

Abstract
Protein tyrosine nitration is a hallmark of oxidative stress related disease states, commonly detected as anti-nitrotyrosine immunoreactivity. The precise reactive oxygen sources, mechanisms of nitration as well as the modified target proteins and functional consequences, however, remain often unclear. Here we explore protein tyrosine nitration under basal conditions and find surprisingly physiologically nitrated proteins. Upon purifying a prominent physiologically nitrotyrosine immunopositive in hearts from mouse, rat and pig, we identify it as lactate dehydrogenase (LDH). Mechanistically, LDH’s degree of basal nitration depended on two canonical sources, NO synthase (NOS) and myeloperoxidase (MPO), respectively. When validating the nitrated amino acid by MALDI-TOF mass spectrometry, we, surprisingly, located LDH nitration not to a tyrosine but the C-terminal tryptophan, Trp324. Molecular dynamics simulations suggested that Trp324 nitration restricts the interaction of the active site loop with the C-terminal α-helix essential for activity. This prediction was confirmed by enzyme kinetics revealing an apparent lower Vmax of nitrated LDH, although yet unidentified concurrent oxidative modifications may contribute. Protein nitration is, thus, not a by definition disease marker but reflects also physiological signaling by eNOS/NO, MPO/nitrite and possibly other pathways. The commonly used assay of anti-nitrotyrosine immunoreactivity is apparently cross-reactive to nitrotryptophan requiring a reevaluation of the protein nitration literature. In the case of LDH, nitration of Trp324 is aggravated under cardiac metabolic stress conditions and functionally limits maximal enzyme activity. Trp324-nitrated LDH may serve both as a previously not recognized disease biomarker and possibly mechanistic lead to understand the metabolic changes under these conditions.
Introduction
Increased levels of reactive oxygen species (ROS), i.e. oxidative stress, is considered a common mechanism of several cardiovascular and other disease states (1). In concert with different nitrogen species (2–4), ROS can lead to protein nitration, e.g. tyrosine residues (NO2Tyr). Tyrosine nitration is the most frequently used and considered the most robust disease marker of oxidative stress (5–7). Its detection relies mainly on NO2Tyr specific antibodies (8–10). With respect to the nitrogen source, anti-NO2Tyr immunopositive signals are generally interpreted as a hallmark of nitric oxide (NO) being scavenged by superoxide and intermediate peroxynitrite (9, 11, 12), although other mechanisms exist (13). Regarding the functional consequences of protein nitration few in vitro data exist suggesting mainly loss of protein function correlating with disease severity (14–17). In vivo data are lacking. Moreover, little is known about the protein factors that govern other nitration sites, e.g. tryptophan, and about the effects of the modification in protein structure and function (18). Here, we address three major knowledge gaps in our understanding of protein nitration: specificity, origin, and disease relevance.
Results
Development of an anti-nitrotyrosine antibody biomarker panel for disease-relevant oxidative stress
We started out by examining a panel of anti-NO2Tyr antibodies (Fig. 1), including those directed against carrier-linked NO2Tyr or against H2O2/NO2− treated proteins (Table S1). We tested these in rodent (rat) and, for later purification, also in porcine tissues. Surprisingly we obtained signals already under physiological conditions. The immunoreaction, however, varied greatly in protein band size and intensity. Two polyclonal antibodies (pAb), pAb1 and pAb2, and one monoclonal antibody (mAb), mAb1, displayed the most robust signals (Fig. S1). The specificity of anti-NO2Tyr antibodies (19–23) is rarely validated, e.g. by antibody pre-absorption with free NO2Tyr (11). In our case, pAb1 signals were partially blocked by pre-absorption with 3 mM NO2Tyr. mAb1 showed very little non-specific background whilst pAb2 exhibited moderate sensitivity with high anti-NO2Tyr specificity.

Experimental outline for an anti-NO2Tyr antibody-based biomarker detection approach. Central panel: Antibody panel screening, an anti-NO2Tyr antibody panel was tested in porcine and rat tissues under physiological conditions. Different antibodies might recognize different nitrated proteins (green and orange antibodies) or one antibody might detect more than one nitrated protein (blue antibody). Right panel: A 38 kDa band was especially prominent and, subsequently, purified in large mammals and identified via MALDI-TOF as LDH. NO2-LDH activity was also assessed. We identified the nitrated residue and simulated the effect of the nitration in the protein structure and mechanism using human LDH. Left panel: Different rodent models, as well as knock-out mouse models were used to elucidate the effects of NO2-LDH in disease, diabetes and myocardial stress, and the enzymatic sources of reactive nitrogen species (RNS) leading to LDH nitration, respectively.
A prominent nitrotyrosine immunoreactive protein band was detected in mouse, rat and pig heart under physiological conditions
To conduct a tissue/species screen, we selected three antibodies, pAb1, pAb2, and mAb1, from our original panel. Three proteins were consistently detected under basal conditions and with apparent molecular weights of 70, 45 and 38 kDa, of which the latter gave the by far the strongest signal in heart (Fig. 2A). Pre-incubation with free NO2Tyr or NO2BSA (3 mM) or nitro-group reduction (22), at least partially, blocked the pAb1 dependent signal. Therefore, subsequent efforts were directed at identifying the nature of the NO2Tyr positive 38 kDa protein and its site of nitration.

