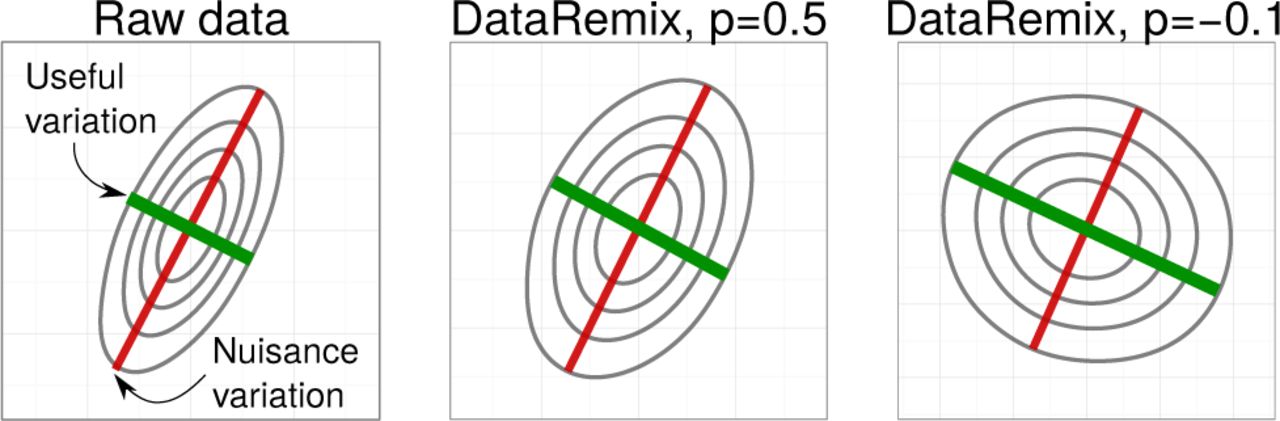
Visual representation of DataRemix transformation. We simulate a 2-dimensional dataset where the nuisance variation contributes more variance than true biological variation. Different power parameters p reweigh the contributions of the two variance axes, making the true biological variation more “visible”.
Intuitively, we expect this approach to succeed because sophisticated normalization methods that use both data structure and some external variables, such as technical covariates, can be thought of as implicit regularizations on the naive SVD-based normalization (which simply removes the first k components), and this formulation simply makes this explicit.
The general workflow of DataRemix is shown in Figure 2. The downstream biological objective depends on your study. Fox example, if you focus on trans-eqtl analysis, the biological objective will be to increase the number of trans-eqtls detected from the DataRemix-normalized gene expression profile and the metric y will be the number of trans-eqtls deemed significant. The parameter optimization step which determines the next point to check is detailed in the next section.

The workflow of DataRemix.
Parameter Optimization
The parameters λ = (k, p, µ) need to be optimized with respect to a particular biological objective. Grid search and random search (Bergstra & Bengio 2012) are among the most popular strategies, but these methods have low efficiency. Most of the search steps are wasted and the optimality of parameters is highly constrained by the step size and available computing power. In order to utilize the search history and keep a good balance between exploration and exploitation, we can formulate parameter search as a dual learning task.
We define a general performance measure y = L(λ, D), with λ representing the parameter tuple (k, p, µ), D as the data, L as the evaluating process and y as the biological objective. Ideally we can determine the optimal point argmaxλ L easily by gradient descent based method, but usually L is derivative-free and it is time intensive. Thus we introduce a surrogate model f (λ) which can directly predict L(λ, D) only given λ. There are two conditions on f : argmaxλ f should be easy to solve and f should have enough capacity.
With these two properties, we can sequentially update f with (λt, yt) and propose to evaluate L at λt+1 = argmaxλ f in the next step. By gradually updating f with newly evaluated samples (λ, y), argmaxλ f approaches the true underlying optimal argmaxλ L as f can gradually fit to the underlying mapping function L. This provides a more efficient approach to explore the parameter space by exploiting the search history. In this work, we model f as a sample from a Gaussian Process with mean 0 and kernel k(λ, λ′), where λ = (k, p, µ)T. It is well known that the form of the kernel has considerable effect on performance. After experimentation we settled on the exponential kernel as the most suited for our application. The exponential kernel is defined as below (note the difference from the squared-exponential or RBF kernel).

