

Abstract
Dynamic trafficking of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid glutamate receptors (AMPARs) to synapses is critical for activity-dependent synaptic plasticity underlying learning and memory, however the identity of key molecular effectors remains elusive. Here, we demonstrate that membrane depolarization and N-methyl-D-aspartate receptor (NMDAR) activation triggers secretion of the chemotropic guidance cue netrin-1 from dendrites. Using selective genetic deletion, we show that netrin-1 expression by excitatory neurons is required for NMDAR-dependent long-term potentiation (LTP) in the adult hippocampus. Further, we demonstrate that application of exogenous netrin-1 is sufficient to trigger the potentiation of excitatory glutamatergic transmission at hippocampal Schaffer collateral synapses via Ca2+-dependent recruitment of GluA1-containing AMPARs, promoting the maturation of immature or nascent synapses. These findings identify a central role for activity-dependent release of netrin-1 as a critical effector of synaptic plasticity in the adult hippocampus.
Glutamatergic synaptic transmission in the adult brain is primarily mediated by AMPARs, with changes in synaptic strength due to alterations in both synaptic structure and receptor composition 1, 2. Long-term potentiation (LTP), an intensively studied experimental model of synaptic plasticity and memory, modifies postsynaptic function through NMDAR-dependent recruitment of AMPARs, cytoskeletal reorganization, and modification of synaptic adhesion 3-6.
Netrin-1 is a secreted protein that directs axon outgrowth and synaptogenesis during development 7-9. The netrin receptor, deleted in colorectal cancer (DCC), directs cell motility by regulating intracellular calcium, RhoGTPases, Src family kinases, focal adhesion kinase, phospholipase C, phosphoinositol 3-kinase, p21-activated kinase, and local protein synthesis, all of which influence adhesion, cytoskeletal organization, and synapse function 10, 11. During neural development, restricted sources of netrin-1 direct the rapid local recruitment of synaptic proteins 7, 12, 13. Further, a form of synaptic consolidation in Aplysia neurons, studied in primary cell culture, requires netrin-1-dependent local protein synthesis, suggesting a critical role for netrin-1 in the modification of synaptic function 14. In the mature mammalian brain, netrin-1 and DCC are enriched at synapses, and DCC co-fractionates with detergent resistant components of the postsynaptic density (PSD). Selective deletion of DCC from excitatory forebrain neurons in the mature brain reduces dendritic spine volume, severely attenuates LTP at Schaffer collateral hippocampal synapses, and impairs hippocampal-dependent learning and memory 15. Several families of ligands have been identified for DCC, including draxins, cerebellins, and netrins 16-18; however, the enrichment of netrin-1 at synapses suggests that it may be a key ligand for DCC to regulate synaptic function.
Here, we show that activity-dependent secretion of netrin-1 from neurons regulates synaptic transmission in the adult hippocampus. Selective genetic deletion of netrin-1 from principal excitatory neurons severely attenuates NMDAR-dependent LTP in the adult mammalian hippocampus. Further, application of exogenous netrin-1 to Schaffer collateral synapses is sufficient to induce a lasting potentiation of evoked synaptic responses through the recruitment of GluA1-containing, GluA2-lacking AMPARs. Taken together, our study identifies the activity-dependent secretion of netrin-1 as a critical component of LTP expression downstream of NMDAR activation.
Results
Netrin-1 is secreted in response to neuronal depolarization and NMDAR activation
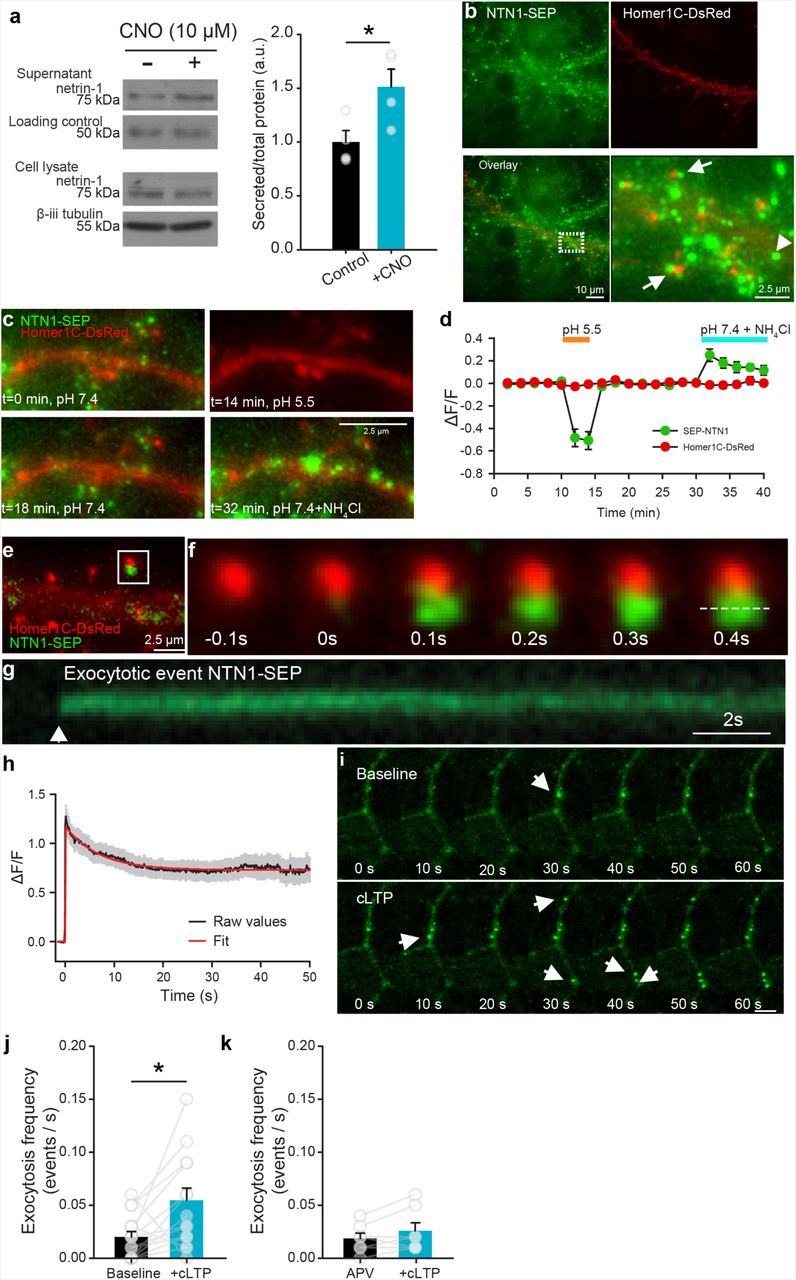
Netrin-1 is highly enriched in intracellular vesicles in synaptosomes 15, suggesting that action potential generation and neuronal depolarization might regulate netrin-1 release at synapses. To initially examine if netrin-1 protein may be secreted from neurons in an activity-dependent manner, we collected and concentrated cell-culture supernatants of embryonic rat neurons (14 DIV) that had been depolarized for 20 min with 20 mM KCl. Immunoblots of the concentrated supernatants revealed significantly increased levels of netrin-1 compared to supernatants from cultures treated with vehicle alone (Supplemental figure 1), indicating that netrin-1 is secreted in response to strong depolarization.