Screening of different native tissue homogenates identifies a 38 kDa band in heart as major physiologically NO2Tyr-immuno positive protein. Further purification and MALDI-MS analysis identified the 38 kDa band as LDH. (A) Immunopositive pig and rat tissues from Fig S1 were further analyzed by SDS-PAGE under reducing conditions (dithiothreitol) and subsequent western blot. The band pattern was different for all three polyclonal (pAb) or monoclonal (mAb) antibodies: pAb1, mAb1 and pAb2. The most intense NO2Tyr immunopositive protein band at approximately 38 kDa (p38nt) was detected using pAb1 in heart. (B) p38nt was purified from porcine heart homogenate by affinity (Blue Sepharose) followed by ion-exchange (Q-Sepharose) chromatography. Purification steps were tracked via protein nitrotyrosine detection using pAb1. (C) Purified cardiac p38nt was cut out from the gel, digested with trypsin and analyzed by mass spectrometry and peptide mass fingerprint analysis. Most peptide ions detected were matched to lactate dehydrogenase: 11 ions matched to LDH (∎) out of a total of 20 detected, with 5 ions matching to the next best protein assignment, malate dehydrogenase (MDH, ☐), identifying p38nt as LDH with a p<0.05. (D) LDH nitration was assessed by quantitative western blot analysis using pAb1 (n=9). (E) Vmax[LDH] remained unchanged up to an equimolar peroxynitrite:LDH ratio, and then decreased at a ratio of 10 (n=8). Data represent means ±SEM.
Upon purification, the 38 kDa nitrated protein was identified as LDH with lower Vmax
Using porcine heart, we isolated the NO2Tyr immunoreactive 38 kDa protein by ion exchange (Q Sepharose, QS) and affinity (Blue Sepharose, BS) chromatography (Fig. 2B). The partially purified 38 kDa protein was stained by colloidal Coomassie Blue and subjected to peptide mass fingerprinting using MALDI-TOF MS. Based on the NCBIprot database most ions present in the spectrum of tryptic peptides matched LDH as the primary protein component of the sample (Fig. 2C).
Next, we tested the effect of nitration on the enzyme activity by nitrating cardiac LDH in vitro with increasing amounts of H2O2/NO2−. Indeed, higher nitration levels of LDH (NO2-LDH) (Fig. 2E) correlated with decreased Vmax (Fig. 2F) and unchanged KM. Our data suggest therefore that protein nitration is not only a physiological event for cardiac LDH, but also an in vivo mechanism modulating enzyme activity by lowering Vmax.
Nitrated LDH is increased in diabetes and myocardial stress
To investigate whether NO2-LDH is further increased in response to metabolic stress, we investigated heart extracts from rat models of streptozotocin-induced short-term (ST) and long-term (LT) diabetes mellitus (24) as well as a doxorubicin (DoxoR) induced mouse model of cardiac stress (25) (Fig. 3A). Diabetic rats were sacrificed 3 (ST) and 16 (LT) weeks after inducing diabetes at week 10. Using the pAb1, NO2-LDH signals were significantly increased in all three models when compared against healthy controls (Fig. 3, B and C). Immunosignals of pAb1 pre-absorbed with NO2Tyr or anti-LDH wildtype antibody did not show any significant change. In both diabetes mellitus models and doxorubicin-treatment animals, levels of cardiac LDH were unchanged versus controls, indicating that indeed the NO2-LDH/LDH ratio was increased.
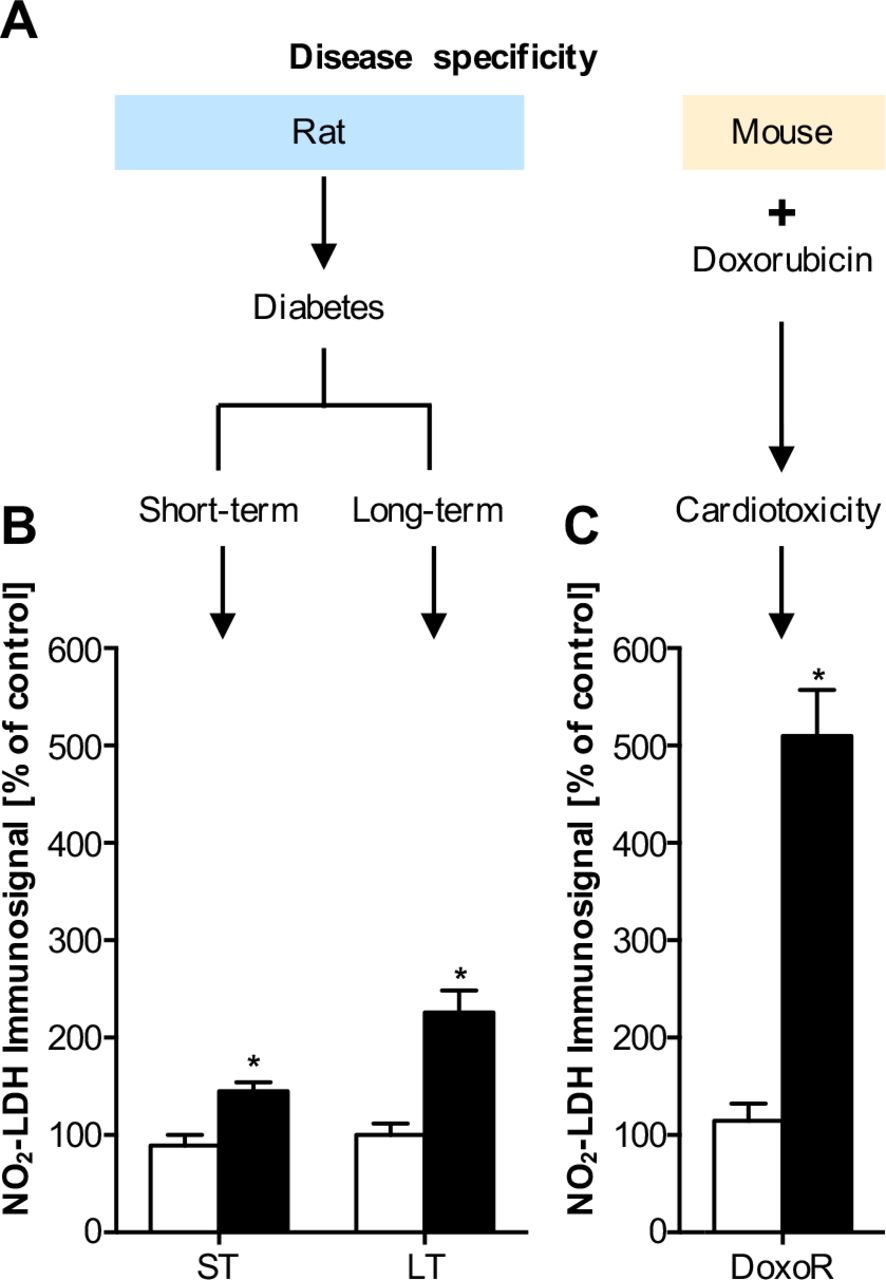
Pathological LDH nitration levels in short-term (ST) and long-term (LT) diabetic disease models and an oxidative stress myocardial dysfunction model. (A) Heart homogenates of ST and LT streptozotocin-treated diabetic rats or doxorubicin-treated mice (DoxoR) were analyzed by SDS-PAGE and immunoblot analysis for LDH nitration using the pAb1 antibody. In all three animal models, the relative NO2-LDH signal intensity was increased. In diabetes, the long-term model yielded higher values than the short-term model, which may indicate either higher nitrosative stress, or, more likely, accumulation of LDH nitration (B). The highest increase was observed in the doxorubicin model (C). Data are expressed as % of untreated control animals (open bars) and represent means ± SEM of n=6-18 experiments.