We observe yt = f (λt) + ∊t, where ∊t ∼N (0, σ2). For Bayesian optimization, one approach for picking the next point to sample is to utilize acquisition functions (Snoek et al.. 2012) which are defined such that high acquisitions correspond to potentially improved performance. An alternative approach is the Thompson Sampling approach (Basu & Ghosh 2017; Agrawal & Goyal 2013; Hernández-Lobato et al.. 2014). After we update the the posterior distribution P (f |λ1:t , y1:t), draw one sample f from this posterior distribution as the optimization target to infer λt+1. Theoretically it is guaranteed that λt converges to the optimal point gradually (Agrawal & Goyal 2013). With this theoretical guarantee, we focus on Thompson Sampling approach to optimize parameters for DataRemix.
Estimation of Hyper-Parameters
First we rely on the maximum likelihood estimation (MLE) to infer the variance of noise σ2 (Rasmussen 2004). Given the marginal likelihood defined by (4), it is easy to use any gradient descent method to determine the optimal σ2
 where
where  and K is the covariance matrix with each entry Kij = k(λi, λj).
and K is the covariance matrix with each entry Kij = k(λi, λj).
Sampling from the Posterior Distribution
Since Gaussian Process can be viewed as Bayesian linear regression with infinitely many basis functions ϕ0(λ), ϕ1(λ), … given a certain kernel (Rasmussen 2004), in order to construct an analytic formulation for the sample f, first we need to construct a certain set of basis functions Φ(λ) = (ϕ0(λ), ϕ1(λ), …), which is also defined as feature map of the given kernel. Then we can write the kernel k(λ, λ′) as the inner product Φ(λ)T Φ(λ′).
Mercer’s theorem guarantees that we can express the kernels in terms of eigenvalues and eigenfunctions, but unfortunately there is no analytic solution given the exponential kernel we used. Instead we make use of the random Fourier features to construct an approximate feature map (Rahimi & Recht 2008). First we compute the Fourier transform p of the kernel (see Supplemental Note for derivation).
 where
where  and
and  Then we draw mt iid samples ω1, …, ωmt ∈ ℝ3 by rejection sampling with () as the probability distribution. Also we draw mt iid samples b1, …, bmt ∈ℝ from the uniform distribution on [0, 2π]. Then the feature map is defined by the following equation.
Then we draw mt iid samples ω1, …, ωmt ∈ ℝ3 by rejection sampling with () as the probability distribution. Also we draw mt iid samples b1, …, bmt ∈ℝ from the uniform distribution on [0, 2π]. Then the feature map is defined by the following equation.
 where the dimension mt can be chosen to achieve the desired level of accuracy with respect to the difference between true kernel values k(λ, λ′) and the approximation Φ(λ)T Φ(λ′).
where the dimension mt can be chosen to achieve the desired level of accuracy with respect to the difference between true kernel values k(λ, λ′) and the approximation Φ(λ)T Φ(λ′).
Thompson Sampling
Any sample f from the Gaussian Process can be defined by f (λ) = Φ(λ)T θ, where θ ∼N (0, I) and Φ(λ)T is defined by (6). In order to draw a posterior sample f, we just need to draw a random sample θ from the posterior distribution 
 where
where  and
and  (see Supplemental Note for more details). The overall algorithm is summarized as the following pseudo code.
(see Supplemental Note for more details). The overall algorithm is summarized as the following pseudo code.

Quality of the correlation network derived from the GTex gene expression study
The GTex datasets (Lonsdale et al.. 2013) is comprised of human samples from diverse tissues, many of which were obtained post-mortem and there are many technical factors which have considerable effects on the gene expression measurements. On the other hand this rich dataset provides an unprecedented multi-tissue map of gene regulatory networks and has been extensively analyzed in this context. It is natural to assume that a dataset that is better at recovering known pathways is likely to yield more credible novel predictions. Thus, we use DataRemix to optimize the known pathway recovery task as a function of the correlation network computed on a Remixed dataset.