Sustained neuronal activity can mobilize vesicular release 19. To determine if increased neuronal spiking induces secretion of netrin-1, we used the designer receptor exclusively activated by designer drugs (DREADD) approach to excite primary hippocampal neurons infected with AAV8-Syn-hM3D(Gq)-mCitrine. Bath application of the synthetic hM3D(Gq) agonist clozapine-N-oxide (CNO; 10 µM) triggered rapid depolarization and spiking of mCitrine-expressing neurons (Supplemental figure 1). We detected a large increase in netrin-1 protein in supernatants from cultures expressing hM3D compared to untransfected control cultures following bath application of CNO (10 µM) for 1 h (Figure 1a), demonstrating that prolonged spiking and depolarization of hippocampal neurons promotes secretion of netrin-1.
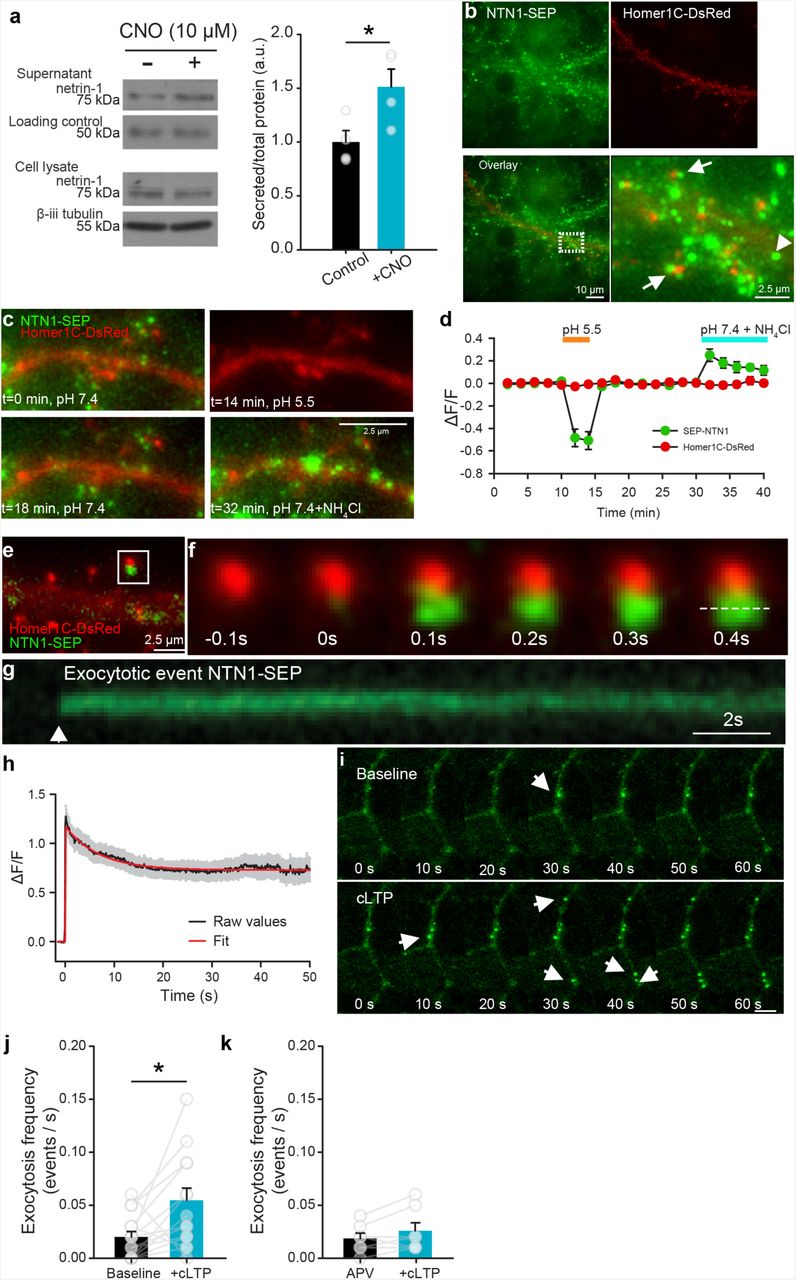
Netrin-1 is secreted by neurons in response to depolarization and NMDAR activation. Western blots (left) and group data (right) of concentrated media supernatants and cell lysates of cultured hippocampal neurons (14 DIV) expressing hM3D(Gq) and treated with vehicle (black) or depolarized with CNO (10 µM, yellow) for 1h (control supernatant: 1.00±0.11, CNO supernatant: 1.51±0.17, n = 4, t6= 2.60, p = 0.04, independent samples t-test, n = 4 independent cultures per condition). Note that an Alexa 633-conjugated secondary antibody (40 ng per tube) was added to the medium as a loading control. (b) Low-(left panels) and high-magnification (far right) of NTN1-SEP (green) appears as puncta at synaptic sites (Homer1C-DsRed-positive, red) in cultured hippocampal neurons (14 DIV). (c) Representative segment of a hippocampal dendrite expressing NTN1-SEP (green) and Homer1C-DsRed (red) during baseline (0 min), following exposure to extracellular solution at pH 5.5 (14 min), recovery (18 min), and bath application of NH4Cl (32 min). (d) Group data show decrease in SEP-NTN1 fluorescence intensity following bath application of extracellular solution at pH 5.5 (orange line), and an increase at intra-dendritic sites following bath application of NH4Cl (blue line, n = 7 cells). (e) High magnification of Homer1C-DsRed (red) positive puncta showing NTN1-SEP (green). (f) Time-lapse of a Homer1C-DsRed puncta (red, in e) prior to and following a NTN1-SEP exocytotic event. (g) Kymograph of NTN1-SEP fluorescence of a Homer1C-DsRed puncta (dashed line in f). (h) Raw group data (average in black, SEM in grey) and fitted curve (red) show amplitude and duration of NTN1-SEP exocytotic events. (i) Time-lapse images of dendritic segment shown in e during baseline (top) and following cLTP induction (bottom). Note the NTN1-SEP exocytotic events (white arrows). (j) Group data show frequency of exocytotic NTN1-SEP events in during base-line and following cLTP induction (baseline: 0.020±0.005, cLTP: 0.055±0.011, t13=2.56, p=0.02, paired-samples t-test, n=14 dendritic segments from 14 independent cultures). (k) Group data show frequency of NTN1-SEP exocytosis during baseline and following cLTP induction in the presence of NMDAR antagonist APV (50 µM) (APV: 0.019±0.005, cLTP: 0.025±0.007, t6=1.70, p=0.14, n=7 dendritic segments from 7 independent cultures, paired-samples t-test).