Cardiac LDH nitration is lowered in NO synthase (eNOS) knockout (KO) and myeloperoxidase (MPO) KO mice
We next wanted to address our second goal, to understand the mechanistic origin of the nitrogen in NO2-LDH. Usually, it is assumed that protein nitration derives from peroxynitrite from the reaction between •NO and O2•− (26). However, another major source, not involving de novo NO synthesis is frequently overlooked, i.e. MPO-catalyzed nitration using nitrite in the presence of hydrogen peroxide (27) (Fig. 4A). To analyze this in vivo, we had to switch to mouse models, i.e. eNOS-KO and MPO-KO lines. Indeed, we observed that NO2-LDH signals in heart tissue were not only reduced in eNOS-KO but also in MPO-KO mice when compared to their respective WT litter mates (Fig. 4B). These results therefore indicated that both pathways contribute to basal nitration of cardiac LDH. Moreover, we also compared LDH nitration to oxidation and showed that oxidation occurred at lower peroxynitrite concentrations and was half-maximal when LDH activity was still unchanged (Fig. S2).

In vivo genetic validation of reactive nitrogen species (RNS) sources. (A) Events leading to oxidized and nitrated LDH. RNS (represented as NO2−/H2O2/ONOO−) can nitrate LDH, resulting in decreased Vmax. (B) Cardiac tissue was obtained from endothelial nitric oxide synthase (eNOS) and myeloperoxidase (MPO) knock-out (KO), and the respective wild-type (WT), mice. Basal nitration of LDH (p38nt) was analyzed by immunoblot, and immunosignals quantified densitometrically. Data are expressed as % of control (i.e., of the respective WT animals) and represent means ± SEM of n=3-5 animals. NO2-LDH depended on both eNOS and MPO
Further MALDI-TOF mass spectrometry analysis of the nitration site identifies the nitrated residue as tryptophan and not tyrosine
A similarly frequent assumption about NO2Tyr immunosignals is specificity with respect to the nitrated amino acid. Other nitration target amino acids, however, have been reported, most prominently tryptophan (28–30). To identify the physiologically nitrated LDH residue(s), heart tissue was homogenized and subjected to 2D-PAGE in-gel digestion prior to MALDI-TOF MS analysis. Surprisingly, signals consistent with a nitrated tryptophan at position 324 of peptide 319-328 (SADTLWGIQK) of LDH (NO2Trp-LDH) were detected (Fig. 5A). We cannot exclude nitration of other peptides of LDH that were below our level of detection. Nevertheless, our results strongly point to NO2Trp being detected by the supposedly anti-NO2Tyr pAb1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Identification of LDH Trp-324 as physiologically nitrated and oxidized. The interaction of the active site loop with the C-terminal α-helix is essential and restricted in NO2Trp-LDH. (A) Rat heart homogenate was subjected to 2D-PAGE and Coomasie staining; spots were subjected to in-gel digestion and MALDI-TOF MS analysis. Leftft panel: Mass spectrum derived from a spot corresponding to rat LDH over the range m/z 600 to 2700. Right panel: An expansion of the same spectrum over the range m/z 1170 to 1230, showing peptide ions assigned primarily to the peptide 319-328 (319SADTLWDIQK328), and its oxidized and nitrated derivatives. Major peptide ions are labeled with their observed m/z values and corresponding LDH mature protein amino acid intervals, as assigned by Mascot™, and additionally by manual comparison to predicted values calculated from the LDH sequence and oxidative and nitrative modifications. The ions correspond to the peptide 319-328 Trp (W)-nitrated species (319-328 + NO2) and its typical high energy decomposition product (319-328 + NO2-O), as well as ions corresponding to the characteristic family of Trp oxidation forms, hydroxytryptophan (319-328 + Ox), didehydro-hydroxytryptophan (319-328 + Ox-2H), dihydroxytryptophan (319-328 + 2Ox), and kynurenin (319-328 + Ox-C). Other peptide ions display single (+ Ox) and double (+ 2Ox) oxidation, or didehydrogenation (− 2H). (B) Distance metrics mapped onto the crystal structure of WT (1I0Z). Color scheme: β-K--β-K loop, dark blue; α-helix, pink; active-site loop, green; α-2G, light blue; pyruvate, grey; NADH, cyan. Residue Trp-324 is shown in space filling. (C) Plot of the differences between T4N and WT backbone RMSF per residue (T4N – WT) in the binary complex. Structures are highlighted using the same color scheme as previously described plus the juxtaposed loop to the C-terminal α-helix which is highlighted in light orange. (D, E) Histograms of the distribution of distances. Blue WT; Red T4N. Metrics m:LH (107:CB-326:CB) showing the juxtaposition of the active-site loop and α-C-terminal helix and m:LO (103:CA-243:CB) showing the open/closed state of the active-site loop.