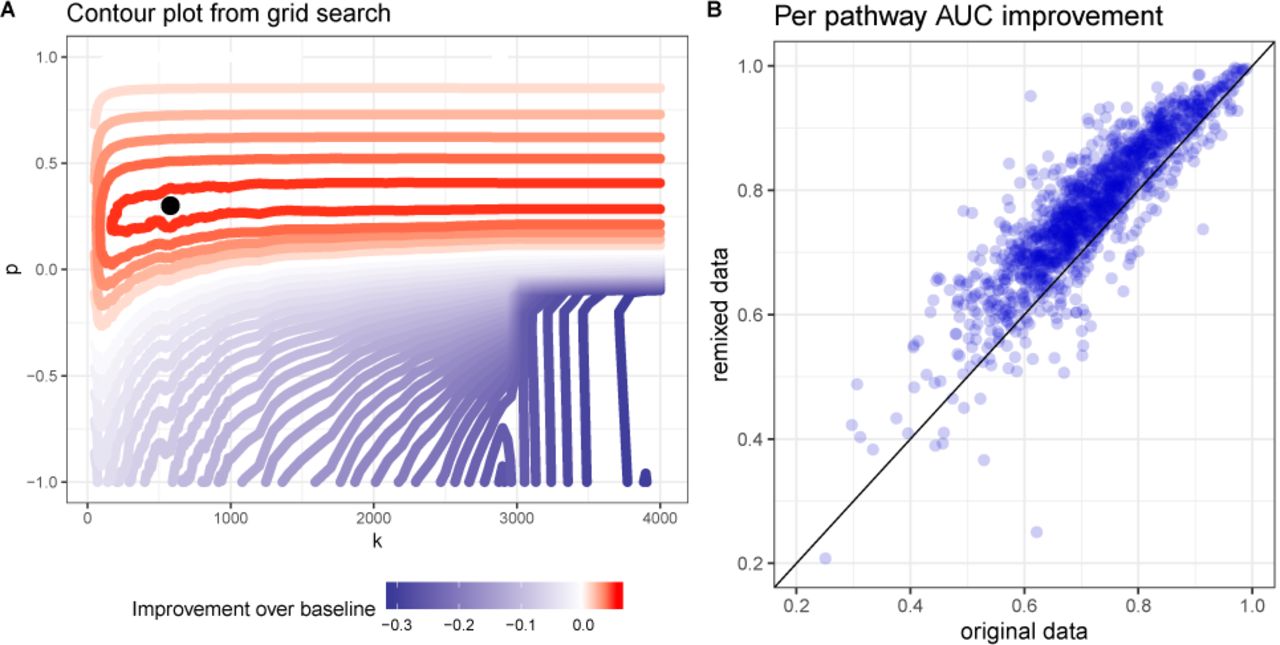
Specifically we start with a quantile normalized TPM data that has not been corrected for technical factors or tissue of origin. We formally define the objective as the average AUC across “canonical” mSigDB pathways (which include KEGG, Reactome and PID) (Subramanian et al.. 2005) using guilt-by-association. Specifically, the genes are ranked by their average Pearson correlation to other genes in the pathway (excluding the gene when the gene itself is a pathway member). Figure 3A depicts the results of grid search for the parameters k, p (with µ fixed at 0.01) and the contour plot shows a clear region of increased performance. Using the optimal transformation found by grid search, we plot per-pathway AUC improvement in Figure 3B and find that the AUC is substantially increased for almost every pathway.

A The improvement in performance of DataRemix transform of the pathway prediction task visualized as a function of k and p parameters (µ is fixed at 0.01). Performance is measured as the mean AUC across all pathways in the “canonical” mSigDB dataset and the red contours indicate improvement over the performance on untransformed data. B Per-pathway performance improvement for the optimal DataRemix transformation.
eQTL discovery in the DGN dataset
We also consider the task of discovering cis- and trans-eQTLs on the Depression Gene Networks (DGN) dataset (Battle et al.. 2014). In the original analysis this dataset was normalized using the Hidden Covariates with Prior (HCP) (Mostafavi et al.. 2013) with four free parameters that were separately optimized for cis- and trans-eQTLs. The rationale behind separate cis and trans optimized normalization can be understood in terms of which variance components represent true biological vs. nuisance variation in the two contexts. Specifically, cis-eQTLs represent direct effects of genetic variation on the expression of a single gene. On the other hand, trans-eQTLs represent network level, indirect effects that are mediated by a regulator. Thus, trans-eQTLs are reflected in systematic variation in the data which becomes a nuisance factor when only direct effects are of interest. It thus follows that the data should be more aggressively normalized for cis-eQTL discovery. The original analysis of this dataset optimized the HCP parameters separately for the cis and trans tasks yielding two different datasets that we refer to as Dcis-optim and Dtrans-optim.