The neuronal specific promoter (synapsin) used in the DREADD experiments indicates that depolarization causes netrin-1 to be released by neurons. However, whether netrin-1 is released perisynaptically by axons or dendrites remained unresolved. To reveal the subcellular localization of netrin-1 secretion, we developed a full-length functional netrin-1 fused to super-ecliptic pHluorin (NTN1-SEP) that increases fluorescence intensity upon shifting from acidic to neutral pH following vesicle fusion with the plasma membrane (Supplemental figure 2). Co-expression of NTN1-SEP and the synaptic marker Homer1C-DsRed in cultured rat hippocampal neurons (14 DIV) revealed NTN1-SEP positive puncta along dendrites at synaptic (Homer 1C-DsRed positive) and extrasynaptic sites (Figure 1b), in a distribution similar to endogenous netrin-1 (Supplemental figure 2). Fluorescence was quenched by a brief pulse of acidic solution (pH=5.5), confirming that it corresponds to extracellular secreted NTN1-SEP. Netrin-1 secreted from dendrites should be initially present in an intra-dendritic endosomal pool. Intracellular distributions of NTN1-SEP were detected by applying an extracellular NH4Cl solution, alkalinizing the intracellular compartment and revealing large clusters of NTN1-SEP throughout dendritic shafts and spines (Figure 1c-d).
To address whether netrin-1 secretion is constitutive or mobilized in response to synaptic activity, we performed wide-field fluorescence video imaging of NTN1-SEP in neurons co-transfected with Homer1C-DsRed. To detect exocytotic events, pre-existing cell-surface fluorescence was first photobleached 20. We then monitored the spontaneous appearance of new bright puncta that reached full fluorescence amplitude, within 1-2 imaging time points, consistent with rapid exocytosis of NTN1-SEP (Figure 1e-f). We observed that NTN1-SEP fluorescence decay frequently lasted for minutes, consistent with NTN1-SEP remaining extracellularly at synapses following secretion (Figure 1g-h).
NMDAR activation is critical for several major forms of activity-dependent synaptic plasticity 21. To determine if NMDAR activation drives netrin-1 secretion, we monitored the dynamic appearance of NTN1-SEP fluorescent puncta in hippocampal neurons following chemical LTP (cLTP), induced by applying an extracellular solution of 0 mM Mg2+, 200 µM glycine, and 30 µM bicuculline that promotes NMDAR conductance and potentiates AMPAR-mediated currents 22, 23. Inducing cLTP increased the frequency of NTN1-SEP exocytosis, and promoted rapid secretion of long-lasting NTN1-SEP puncta (Figure 1i-j, Supplemental movie 1). To examine whether the increase in frequency of NTN1-SEP exocytosis requires NMDAR activation, we repeated these experiments in the presence of 2-amino-5-phosphonovaleric acid (APV, 50 µM), a potent and specific NMDAR antagonist. cLTP stimulation in the presence of APV failed to increase the frequency of NTN1-SEP exocytotic events (Figure 1k), indicating that the increase in secretion requires NMDAR activation. Together, these findings demonstrate that NMDAR activation promotes netrin-1 secretion from dendrites.