NO2Trp-LDH molecular dynamics simulations suggested higher restriction of the activity-essential interaction of the active site loop with the C-terminal α-helix
Since we did observe a Vmax effect on NO2-LDH, we wanted to investigate whether there is a likely structural correlate for this via Trp-324. We therefore modeled the nitration into the structure of tetrameric and binary human LDH, and then performed molecular dynamics simulations. For distance metrics of human LDH and key structural components refer to Fig. 5B. The root-mean squared fluctuations (RMSF) of all atoms in each residue were averaged across the four subunits in each combined 3.5 μs trajectory (Fig. S3). Subtracting the wildtype tetramer (WT) RMSF from the T4N (tetramer with NO2Trp-324 residue on each subunit) highlighted differences in mobility between the dynamics of the WT compared with NO2Trp-LDH (Fig. 5C). Positive values in the resulting graphs indicated greater mobility of residues in T4N compared with WT and vice versa. Moreover, distance histograms (Fig. 5, D and E) show an increased mobility in the binary complexes compared with the ternary, expressed as a broadening of the histograms.
We found that NO2Trp-LDH weakened the interaction between α-H and the active site loop. This decoupling led to a greater population of closed and “over-closed” states of the T4N active-site loop. By corollary, in WT LDH, the open active-site loop states are partly stabilized by interaction with the α-H helix. Therefore, nitration of LDH Trp-324 is expected to decrease LDH activity, consistent with our enzyme-kinetic findings (Fig. 2, D and E).
Discussion
We here close three major knowledge gaps in our understanding of protein nitration: specificity, origin, and disease relevance. With respect to the specificity of protein nitration, a vast heterogeneity of proteins are detected by supposedly uniform anti-NO2Tyr antibodies. Moreover, tryptophan nitration (NO2Trp) was a cofounding factor of our apparent NO2Tyr-immunosignals. Protein NO2Trp formation has been described in isolated proteins treated with H2O2/NO2− in vitro (31–34), peroxynitrite-exposed cells (35), and as physiological modification during aging in rat heart mitochondria (29). Trp is an alternative target of nitration that deserves more attention (36, 37) and immunosignals by anti-NO2Tyr antibodies should rather be interpreted as NO2Tyr/ NO2Trp signals unless purified and chemically validated. Rebrin et al. (29) reported Trp nitration of succinyl-CoA:3-ketoacid CoA transferase (SCOT), which could not be reproduced by Wang et al. (38). Whether Trp nitration has a functional consequence on SCOT remained unclear because both activation and inactivation were observed (29). In hindsight, SCOT nitration is also the first documented example where Trp nitration has been mistaken as Tyr nitration based on cross-reactivity of a supposedly NO2Tyr specific antibody (39), and many more of these examples may exist in the literature.
With respect to the origin of the nitrating nitrogen in NO2Tyr/ NO2Trp positive proteins, we show that both eNOS/NO and MPO/nitrite pathways, and possibly others, need to be equally considered and that the a priori assumption that NO2Tyr immunosignals indicate NO scavenging by superoxide is not valid. Nevertheless, the effects of KO of eNOS and MPO are significant but not great. Previous data shows that NO modifications remains present when NO synthesis and dietary nitrite/nitrate intake (e.g., in eNOS−/− animals on a low nitrite/nitrate diet and under the acute action of L-NIO, a NOS-inhibitor) is suppressed (40). Moreover, NO levels remain essentially unchanged upon deletion of each NOS isoform, suggesting a compensatory mechanism maintains basal levels of NO metabolites in tissues.
With respect to disease relevance, probably our most relevant finding is that protein tryptophan nitration is a physiological mechanism modulating an enzyme, cardiac LDH, activity. This represents a similar dogma shift as for ROS formation, which also should not be a priori considered as oxidative stress and a disease marker. Thus, both ROS and reactive nitrogen and protein nitration represent physiological events, even though they may be upregulated in disease as we show for diabetes and cardiac stress (Fig. 3).
Not many specific functional consequences have been described for protein nitration. Assuming Trp nitration and activity modulation of both LDH (decreasing activity) and SCOT (increasing activity) would occur, this may be a protective mechanism, allowing the heart to better utilize ketone metabolism for energy production at a time when other metabolic processes may be diminished (28, 29). Molecular dynamics simulations were performed on NO2-LDH, aiming to understand the impairment of the enzyme activity (Fig. 5). While differences in activity between WT and T4N of a factor of two is significant in physiological terms, it corresponds to minor changes in the free energy states in the catalytic process. The catalytic cycle of LDH is a well understood ordered bi-bi reaction mechanism where substrate and product enter and leave the active-site, with concomitant closing and opening of the active-site loop, after cofactor binding. Detailed kinetic characterization of LDH from Bacillus stearothermophilus (41) and Plasmodium falciparum (42) has revealed this loop movement to be a rate limiting step in the reaction mechanism. Any perturbation of this delicately balanced conformational change is expected to compromise the catalytic activity of the enzyme. We suggest that conformational movement of the active-site loop in cardiac LDH is also a rate limiting step and the weakening of the interaction between this loop and the α-H helix in T4N (as described in our results) is responsible for the impaired catalytic activity. Whether tryptophan nitration effects the conversion of either pyruvate or other 2-oxo/-2-hydroxy acids warrants further investigation.