The HCP model takes various technical covariates as input, and of the covariates used in the original study 20 cannot be inferred from the gene-level counts. In order to investigate how much improvement can be achieved via DataRemix in the absence of access to these covariates we also consider a “naively” normalized dataset, quantile normalization of log-transformed counts, or DQN.
cis-eQTLs
In this task we focus on optimizing the discovery of cis-eQTLs. We define cis-eQTLs as a SNP-gene interaction where the SNP is located within 50kb of the gene’s transcription start site. The interaction is quantified with Spearman rank correlation and deemed significant at 10% FDR (Benjamini-Hochberg correction for the total number of tests).
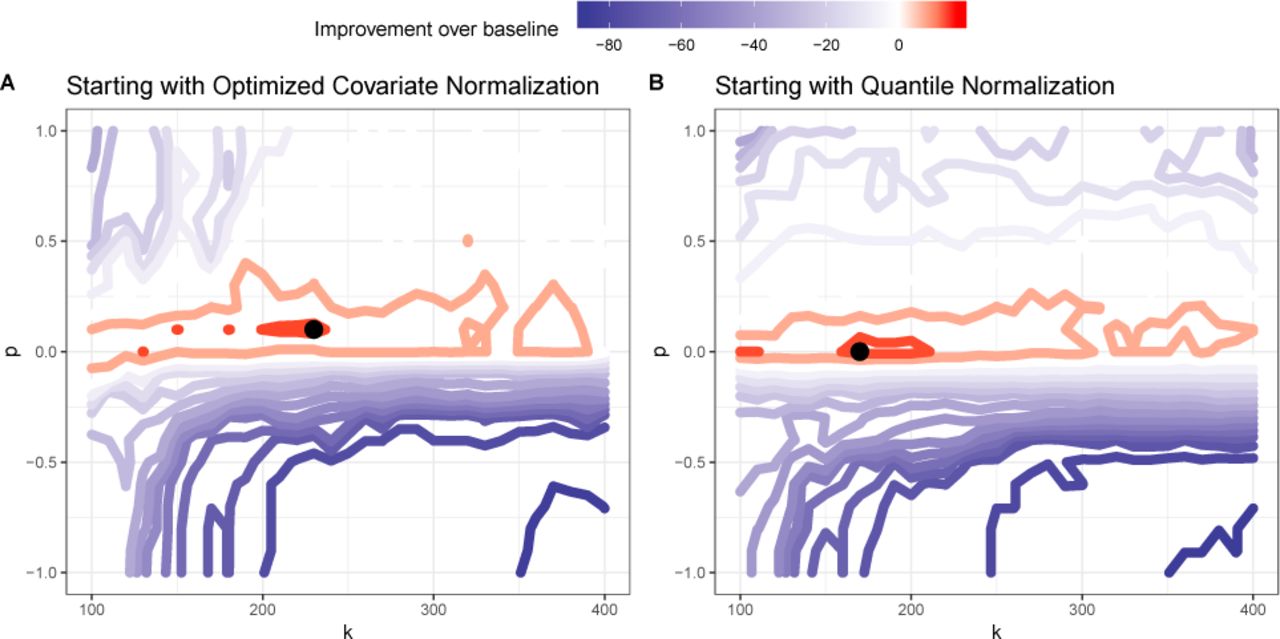
We perform our analysis in a cross-validation framework, whereby we can optimize DataRemix parameters (using grid search or Thompson Sampling) using SNPs on the odd chromosomes only and then evaluate the parameters on the held-out even chromosome set. We visualize the effect of varying the k and p parameters on the performance of the DataRemix transform in Figure 4. Red regions indicate improvement over the number of cis-eQTLs discovered with the Dcis-optim dataset. We find that both versions of the dataset can be improved via the DataRemix transform to a similar degree. We also find that on this task the optimal p parameter is negative and the result is relatively insensitive to the choice of k. The last observation can be interpreted when we consider the interaction between p and µ (the multiplier for the residual part including k +1 through max(k) components). If we wish to bring forward small-variance components, as is the case with cis-eQTL discovery, we would like the diagonal values of µΣk+1:rank of X, representing the contribution of the later components, to be in the same range or larger than max which is the largest contribution of the high variance components. This can be achieved by picking different values of k.
which is the largest contribution of the high variance components. This can be achieved by picking different values of k.