CA1 neurons in the adult hippocampus express netrin-1
RNA-seq analysis previously detected netrin-1 mRNA in adult mouse hippocampal CA1 neurons 24. Using a validated netrin-1 specific monoclonal antibody 25, immunohistochemical analyses of hippocampal sections from Thy1-GFP L15 mice revealed a punctate distribution of endogenous netrin-1 protein across all layers of the CA1 region, including strata oriens, pyramidale, and radiatum (Supplemental figure 3). These findings indicate that ntn1 is expressed by hippocampal neurons, and that endogenous netrin-1 protein can be detected in all layers of the adult hippocampus.
The subcellular distribution of netrin-1 was then assessed throughout stratum radiatum in adult Thy1-GFP L15 mice using super-resolution confocal microscopy (Figure 2a, Supplemental figure 3). The apical dendrites of CA1 pyramidal neurons, visualized by membrane-targeted GFP expression, show punctate endogenous netrin-1 immunoreactivity throughout the neuropil (Supplemental figure 3). Immunostaining for netrin-1 shows clusters of endogenous protein throughout the dendritic shafts and spines. Many of these netrin-1 clusters are contained within dendritic spines, as revealed by opaque surface renderings of the membrane-targeted GFP (Figure 2a). Consistent with NTN1-SEP live imaging (Figure 1), these results localize endogenous netrin-1 protein within dendrites and spine heads of CA1 pyramidal neurons.
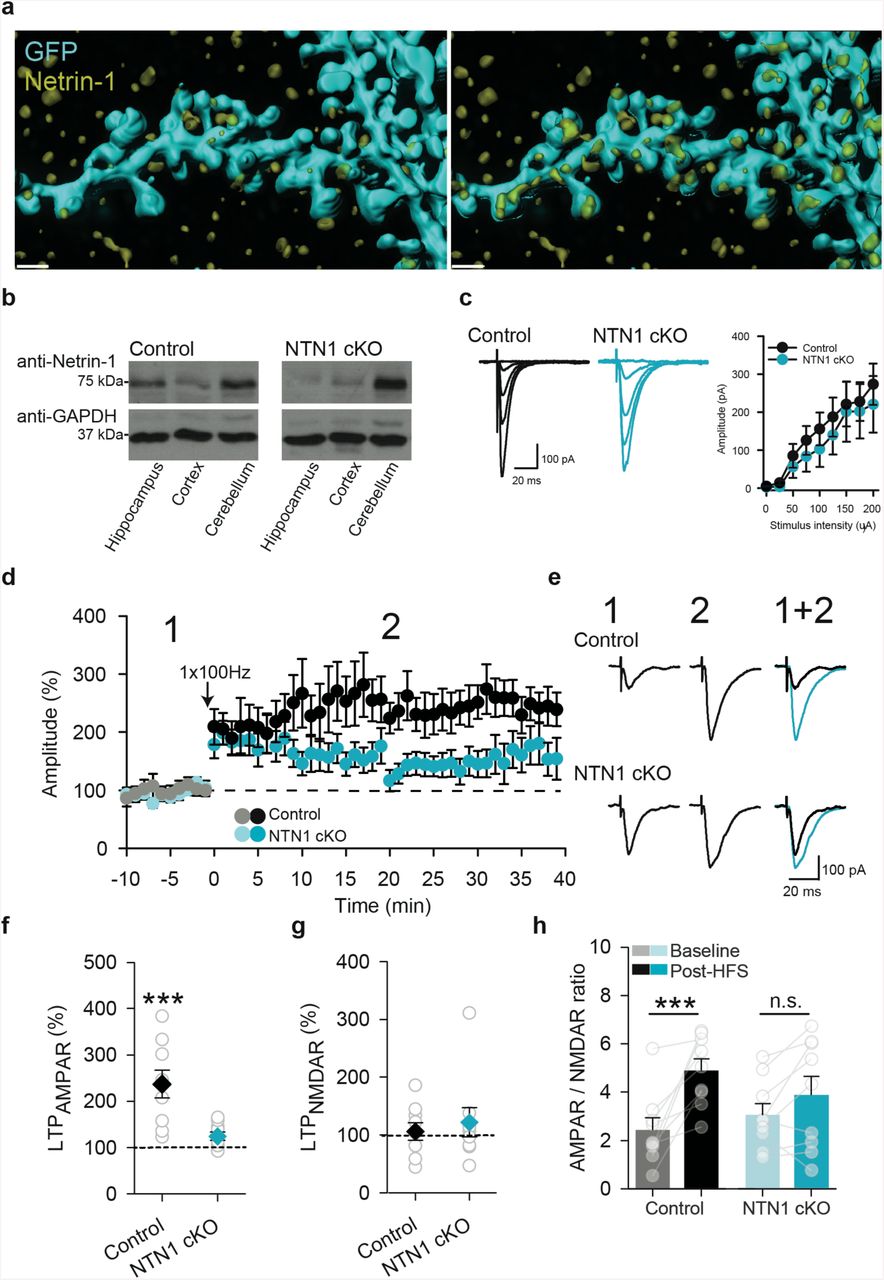
Neuronal netrin-1 is required for hippocampal LTP. (a) Dendrite of hippocampal CA1 neuron expressing GFP (cyan), counterstained with anti-netrin-1 (yellow). Full opacity (left) of membrane linked GFP shows netrin-1 immunoreactivity associated with the dendritic arbor. GFP transparency (right) reveals endogenous netrin-1 within the dendritic shaft and spines. Scale bar = 1 µm. (b) Representative western blots of whole hippocampal, cortical, and cerebellar homogenates from control wildtype littermates (left) and CaMKIIα∷Cre/NTN1fl/fl (right) mice. Note that the cerebellum, which does not express CaMKIIα∷Cre, shows comparable levels of netrin-1 protein in both genotypes. (c) Representative traces (left) and group data (right) showing evoked EPSC amplitude in CA1 pyramidal neurons in response to incremental stimulation intensity of Schaffer collaterals from wild-type control littermates (black; Control) and CaMKIIα∷Cre/NTN1fl/fl mice (blue; NTN1 cKO). (d) Effect of Schaffer collateral high-frequency stimulation (arrow, HFS; 1sX100 Hz) on evoked responses in CA1 pyramidal neurons in acute hippocampal slices from wild-type control littermates (black circles; control, n = 9 cells from 6 mice) and CaMKIIα∷Cre/NTN1fl/fl mice (blue circles; NTN1 cKO, n = 9 cells from 6 mice). (e) Representative evoked EPSCs in a CA1 hippocampal neuron in response to Schaffer collateral stimulation before (1 in d), and after (2 in d) high-frequency stimulation (HFS; 1s of 1×100 Hz) in wild-type control littermates (black) and CaMKIIα∷Cre/NTN1fl/flmice (blue). (f-h) Group data show AMPAR-mediated current (f) (control: 237±30% of baseline, p<0.001; NTN1 cKO: 124±9% of baseline, p=0.23; 2-way ANOVA: Interaction of genotype X LTP: F1,16=4.57, p = 0.04), NMDAR current (g) (control: 106±15% of baseline, NTN1 cKO: 125±24% of baseline; 2-way ANOVA: Main effect of genotype, F1,16 =0.09, p = 0.77; Main effect of simulation, F1,16=0.02, p=0.87; Interaction of genotype X stimulation, F1,16=1.27, p=0.28), and AMPAR-to-NMDAR ratio (h) (control: 2.43±0.49 in baseline vs. 4.88±0.48 following HFS, p<0.001; NTN1 cKO: 3.16±0.45 in baseline vs. 4.08±0.76 following HFS, p=0.09; 2-way ANOVA: Interaction of genotype X stimulation, F1,16=4.48, p=0.049) in wild-type control littermate (n=9 cells from 6 mice; black) and CaMKIIα∷Cre/NTN1 −1fl/fl mice (n=9 cells from 6 mice; blue) before and 20 min after high-frequency stimulation (grey bars).