Like many enzymes, LDH post-transcriptional activity is regulated by phosphorylation and acetylation of amino acid residues. The tyrosine kinase FGFR1 has been shown to directly phosphorylate LDH at Y10 and Y83 (43). Y10 phosphorylation of LDH promotes active, tetrameric LDHA formation, whereas phosphorylation of Y83 promotes NADH substrate binding. Our finding obviously promotes the opposite, suggesting an antagonistic effects of tyrosine phosphorylation and tryptophan nitration. Inhibition of LDH reduces aerobic glycolysis, gene transcription, channel complex regulation, molecular chaperones and cell cycle regulation. Cells are then forced to use oxidative phosphorylation and pyruvate enters the mitochondria. This leads to reactive oxygen species (ROS) generation (44). Since the nitration is already a ROS-induced event, this may be a mechanism of ROS-induced ROS formation (45), which in heart is protective triggering for example capillary density and angiogenesis (46, 47).
In conclusion, cardiac LDH appears to be the first protein basally regulated by nitration and not only a loss-of-function biomarker of metabolic stress. NO2-LDH has a lower Vmax and the nitration is further increased under cardiac metabolic stress. Furthermore, in vivo NO2Tyr immunoreactivity not necessarily reflects Tyr nitration or solely NO scavenging. Instead, it may indicate nitrite/MPO-dependent Trp nitration and physiological signaling. This has wide implications on re-interpreting previous data using NO2Tyr immunoreactivity as a biomarker and on our understanding of nitration as a physiological protein modification and signaling mechanism.
Materials and Methods
Chemicals
Immunoblotting reagents and column materials were obtained from Amersham Pharmacia Biotech (Freiburg, Germany); membranes, from Bio-Rad Laboratories (Hercules, CA, USA). Primary antibodies against nitrotyrosine were generous gifts from L.O. Uttenthal and J.S. Beckman or purchased from HyCult Biotechnology (Uden, Netherlands), Calbiochem-Novabiochem GmbH/Merck Millipore (Bad Soden/Darmstadt, Germany), Alexis/Axxora GmbH (Grünberg/Lörrach, Germany) and Upstate Inc./Millipore (Lake Placid, NY, USA/Darmstadt, Germany). As secondary antibodies, we used horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulins for polyclonal antibodies or anti-mouse immunoglobulins for monoclonal primary antibodies from DAKO Diagnostika GmbH/Agilent Technologies (Hamburg, Germany). For gel loading we used Rotiload buffer from Carl Roth GmbH + Co. KG (Karlsruhe, Germany). The Oxyblot kit was supplied by Intergen Company/Millipore (Purchase, NY, USA/Darmstadt, Germany). Nonfat dry milk was obtained from Nestlé Inc. Carnation (Solon, Ohio, USA); Cell culture medium M199, from PAA Laboratories GmbH (Martinsried/Cölbe, Germany); Nitrating reagent, H2O2/NO2−, from Alexis/Axxora GmbH; Complete Protease Inhibitor Cocktail, from Roche Diagnostics GmbH (Mannheim, Germany). Porcine heart LDH was purchased from MyBiosource. All other chemicals were of the highest purity grade available and purchased from either Sigma-Aldrich Chemie GmbH (Taufkirchen, Germany) or Merck KG (Darmstadt, Germany).
Animals
Adult male Wistar Unilever rats (270-320 g) were purchased from Harlan-Winkelmann (Borchen, Germany). eNOS and MPO KO mice were generated and bred as described previously (20). Rats and mice were housed under standard or SPF conditions, respectively, at a temperature of 18-20°C and a day-night rhythm of 12 h, with access to water and food ad libitum. Porcine hearts were obtained from a local abattoir.
Nitration of proteins by peroxynitrite
If not stated otherwise, nitration was performed by continuous mixing protein and nitrating reagent (H2O2/NO2−) in a 1:5 molar ratio in phosphate buffer pH 7.3 (15 mM KH2PO4, 80 mM Na2HPO4) for 5 min at room temperature. Due to the strong alkaline storage conditions of H2O2/NO2−, the pH in the samples was immediately readjusted to neutral pH with HCl. No further purification or dialysis steps were conducted. For longer storage, samples were aliquoted and frozen at −20°C and used within 1 month or mixed with gel loading buffer and boiled for 10 min at 95°C for daily use. In this case, no change in signal characteristics or intensities was observed over time.
SDS-PAGE, western and dot-blot analysis
Protein samples were separated by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions using 10% polyacrylamide gels. Alternatively, pre-cast gels (NuPAGE Novex 10% bis-Tris Midi Gel 1,0 mm × 26 well, Invitrogen) were used according to the manufacturer’s protocol with MES running buffer XCell SureLock Midi-Cell running tanks. For mass spectrometry proteins were stained with colloidal Coomassie blue. Proteins were immobilized on nitrocellulose membranes using standard dot-blot or semi-dry blotting techniques (21). Efficiency of protein immobilization was evaluated by staining the membrane in 0.5 % (w/v) Ponceau S in 3 % (w/v) trichloroacetic acid (TCA). Membranes were treated with 5 % (w/v) nonfat dry milk in Tris-buffered saline (TBS, 20 mM Tris/HCl pH 7.5, 150 mM NaCl) containing 0.1 % (v/v) Tween 20 (TBS-T) for 1 hour to block nonspecific protein binding. Then the membrane was incubated overnight at 4°C with the first antibody in 5% (w/v) nonfat dry milk in TBS-T and afterwards washed 3 × 10 min with TBS-T. To test the specificity for NO2Tyr, antibodies were pre-incubated with 3 mM NO2Tyr (footnote1). Alternatively, in order to reduce NO2 groups of nitrated proteins to NH2 groups, blots were first incubated with the reducing agent dithionite (1 mM) in 100 mM sodium borate pH 10 for 5 min at room temperature, washed 3 × 10 min with TBS-T and then incubated with anti-NO2Tyr antibody (48). As second antibody, we used horseradish peroxidase (HRP)-coupled antibodies against rabbit immunoglobulins (Ig) for polyclonal first antibodies or against mouse Ig for monoclonal first antibodies. HRP-labeled conjugates against the first antibodies were dissolved in 5% (w/v) nonfat dry milk in TBS-T. After washing 3 × 10 min with TBS-T the immunosignals were developed using the ECL detection reagent by Amersham Biosciences and blots were analyzed using a Kodak chemiluminescence Imager (Scientific Imaging System, Eastman Kodak Company, New Haven, USA). The resolution of the imager was 752 × 582 pixels and the linear area of signal quantification was verified by repeated measurements using increasing protein amounts. Light signals were directly quantified employing the Kodak 1D Image analysis software (version 3.5.2).