Contour plot representing the effects of the k and p parameters on the performance of DataRemix on cis-eQTL discovery on 50,000 randomly selected SNPs on odd chromosomes (training set). Red contours represent parameter combinations that increase the number cis-eQTLs beyond what can be achieved using the Dcis optim dataset. Panel A shows the results starting withDcis optim while DQN is used for panel p. Improvement can beachieved starting with either dataset. We note that the optimal p parameter is negative (though slightly different) for both datasets.
The final results for both the train and test set are depicted in Figure 5. We find that the optimal parameters are indeed generalizable as we achieve a similar level of improvement on the train and test datasets. Importantly, we find that while the quantile-normalized dataset DQN performs considerably worse that Dcis-optim the two datasets achieve comparable performance after applying DataRemix. Moreover, the final performance of the Remixed DQN dataset is an improvement of the baseline Dcis-optim demonstrating the near optimal normalization is possible without access to technical covariates. We do note, on this task, the final performance of the Remixed Dcis-optim is slightly better than that of DQN and thus it is still advisable to include such covariates in the normalization pipeline if they are available.

Final results from DataRemix parameter search using a cross-validation framework. Optimal parameters are determined using the odd chromosome SNPs only and then tested on the even chromosome SNPs. We find that the DataRemix transform does not overfit the objective as the degree of improvement is similar across the test and train SNP sets (note: the starting value of the baseline (DataRemix=”None”) datasets differ between the test and train SNP set). Moreover, we find that Thompson Sampling is able to match grid search results using only 100 evaluations.
Contour plot representing the effects of the k and p parameters on the performance of DataRemix on trans-eQTL discov-ery on 50,000 randomly selected SNPs on odd chromosomes (training set). Red contours represent parameter combinations that increase the number of trans-eQTLs beyond what can be achieved using the Dtrans optim dataset. Panel A shows the results starting with Dtrans optim while DQNis used for panel p. Improvement can be achieved starting with either datasets. We note that the performance is more sensitive to the choice of k.
trans-eQTLs
In our third task, we optimize the discovery of trans-eQTLs in the same DGN dataset. Ideally, trans-eQTLs represent network-level effects and thus give some insight about the regulatory structure of gene expression. However, in practice trans-eQTLs are simply defined as SNP-gene associations where the SNP and the gene are located on different chromosomes. While this is a useful heuristic definition, it doesn’t guarantee that the association is mediated at the network level. One possible source of bias is mis-mapped RNAseq reads which contaminate the quantification of the apparently trans-associated gene with reads from a homologous locus that has cis association. Even in the absence of technical artifacts, direct interchromsomal interactions have been observed (see Williams et al.. 2010 for a comprehensive review). In order to focus on potential indirect effects, we apply an additional filter to trans-eQTL discovery. Specifically we require SNPs involved in a trans effect to be associated with more than one gene at a FDR of 20% (Benjamini-Hochberg correction for the total number of test (approximately 8×109). We term these SNPs trans-SNPs+. In comparison with same chromosome cis-eQTLs, inter-chromosome trans-eQTLs are rare and trans-SNPs+ (as defined above) are more rare still. In fact, using the odd chromosome SNPs subsampled at 20%, we find only 88 such SNPs using Dtrans-optim dataset and this is the default value we wish to improve.
As is the case with cis-eQTLs, we investigate the k, p performance surface of the DataRemix transform at the grid-search optimal µ = 0.01. Given that the relevant variance components that would maximize the trans-eQTL objective are different, it is not surprising that we find that the performance surface differs as well. In particular, we find that the optimum value of p is positive but close to 0 and thus the first k variance components are weighted equally with a weight close to 1. Consequently, at µ = 0.01 and p≈0 the contribution of the first k components is considerably larger than that of the remaining ones and we find that the performance is more sensitive to the exact value of k.