Exogenous netrin-1 potentiates synaptic transmission in adult mouse hippocampus by increasing postsynaptic AMPA receptor-mediated current. (a) Effect of 5 min bath application of netrin-1 (blue) on evoked synaptic responses in CA1 pyramidal neurons in response to Schaffer collateral stimulation (100 ng/ml, white circles: n = 8 cells from 5 mice; 200 ng/ml, black circles: n=16 cells from 10 mice). (b) Representative evoked responses (top, 50 ms ISI) in a CA1 pyramidal neuron before (corresponding to 1 in a, black trace in overlay) and after (corresponding to 2 in a, blue trace in overlay) perfusion with netrin-1 (200 ng/ml). Group data show that paired-pulse ratio is not significantly changed from baseline following 5 min bath application of netrin-1 (200 ng/ml) across a range of ISIs. (Main effect of condition: F1,6=4.05, p=0.09; main effect of ISI: F1,6=5.22, p<0.001; Interaction between condition X ISI: F6,36=0.35, p=0.91; n=7 cells from 5 mice; 10 ms, 1.52±0.19 in baseline vs. 1.30±0.12 in netrin-1; 20 ms, 1.76±0.19 in baseline vs. 1.62±0.13 in netrin-1; 30 ms, 1.79±0.16 in baseline vs. 1.71±0.09 in netrin-1; 50 ms, 1.73±0.12 in baseline vs. 1.55±0.11 in netrin-1; 100 ms, 1.41±0.08 in baseline vs. 1.30±0.08 in netrin-1; 150 ms, 1.17±0.18 in baseline vs. 1.28±0.09 in netrin-1; 200 ms, 1.32±0.10 in baseline vs. 1.19±0.12 in netrin-1). (c) Representative evoked synaptic responses during baseline (left, 1 in a, black trace in overlay) and after netrin-1 (middle, 2 in a, blue trace in overlay) at −70 mV (bottom) and +40 mV (top). (d-e) Group data show that the AMPAR-mediated current (d; 255±44% of baseline, n=16, p<0.001, Wilcoxon Signed Rank) is significantly increased following bath application of netrin-1 (200 ng/ml), while the NMDAR-mediated current remains unchanged (e; 110±11% of baseline, p = 0.762). (f) Group data show that the AMPAR-to-NMDAR ratio is significantly increased 20 min after bath application of netrin-1 (2.40±0.42 in baseline vs. 4.79±0.61 following netrin-1, t15=4.99, p<0.001, paired-samples t-test).
Netrin-1 is necessary for long-term potentiation at Schaffer collateral synapses
High-frequency stimulation of the Schaffer collaterals induces NMDAR-dependent LTP in the adult hippocampus 26. Since NMDAR activation by cLTP stimulation increases NTN1-SEP exocytosis at synaptic sites, we hypothesized that the endogenous release of netrin-1 may contribute to LTP induced by high-frequency stimulation of the Schaffer collateral hippocampal pathway. To test this, we selectively deleted a floxed ntn1 allele from excitatory neurons by Cre expression regulated by the CaMKIIα promoter (T29-CaMKIIα∷Cre/NTN1fl/fl). Cre recombinase is first expressed at ∼2.5 weeks postnatally in these mice, and is detected throughout all subfields of the hippocampus, exclusively in glutamatergic neurons, by one month of age. Critically, expression occurs after developmental axon guidance is complete 15, 27. Hippocampal homogenates from 3-6 month old T29-CaMKIIα∷Cre-NTN1fl/fl (NTN1 cKO) mice showed reduced levels of netrin-1 protein compared to control littermates (Cre-negative NTN1fl/fl), consistent with deletion of ntn1 expression from excitatory forebrain neurons (Figure 2b). Further, immunohistochemical analyses demonstrated that netrin-1 immunoreactivity is substantially reduced in stratum radiatum of NTN1 cKO mice compared to control littermates (Supplemental figure 3).