Screening of tissues and determination of nitrated proteins
Several tissues from rat and pig were homogenized in 10 volumes (w/v) of lysis buffer, containing 25 mM triethanolamine HCl (TEA) pH 7, 1 mM EDTA, 5 mM dithiothreitol, 50 mM NaCl, 10 % (v/v) glycerol and Complete™ Protease Inhibitor Cocktail (Roche, Mannheim, Germany). For long-term storage, the homogenates were shock frozen in liquid nitrogen and stored at −20 °C. For daily use, gel loading buffer was added to the samples and heated to 95°C for 10 min. Any remaining particles were pelleted by centrifugation in a desktop centrifuge (10000 × g, 10 min, 4°C). The samples were first screened by use of all antibodies listed in Tab. 1 by dot blot analysis. Tissues that yielded more than 4 hits by all antibodies were subjected to further analysis by SDS-PAGE and western blot as described above.
Characterization and isolation of a 38 kDa protein in pig heart tissue
A column packed with 2 ml of Blue Sepharose resin (Amersham Bioscience) was washed and pre-equilibrated with 10 mL lysis buffer. Pig heart supernatant (1 mL) was applied to the column. The flow-through was subsequently applied to 1 mL Q-Sepharose column that was also washed and equilibrated with lysis buffer. This column was then washed again with lysis buffer (5 mL) and the bound proteins were eluted with 2 mL of lysis buffer containing 1 M NaCl. The partially purified 38-kDa protein was mixed with gel loading buffer, boiled at 95°C for 10 min and subjected to SDS-PAGE. It was stained with colloidal Coomassie blue (23). The stained protein band at 38 kDa was cut out, destained and enzymatically digested. Tryptic fragments were separated by high pressure liquid chromatography (HPLC). Fractions containing tryptic peptides were subjected to matrix-assisted laser-desorption/ionization mass spectrometry (MALDI-MS, Bruker Reflex III™, Bruker Daltonics, Bremen, Germany). Peptide sequences were compared with proteins in the NCBIprot database.
2D-PAGE analysis of rat heart tissue
2D-PAGE analysis was conducted using standard conditions (49). Briefly, 7 cm ReadyStrips™ IPG strips (BioRad) of pI 3-10 were subjected to active rehydration overnight with heart homogenate in Destreak™ IEF buffer, supplemented with 0.02% pI 3-10 ampholites (BioRad). Isoelectric focusing was performed in a Protean™ IEF cell (Bio-Rad) using a ramped voltage up to 4000 V, until a total of approximately 10,000 Vh had elapsed. Strips were subjected to reduction, alkylation, and equilibration for the second dimension, using Equilibration Buffers I & II (BioRad) for 30 min each, then SDS-PAGE, using 10% polyacrylamide NuPage™ Novex Bis-Tris gels (Invitrogen). Gels were subsequently Coomassie stained (Gelcode™ Blue, Pierce).
In-gel digestion of rat heart samples
In-gel digestion was conducted as previously described (50). Briefly, the well-resolved protein spot of interest was punch-excised using wide-orifice pipette tips. Gel pieces were destained with 100 mM ammonium bicarbonate, pH 9/50% acetonitrile, and were washed three times with wash solution 1 (100 mM ammonium bicarbonate, pH 9), wash solution 2 (100 mM ammonium bicarbonate, pH 9/50% acetonitrile), followed by wash solution 3 (100% acetonitrile). Gel pieces were swelled in 50 mM ammonium bicarbonate, pH 9/10% acetonitrile, containing approximately 10 ng Trypsin Gold (Promega) and incubated overnight at 37°C. Peptides were extracted (each time for 20 min) once with 20 mM ammonium bicarbonate, pH 9, twice with 1% trifluoroacetic acid/50% acetonitrile, followed by once with 100% acetonitrile. The entire extraction process was performed in duplicate, and all extraction supernatants were pooled and spun to dryness in a SpeedVac™ (Thermo-Savant, Waltham, MA).
MALDI-TOF MS of rat heart samples
Peptides were desalted and prepared for MS using ZipTips™ (Millipore). Mass spectra were obtained after purified peptides were co-crystallized with the matrix 2,5-dihydroxybenzoic acid onto AnchorChip™ targets (Bruker Daltonics, Billerica, MA) using the dried-droplet technique (29, 51) and a Reflex IV™ MALDI-TOF MS instrument (Bruker-Daltonics) in positive ion, reflectron mode, over the range 400-8000. The laser intensity was adjusted to be ~3% above the threshold value for peptide ion signals. Signals from 100 to 200 laser shots were summed for each mass spectrum. External and internal calibration was achieved to within 50 ppm using the masses of known purified peptide standards and trypsin autolysis peptides.