Despite the difference in the performance landscape, we find that the DataRemix transform behaves similarly on this objective. Specifically, either starting dataset can be improved to similar final performance, though the optimal parameters are slightly different. As is the case with the cis-eQTL objective, the crossvalidation procedure gives consistent results and no overfitting is observed for either grid search or Thompson Sampling (Figure 7).

Final values for the eQTL statistics obtained from two versions of datasets. Here we make a comparison between quantile normalized DQN and HCP normalized Dtrans-optim with parameters optimized for trans-eQTL discovery. We find DataRemix is able to improve upon either of starting datasets and the improvement on both the train and test dataset are comparable which indicates that overfitting is not a problem
Since trans-eQTLs are likely to reflect pathway level effects, we expect that a dataset that is optimally transformed for trans-eQTL discovery should also produce better correlation networks. We thus investigate if optimal DataRemix transform is transferable between tasks by checking if Remixed dataset optimized with respect to trans-eQTL discovery also improves the network quality criterion. Similar to our analysis of the GTex datasets, we use the correlation network to perform guilt-by-association pathway predictions and evaluate the results over 1,330 MSigDB canonical pathways. Figure 8 shows scatter plots of per-pathway AUPR (area under precision-recall curve) for several comparisons with respect to the baseline Dtrans-optim dataset. In the first panel we contrast the performance to DQN and we observe that Dtrans-optim brings a considerable improvement over the quantile normalized dataset. In the second panel we contrast Dtrans-optim with the Remixed version of DQN (optimized for trans-eQTL discovery with Thompson Sampling). We find that the pattern becomes opposite and the Remixed DQN dataset performs consistently better that Dtrans-optim. The final panel shows the results of Remixing Dtrans-optim itself which also improves the performance. Overall, we find that DataRemix improves multiple criteria of biological validity as optimizing for the trans-eQTL objective also results in improved correlation networks. Interestingly, we find that while the Remixed Dtrans-optim is no better than Remixed DQN on trans-eQTL discovery, it performs slightly better on the pathway prediction task. Taking the two objectives into account, we conclude that starting with a properly covariate-normalized dataset is superior overall, which is also the our finding regarding the cis-eQTL objective.