Previous work has suggested that DCC may function as a dependence receptor, and therefore reducing the level of netrin-1 might increase apoptosis 28. However, levels of cleaved caspase-3 were not found to be significantly different between 3-6 month old NTN1 cKO and control littermates (Supplemental figure 3).
To determine if netrin-1 contributes to basal synaptic transmission, we measured evoked excitatory postsynaptic currents (eEPSCs) from CA1 pyramidal neurons following Schaffer collateral stimulation of acute hippocampal slices from 3-6 month old NTN1 cKO and control littermates in the presence of picrotoxin (PTX; 100 µM) to block GABAA-mediated synaptic transmission. We detected no significant differences in AMPAR or NMDAR eEPSC amplitudes, paired pulse ratios, rectification, and AMPAR-to-NMDAR ratios indicating that basal evoked synaptic transmission was not affected by deletion of ntn1 (Supplemental figure 3). To assess whether conditional deletion of netrin-1 impaired the relative strength of individual synapses, we recorded AMPAR-mediated miniature EPSCs (mEPSCs) in CA1 pyramidal neurons from control and NTN1 cKO brain slices in the presence of PTX (100 µM) and tetrodotoxin (TTX; 1 µM). The amplitude of mEPSCs was slightly but significantly reduced in CA1 pyramidal neurons from NTN1 cKO compared to littermate controls, whereas no changes were detected in mEPSC event or instantaneous frequency (Supplemental figure 4). These results provide evidence that conditional deletion of ntn1 from principal excitatory neurons lead to a modest weakening of synapses onto CA1 pyramidal neurons.
To assess whether netrin-1 may contribute to NMDAR-dependent LTP, we recorded eEP-SCs in CA1 pyramidal neurons from adult NTN1 cKO and control littermates in the presence of the GABAA receptor antagonist, picrotoxin (100 µM). Brief high-frequency stimulation (HFS; 1-s at 100 Hz) resulted in significant short-term facilitation of eEPSCs in CA1 pyramidal neurons from both NTN1 cKO and control littermates. The amplitude of eEPSCs remained significantly potentiated in slices from control littermates after 20-25 min compared to baseline values. However, the amplitude of synaptic potentiation was significantly attenuated in slices from NTN1 cKO mice (Figure 2d-e). Moreover, we observed significant increases in AMPAR-mediated current and AMPAR-to-NMDAR ratio in slices from control littermates following tetanic stimulation, but not in slices from NTN1 cKO mice (Figure 2f-h). These findings demonstrate that deletion of ntn1 from principal excitatory neurons significantly attenuates NMDAR-dependent LTP, and indicate that netrin-1 plays a central role in synaptic plasticity in the adult hippocampus.
Netrin-1 potentiates CA3-CA1 Schaffer collateral synapses via an NMDAR-independent postsynaptic mechanism
Genetic studies in C. elegans provided initial evidence that netrin directs the local enrichment of synaptic proteins during synaptogenesis 12, 13, 29. In mammalian neurons, synaptic proteins, such as PSD95, are rapidly recruited to synaptic sites in response to a local source of netrin-1 7. The impaired LTP detected in T-29-CaMKIIα∷Cre/NTN1fl/fl mice suggested that netrin-1 secreted by neurons may be a critical effector of activity-dependent LTP in the adult hippocampus that functions in parallel to or downstream of NMDAR activation. To determine if netrin-1 might be sufficient by itself to elicit changes in synaptic strength, we assessed netrin-1 gain-of-function by perfusing hippocampal slices from adult wild-type C57/B6 mice (2-3 month old) with exogenous recombinant netrin-1. Schaffer collateral-evoked AMPAR-mediated eEPSCs were measured using whole-cell patch clamp recordings from CA1 pyramidal neurons held at −70 mV in the presence of PTX (100 µM). Bath perfusion of netrin-1 (100-200 ng/ml, 5 min) without tetanic stimulation elicited a dose-dependent potentiation of synaptic responses that persisted for at least 40 min (Figure 3a).
To determine if netrin-1 potentiation of eEPSCs was associated with facilitation of presynaptic release, we assessed the paired-pulse ratio across a range of inter-stimulus intervals (10-200 ms ISI) prior to and 20 min after netrin-1 application. We detected no change, consistent with the lack of an effect on presynaptic release properties (Figure 3b). Further, individual paired comparisons failed to detect any change in paired-pulse ratio. These findings suggest that exogenous netrin-1 facilitates AMPAR-mediated synaptic currents in CA1 pyramidal neurons independently of changes in presynaptic glutamate release.
Glutamatergic synapses can be potentiated by the postsynaptic recruitment of AMPARs 1. We found that AMPAR-mediated current (measured at −70 mV) was significantly increased following perfusion of netrin-1, whereas NMDAR-mediated current (measured at +40 mV, 50 ms post-tetanus) was unchanged, resulting in an increased AMPAR-to-NMDAR current ratio (Figure 3c-f, Supplemental figure 5). These findings provide evidence that the addition of netrin-1 is sufficient to rapidly and selectively facilitate glutamatergic synaptic transmission in the hippocampus via the postsynaptic insertion of AMPARs into hippocampal synapses.