MS data analysis and peptide mass fingerprinting
Peak lists from MALDI-TOF mass spectra (derived using MoverZ™ software, Genomic Solutions, Ann Arbor, MI) were submitted on-line to the Mascot™ search engine (Matrix Science, London, UK) for peptide mass fingerprinting (PMF) analysis against the SwissProt or NCBI non-redundant protein databases, using the following restrictions: a) Rattus sp., b) trypsin digestion with up to 3 missed cleavages, c) +/− 50 ppm error, and d) cysteine carbamidomethylation and methionine oxidation as fixed and variable modifications, respectively. LDH was unambiguously identified by PMF, with a Mascot™ Score of 150 (corresponding to an E value of 6 × 10−12, the likelihood of a false assignment) and a sequence coverage of 65% from 35 matched peptide ions, given search parameters. Additional peptide ion assignments (as well as validation of Mascot™ assignments) were made by manual inspection of spectra using theoretical digest and post-translational modification mass values for LDH.
LDH activity, oxidation and nitration by H2O2/NO2−
Commercial LDH was treated with different amounts of peroxynitrite as described above. Aliquots of H2O2/NO2− were diluted with 2 M NaOH to reach the desired concentrations indicated in Fig. 2. LDH activity and kinetic parameters of exogenously nitrated LDH were measured using a fluorescence-based assay, using excitation/emission setting of 340/470 nm, respectively. The assay used uses the fluorescent properties of NADH which can be excited at 340 nm and emission is measured at 440 nm. The oxidized form of NADH, NAD+, does not have these properties. Measurements were collected every 20 seconds during 5 minutes. The final concentrations in the reaction were LDH, 2 mU/mL, NADH 0.12 mM and Sodium pyruvate 1.13 mM. Nitration of LDH was measured as the intensity of the NO2Tyr immunosignal in arbitrary units.
Molecular Dynamics Methods
System setup
Two simulation systems were setup based on the crystal structure of the ternary complex of human-heart LDH (pdb 1I0Z). One being the wild type tetramer (WT) and the other being the tetramer with a nitro group substituted at each Trp-324 residue (T4N). The oxamate molecules were converted to pyruvate and acpype (52) used to generate Amber GAFF parameters for the substrate and cofactor molecules. A combination of GAFF parameters for nitrotryptophan and analogy was used to generate a residue library entry for a nitrotryptophan in the Gromacs AmberSB99-ildn forcefield (53). Gromacs tools were used to add hydrogen atoms to all residues consistent with pH 7 and to protonate the catalytic histidine (His-193). The complexes were solvated in boxes of TIP3P water containing 0.15 M sodium chloride ions. Calculations were performed at NTP under period boundary conditions and the PME method used for long range electrostatics. The pressure was 1 Barr and the temperature 300 K for all molecular dynamics (MD) simulations. Both systems were relaxed by 2000 steps of steepest descent energy minimisation followed by 0.2 ns MD while restraining the protein atoms to their initial positions.
Another pair of simulation systems were prepared in the same fashion, with the pyruvate removed. This gave a total of four simulation systems, referred to as WT-ternary, WT-binary, T4N-ternary and T4N-binary.
Trajectory development and analysis
Three 200 ns simulations (initialized with different velocities) were performed for each of the four simulation systems. These simulations were performed on the University of Bristol HPC machines BlueCrystal and BlueGem. Structures were extracted every 5 ns from each trajectory and energy minimized. This gave 120 starting structures for each of the four systems for further parallel trajectory development. Each new trajectory was initialized with a fresh set of random velocities and run for 28-31 ns on the UK National HPC machine Archer, giving around 3.5 μs combined simulation data for each system. All simulations were run and analyzed using the Gromacs 5.1.2 suite of software (54) and visualizations performed with VMD 1.9.2 (55) and Chimera 1.11 (56).
Molecular Dynamics Analysis
The active site loop (residues 98-110) was found to be less mobile in T4N than WT in the binary complex (LDH+NADH), while the C-terminal helix (α-H) and the juxtaposed loop (residues 277-287) were more mobile in T4N. The lower mobility of the active site loop is also detectable in the ternary complex (LDH+NADH+pyruvate) albeit greatly attenuated, while the C-terminal helix mobility is similar to the WT. The other main features in these plots are the differences in mobility of the 205-225 loop. However, as this is one of the most flexible parts of the protein and is not located near the active site we do not consider these differences to impact the activity of the two proteins. By contrast, the C-terminal side of the active site loop juxtaposes the C-terminal end of the final helix in the protein, α-H (residues 309-332). A crucial contact between these two structural elements is made by Leu-107 and Ile-326. Hence, we define one metric reporting the association of the active site loop with α-H via the distance between CB atoms of these residues, as m:LH. Likewise, two residues spanning the mouth of the active site Gly-103 and Lys-243 were chosen to monitor openness of the active site loop, or m:LO. Distance histograms show the distribution of these distances over each of the four 3.5 μs combined trajectories. They indicate an increased mobility in the binary complexes compared with the ternary, expressed as a broadening of the histograms.
The distance between the active site loop and α-H (m:LH) is slightly shorter on average for WT than T4N in the ternary complex and this difference is amplified in the binary complex where about half of the WT loops interact more tightly with α-H than T4N. Comparison with the second metric monitoring active site loop closure (m:LO) in the binary complex shows that the T4N loop spends more time at a distance comparable with the ternary complex than the WT and also is an “over-closed” state. The results of the ternary simulations are again more similar, but a tendency towards and “over-closed” state persists.