DataRemix-transformed datasets improve the pathway prediction objective which is not explicitly optimized. Each plot is a per-pathway AUPR (area under precision-recall curve) from various datasets (y-axis) contrasted with the results from the opti-mal covariate-normalized dataset Dtrans-optim which serves as the baseline (x-axis). Panel A shows the contrast between Dtrans-optim and DQN. The performance of Dtrans-optim is considerably better. Panel B shows the results of the Remixed DQN datasets (optimized for trans-eQTL discovery with Thompson Sampling). Even though DQN starts out as considerably worse, the Remixed version is able to outperform Dtrans-optim. Panel C shows the results of Remixed Dtrans-optim We choose to use AUPR instead of AUC because we find that Remixed version matches but doesn’t further improve the AUC performance of Dtrans-optim

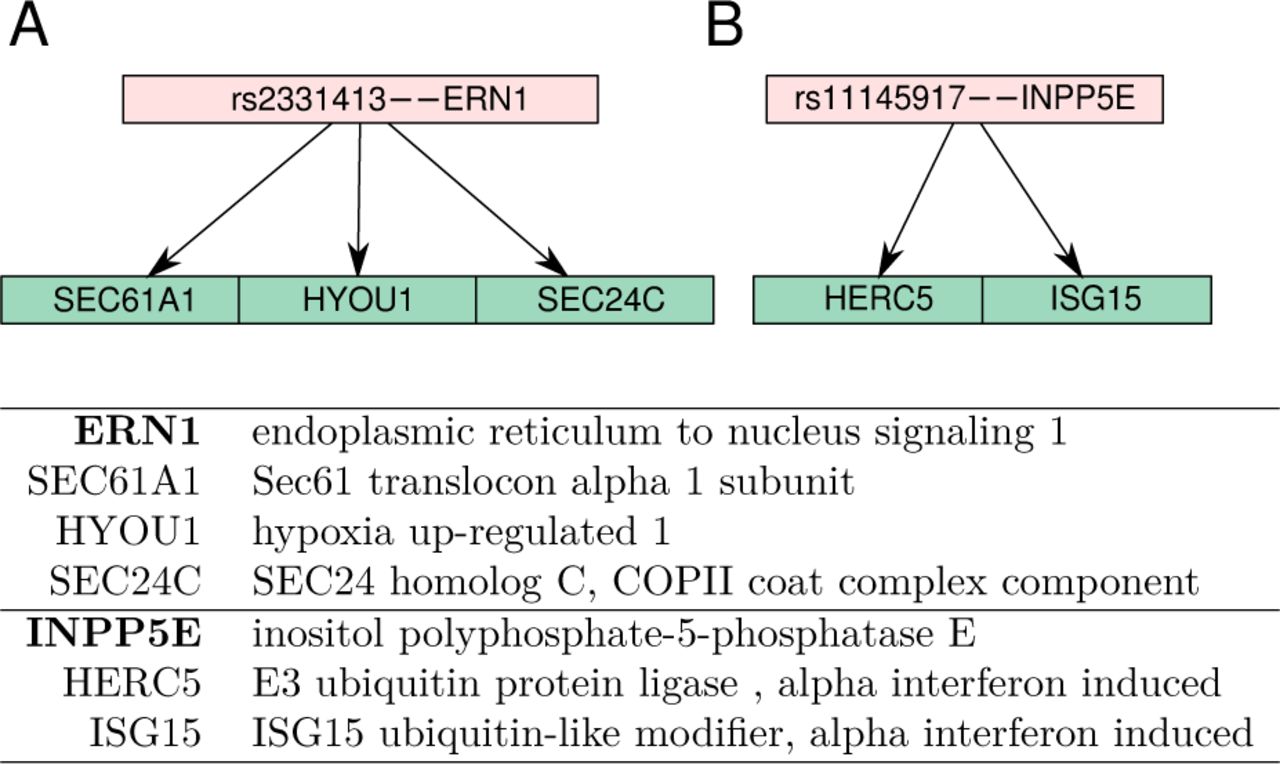
Clusters of trans-eQTLs detected by DataRemix that were not significant in the original dataset. Panel A. Both the cis and trans genes are involved in ER biology and specifically unfolded protein response. Panel B. Both of the trans genes are canonical targets of alpha interferon. The upstream cis gene, INPP5E, is a signaling molecule that mediates cell responses to various stimulation and its locus has been implicated in a variety of autoimmune diseases as well as blood immune-cell composition phenotypes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Objective evaluations as a function of iteration number for the trans-eQTL and cis-eQTL objectives using the quantile normalized DQN dataset. Red lines indicate the maximum value that was obtained by grid-search and blue lines indicate the cumulative maximum of Thompson Sampling.
A major finding of our study is that for the eQTL and pathway prediction tasks, the starting point of normalizing DGN datasets appears to matter relatively little. Even though the quantile-normalized dataset performs considerably worse in the beginning, after Remixing its performance matches that of the optimal covariate-normalized datasets. Of course, if covariates are available, it is preferable to use them and in the case of DGN, slightly further improvement can be achieved. However our results indicate that in some cases datasets can be effectively normalized even in the absence of meta-data about quality control or batch variables which is an important consideration for many legacy datasets where such information is not available.
Novel Biological Findings
At the optimal DateRemix parameters for DQN, we find an additional 24 loci that have significant associations with more than one gene and are not in linkage disequilibrium with those significant hits in the Dtrans-optim. We highlight two examples of new regulatory modules recovered via DataRemix that appear to be biologically credible based on the known functions of the genes involved. One of the newly significant interactions involves the SNP rs2331413 located in proximity of the ERN1 gene, which functions as a sensor of unfolded protein in the endoplasmic reticulum and triggers an intracellular signalling pathway termed the unfolded protein response. Three downstream genes associated with rs2331413 are like-wise endoplasmic reticulum proteins. The ERN1 locus has been associated with several phenotypes in GWAS studies, most notably drug induced hepatotoxicity (Petros et al.. 2017).