AMPAR insertion into immature spines can lead to synaptic maturation and contribute to potentiation of synaptic responses in the hippocampus 30, 31. To determine if bath application of netrin-1 increases the number of functional synapses by recruiting immature synapses, we recorded AMPAR-mediated mEPSCs in CA1 pyramidal neurons from adult mouse acute hippocampal slices in the presence of PTX (100 µM) and TTX (1 µM) (Figure 4a). Bath application of netrin-1 (200 ng/ml, 5 min) significantly increased mEPSC frequency but not amplitude, consistent with insertion of AMPARs into nascent synapses (Figure 4b-c). To directly assess whether netrin-1 leads to synaptic maturation through the insertion of AMPARs, we recorded evoked AMPAR-mediated currents in response to minimal stimulation. During baseline conditions, low levels of stimulation frequently failed to elicit an evoked inward synaptic current (Figure 4d). Following bath application of netrin-1, the evoked EPSC failure rate was significantly reduced, consistent with an increase in the number of functional glutamatergic synapses recruited (Figure 4e-f). These results provide evidence that netrin-1 recruits AMPARs into immature synapses in adult hippocampal networks.

Netrin-1 recruits new synapses in CA1 pyramidal neurons during activity-dependent long-term potentiation. (a) Representative voltage-clamp recordings of mEPSCs from CA1 pyramidal neurons held at −70 mV in an acute adult mouse hippocampal slice in the presence of tetrodotoxin (1 µM) and picrotoxin (100 µM) before and after 5 min bath application of netrin-1 (200 ng/ml) (n=9 cells from 5 mice). (b-c) Cumulative distribution plots (left) and group data (right) of mEPSC amplitude (b; 16.5±1.1 pA in baseline vs. 16.4±1.3 pA following netrin-1, t8=0.35, p=0.73, paired samples t-test) and frequency (c; 4.2±1.8 Hz in baseline vs. 7.1±1.7 Hz following netrin-1, t8=3.37, p<0.01, paired samples t-test). Kolmogorov-Smirnov test for cumulative distribution data. (d-f) Synaptic responses evoked by minimal stimulation (raw traces in grey, averaged in black, d) and individual response amplitudes (e) during baseline and 20 min following netrin-1 (blue in e). Group data show a significant decrease in failure rate following bath application of netrin-1 (200 ng/ml). (f; 64±11% in baseline vs. 24±11% following netrin-1, t5=3.46, p=0.018, paired samples t-test; n=6 cells from 6 mice).
Exogenous netrin-1 occludes activity-induced LTP
The rapid and long-lasting potentiation of synaptic responses after bath application of netrin-1, as well as the attenuation of potentiation induced by high-frequency stimulation in hippocampal slices from in T-29-CaMKIIα∷Cre/NTN1fl/flmice, suggest that activity-dependent secretion of netrin-1 may contribute to the expression of LTP induced by high-frequency stimulation. To assess whether netrin-1 is linked mechanistically with high-frequency stimulation induced-LTP, we first examined the effect of netrin-1 on previously-potentiated synapses. Following a 5 min baseline period, LTP was induced by brief HFS (1-s at 100 Hz). Synaptic responses were significantly potentiated compared to baseline values and resulted in a significant increase in the AMPAR-to-NMDAR ratio, consistent with insertion of AMPARs at Schaffer collateral synapses in the adult mouse brain following HFS. Bath application of netrin-1 failed to elicit additional enhancement of AMPAR-mediated evoked synaptic responses, nor alter AMPAR-to-NMDAR ratios compared to post-HFS levels (Figure 5a-e). These data indicate that activity-dependent HFS LTP can successfully occlude netrin-1-mediated potentiation.

Netrin-1 occludes HFS LTP in the adult hippocampus. (a) Representative traces of EP-SCs during baseline (1, black), 20 min following HFS (2, red, 1-s at 100 Hz arrow in b), and following 5 min bath application of netrin-1 (200 ng/ml) (3, blue). (b-e) Group data show that HFS results in potentiation of AMPAR-mediated synaptic responses (b), but does not occlude further potentiation by 5 min netrin-1 application (c; 218±34% of baseline following HFS vs. 290±51% following netrin-1, F2,23=21.84, p<0.001, One-way RM-ANOVA; pairwise comparisons, baseline vs. HFS: p=0.001, baseline vs. netrin-1: p=0.002, HFS vs. netrin-1: p=0.106). NMDAR-mediated EPSCs were not significantly changed between all conditions (d; 105±12% of baseline following HFS vs. 120±15% following netrin-1, F2,23=1.05, p=0.374, One-way RM-ANOVA), whereas AMPAR-to-NMDAR ratios were significantly elevated following both HFS and netrin-1 (e; Baseline: 2.8±0.7, HFS: 5.4±1.0, netrin-1: 6.6±1.5, F2,23=11.01, p=0.006, One-way RM-ANOVA; pair-wise comparisons, baseline vs. HFS: p=0.018, baseline vs. netrin-1: p=0.029, HFS vs. netrin-1: p=0.311). n=8 cells from 5 mice. (f) Representative traces of evoked synaptic responses during baseline (1, black), 25 min following bath application of netrin-1 (2, blue, 5 min at 200 ng/ml), and following HFS (3, red, 1-s at 100 Hz arrow in g). (g-j) Group data show that bath application of netrin-1 (blue in g) leads to significant potentiation of AMPAR-mediated synaptic responses, but that HFS (1-s at 100 Hz, arrow in g) does not further increase evoked amplitude (h; 201±26% of baseline following netrin-1 vs. 237±31% of baseline following HFS, F2,13=11.33, p<0.001, One-way RM-ANOVA; pairwise comparisons, baseline vs. netrin-1: p=0.001, baseline vs. HFS: p<0.001, netrin-1 vs. HFS: p=0.92, n=8). NMDAR-mediated synaptic responses were not significantly affected by netrin-1 (99±8% of baseline following netrin-1, 98±8% following HFS, F2,13=1.57, p=0.233), whereas AMPAR-to-NMDAR ratio (i) was significantly elevated following netrin-1, but was not further increased following HFS (baseline: 3.9±0.5, netrin-1: 7.5±1.4, HFS: 8.5±1.2; F2,13=17.28, p<0.001; baseline vs. netrin-1: p=0.004, baseline vs. HFS: p<0.001, netrin-1 vs. HFS: p=0.39, pairwise comparisons). n=14 cells from 9 mice. All pairwise comparisons using Tukey’s multiple comparison test.