Synthesis and Characterization of Nitrotryptophan
Nitrotryptophan (NO2Trp) was synthesized according to Moriya, et al. (15). 20 g L-Trp and 0.25 g urea were suspended in 350 mL glacial acetic acid. Under vigorous stirring at 4°C, 10 mL 24 M nitric acid in 30 mL glacial acetic acid were added. After the suspension changed to a yellow clean solution, it was stirred at 4°C until it changed again to a suspension. Then an additional 10 mL fuming nitric acid in 30 mL glacial acetic acid were added drop by drop at 15°C. After stirring overnight at room temperature, the resulting yellow precipitate was collected by filtration and washed with glacial acetic acid. The crude product was recrystallized in 100 mL 2 M nitric acid at temperatures below 50°C and converted to the free amino acid in 100 mL hot water by the addition of sodium carbonate until the pH was neutral. As analyzed by its infrared (IR) and nuclear magnetic resonance (NMR) spectra, no 4-NO2Trp was detectable. Because 7-NO2Trp cannot be synthesized by the action of 24 M nitric acid on the tryptophan ring (16), the products of the synthesis with respect to UV spectrum are a mixture of 5/6-NO2Trp. NO2Trp crystallized as yellow needles with a melting point at 265°C (decomp.). The compound was characterized according to: UV (in 1 N NaOH) 330 nm, 380 nm; IR (KBr) (−NH3+) 3190 cm−1, v(C=C) and amide I 1600-1650 cm−1, amide II and δ(−NH3+) 1550-1600 cm−1, vas(−NO2) 1495 cm−1, vs(−NO2) 1330 cm−1, predominantly 5/6-NO2 substitution at 818 cm−1 instead of 4-NO2 substitution at 780 cm−1; NMR (D2O-NaOH, δ 4.7 HDO) δ 2.−3.1 (2H, m), 3.4−3.6 (1H, m), 7.35 (1H,s), 7.5 (1H, d, J= 9 Hz), 7.7 (1H, dd, J= 2 and 9 Hz), 7.95 (1H, d, J= 2 Hz).
Animal models of disease
To examine LDH nitration under pathophysiological conditions, two in vivo models involving nitrative stress were assessed, including a short- and longer-term streptozotocin diabetes model in the rat, and a murine model of doxorubicin-induced cardiotoxicity.
Streptozotocin model of diabetes mellitus
At an age of 10 weeks, rats (n=12) received a single intraperitoneal injection of 60 mg/kg BW of streptozotocin (STZ) dissolved in citrate buffer 50mM pH 4,5. Age matched control rats (n=12) received vehicle injections with citrate buffer. Hyperglycemia was defined as a positive test of glucose in urine (Haemo-Glukotest, Boehringer Mannheim, Germany) and a non-fasting blood glucose level higher than 11 mmol/L (hexokinase method) one week after STZ application. Rats were sacrificed 3 (n=12) and 16 weeks (n=12) after diabetes induction by CO2-inhalation. After thoracotomy, hearts were excised and freed of connective tissue while immersed in modified Krebs-Hepes buffer pH 7.35 containing 99 mM NaCl; 4.7 mM KCl; 1.9 mM CaCl2; 1.2 mM MgSO4; 25 mM NaHCO3; 1 mM K2HPO4; 20 mM Na-Hepes; 11.1 mM D-glucose. Hearts were shock frozen in liquid nitrogen, pulverized, and homogenized in lysis buffer. For SDS-PAGE and western blot, gel loading buffer was added and protein concentrations were determined after TCA precipitation by the method of Lowry.
Doxorubicin cardiotoxicity model
Doxorubicin (DOX, Adriamycin) is a broad-spectrum anthracycline antibiotic that is used to treat a variety of cancers. However, the clinical use of DOX is limited because of its serious cardiotoxicity, possibly involving increased oxidative stress, alteration of cardiac energetic status and direct effects on DNA. Hearts from DOX-treated mice were obtained as described elsewhere (57), and homogenized and shock-frozen as described above.
Endogenous LDH nitration in eNOS-KO and MPO-KO mice
Hearts from eNOS-KO and MPO-KO mice were excised, washed in perfusion buffer, blotted dry on filter paper, weighed, and homogenized immediately in ice-cold 50 mM PBS, pH= 7.4, containing 2.5 mM EDTA, 1 mM DTT, 1% NP-40, phosphatase inhibitors (10 mM sodium fluoride, 10 mM sodium pyrophosphate, 2 mM sodium orthovanadate, and 50 mM β-glycerophosphate), and Complete™ protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Homogenates were incubated on ice for 30 min to facilitate solubilization, after which they were clarified by centrifugation at 12000 × g for 15 min at 4°C, aliquoted and stored at −80°C. Prior to use, samples were thawed on ice and assayed for protein concentration using the BCA method.
Funding
Work in A.J. laboratory was supported by BBSRC, BHF and MRC; in P.B. laboratory by NKFIH (K123975); in H.H.H.W.S. laboratory by an ERC Advanced Investigator Grant (294683 - RadMed), an ERC proof-of-concept grant (139-101052 - SAVEBRAIN) and the EU Horizon 2020 programme, REPO-TRIAL.
Author contributions
H.H.H.W.S. designed research; J.F., D.T., T.N., C.I.A., A.S., P.B., C.S., V.S., P.R., A.J., A.G., D.H.P., R.B.S. and M.F. performed research; A.S., A.G., A.K., S.B., D.H.P., M.F. and H.H.H.W.S. contributed new reagents/analytic tools; J.F., D.T., D.H.P., and H.H.H.W.S. analyzed data; and C.N., J.F., C.S., D.H.P., M.F., A.I.C. and H.H.H.W.S. wrote the paper.
Competing interests
The authors declare no conflict of interests.
Data and materials availability
Data and materials available upon reasonable request.
Acknowledgments
The authors would like to thank G. Jansen for technical help, and Dr. P.M.H. Schiffers for carefully reading earlier stages of the manuscript. We also thank the Advanced Computing Research Centre at Bristol and BrisSynBio for access to the HPC machines BlueCrystal and BlueGem. We thank the HECBiosim Consortium for access to the UK National HPC facility Archer.
Footnotes
One Sentence Summary: We here expand protein nitration from mainly affecting tyrosine moieties and being a disease marker of oxidative stress to physiological nitration of a tryptophan in cardiac lactate dehydrogenase downregulating its maximal activity under basal conditions and metabolic stress.
References and Notes