We also find an SNP rs11145917 located near INPP5E gene which is associated with two genes in the alpha interferon response. Even though only two genes show genome-wide significance, several other canonical members of the alpha interferon response are just slightly short of the significance threshold suggesting that the locus affects the upstream signaling components. The INPP5E locus has been implicated in a variety of autoimmune diseases as well as blood immune-cell composition phenotype (de Lange et al.. 2017; Astle et al.. 2016), though to our knowledge no mechanism has been proposed. Our analysis suggests that INPP5E may affect baseline activity of the alpha interferon pathway, which is a testable prediction with potential clinical importance.
Thompson Sampling Performance
We find that Thompson Sampling matches the best grid-search performance in under 100 steps giving a 40-fold reduction in the number of evaluations. We also note that it is possible for the Thompson sampling to surpass the grid-search results since the parameter combinations are not constrained by the choice of grid.
Discussion
We have proposed DataRemix, a new optimizable transformation for gene expression data. The transformation is able to improve the biological validity of gene expression representations and can be used for effective normalization in the absence of any knowledge of technical covariates. One limitation of the DataRemix approach is that it works best on data that is well approximated by a single Gaussian. However, it is relatively straightforward to adapt the approach to matrix decompositions different from SVD that are more suitable for non-Gaussian data, such as independent component analysis. We also note that it is possible to introduce additional parameters that specify more complex weighting schemes. However, as the number of parameters is increased, there is a potential for over-optimization of a specific objective above others. We emphasize that in our simple parametrization, we observe that multiple metrics of biological validity improve when only one is explicitly optimized. Specifically we find that optimizing for trans-eQTL discovery also improves the correlation network as measured by guilt-by-association pathway prediction. This property is less likely to be preserved as the number of parameters is increased.
Methods
GTex Dataset
We downloaded the complete gene-level TPM data (RNASeQCv1.1.8) from the GTex consortium (Lonsdale et al.. 2013). These data were quantile normalized.
DGN Dataset
Depression Gene Networks (DGN) dataset contains whole-blood RNA-seq and genotype data from 922 individuals. The genotype data was filtered for MAF>0.05. The genomic coordinate of each SNP was taken from the Ensembl Variation database (version 90, hg19/GRCh37). SNP identifiers that were not present in that release were excluded. After filtering there were 649,875 autosomal single nucleotide polymorphisms (SNPs). Data is available upon application through NIMH Center for Collaborative Genomic Studies on Mental Disorders. For gene expression we used the gene-level quantified dataset. The dataset comes already filtered for expressed genes and was further filtered for gene symbols that were not present in Ensembl 90 leaving 13,708 genes. The dataset comes in two covariate normalized versions with normalization parameters optimized for cis- and trans-eQTL discovery separately. To create the naive-normalized dataset, we applied a log transformation, log(x + 1), to the raw counts and quantile normalized the results.
eQTL mapping
eQTL association mapping was quantified with Spearman rank correlation. For cis-eQTLs, testing was limited to SNPs which locate within 50kb of any of the gene’s transcription start sites (Ensembl, version 90). cis-eQTl is deemed significant at 10% FDR with Benjamini-Hochberg correction for the total number of tests. For trans-eQTLs, the significance cutoff is 20% FDR with Benjamini-Hochberg correction for the total number of tests. Since the Benjamini-Hochberg FDR is a function of the entire p-value distribution in order to ensure consistency comparisons, the rejection level was set once based on the p-value that corresponded to 10% or 20% FDR in the original cis-optimized Dcis-optim and trans-optimized Dtrans-optim dataset respectively. To reduce the computational cost of grid evaluations, all the optimization computations were performed on a set of 100,000 subsampled SNPs.
Correlation network evaluation
We evaluated the quality of the correlation network derived from a particular dataset using guilt-by-association pathway prediction. Specifically, the genes were ranked by their average Pearson correlation to other genes in the pathway (excluding the gene when the gene itself is a pathway member). The resulting ranking was evaluated for performance using AUC or AUPR metric. For pathway ground-truth we used the “canonical” pathways dataset from MSigDB, comprising 1,330 pathways (Subramanian et al.. 2005).
Software Access
DataRemix is an R package which is freely available at GitHub (https://github.com/wgmao/DataRemix).
Competing interests
The authors declare that they have no competing interests.
Author’s contributions
Text for this section …
Acknowledgements
Text for this section …
References