We then assessed whether netrin-1-induced potentiation can occlude the expression of LTP induced by high-frequency stimulation. Bath application of exogenous netrin-1 resulted in rapid potentiation of synaptic responses. Twenty min following the application of netrin-1, we attempted to induce LTP using HFS. A single burst of high-frequency stimulation resulted in a brief increase in eEPSC amplitude, consistent with a post-tetanic short-term facilitation of Schaffer collateral inputs. However, by 15 min after high-frequency stimulation we observed no significant potentiation compared to pre-tetanus eEPSC amplitudes (Figure 5f-j). This occlusion suggests that the application of exogenous netrin-1 saturates a mechanism required for high-frequency stimulation-induced LTP. Failure to induce LTP may also have been due to whole-cell dialysis at the induction time point. We therefore applied HFS 30 min after whole-cell break-in and found that it was sufficient to induce LTP (Supplemental figure 6). Together, these findings support the conclusion that high-frequency stimulation promotes netrin-1 secretion, which in turn activates downstream effectors that recruit AMPARs to hippocampal synapses.
Netrin-1 facilitates synaptic responses via NMDAR-independent increases in intracellular Ca2+ levels and activation of CaMKII
NMDAR activation triggers activity-dependent LTP of AMPAR currents 21. To assess the role of NMDARs in netrin-1 potentiation of synaptic responses, we bath applied exogenous netrin-1 in the presence of the NMDAR antagonist, APV (50 µM). Netrin-1 resulted in strong potentiation of evoked AMPAR-mediated current (Figure 6a-c), indicating that NMDAR activation is not required for netrin-1 potentiation of synaptic responses. Taken together with the demonstration that NMDAR-dependent cLTP stimulation increases netrin-1 secretion, we conclude that NMDAR activation promotes the release of netrin-1, and that netrin-1 triggers downstream mechanisms that are sufficient to potentiate synaptic responses independent of NMDAR activation.

Netrin-1 potentiation of AMPA current requires increased intracellular Ca2+ and CaMKII activation independent of NMDAR function. (a-c) Evoked synaptic responses in CA1 pyramidal neurons from acute mouse hippocampal slices after bath application of netrin-1 (200 ng/ml, blue) in the presence of APV (a; 50 µM, purple; n=9 cells from 5 mice). Representative traces (b) and group data (c) prior to (1 in a) and following bath application of netrin-1 (2 in a) in the presence of APV (259±53% of baseline; t8=4.66, p=0.001, paired samples t-test). (d-f) Effect of netrin-1 (d; blue bar) on synaptic responses in CA1 pyramidal neurons from acute mouse hip-pocampal slices from aged DCC cKO (>6 months old CaMKIIα∷Cre/DCCfl/fl, red, n=7 cells from 5 mice), young DCC cKO (4-6 weeks old CaMKIIα∷Cre/DCCfl/fl, green, n=5 cells from 3 mice), and control aged littermates mice (>6 months old CaMKIIα∷Creneg/DCCfl/fl, black, n=7 cells from 5 mice). (e) Group data show that netrin-1 increases amplitude of AMPAR-mediated current in aged littermates and young DCC cKO mice but not in aged DCC cKO (aged control littermate: 200±33% of baseline, p=0.002; young DCC cKO: 153±15% of baseline, p=0.04; aged DCC cKO: 89±8% of baseline, p=0.44; Two-way RM-ANOVA: Interaction between genotype X LTP: F2,17=5.54, p = 0.014) (f) AMPAR-to-NMDAR ratio was significantly increased in aged littermates and young DCC cKO following bath application of netrin-1, but not in aged DCC cKO (aged control littermate: 3.0±0.6 in baseline vs. 5.5±0.9 following netrin-1, p<0.001; young DCC cKO: 2.1±0.2 in baseline vs. 3.8±0.4 following netrin-1, p=0.005; aged DCC cKO: 2.8±0.5 in baseline vs. 2.7±0.3 following netrin-1, p=0.91; Two-way RM-ANOVA Interaction between genotype X LTP: F2,17=6.22, p = 0.009) (g-i) Effect of netrin-1 (blue) on average synaptic responses (g) and representative evoked EPSCs (h) recorded in CA1 pyramidal neurons in acute mouse hippocampal slices with intracellular BAPTA (20 mM, dark blue, n = 6 cells from 4 mice). Group data show that netrin-1 did not significantly potentiate AMPAR-mediated synaptic responses (i, left; 118±22% of baseline, t5=0.66, p=0.54, paired samples t-test) or increase AMPAR-to-NMDAR ratio (j; 2.9±0.4 in baseline vs. 3.8±0.9 following netrin-1; t5=1.47 p=0.20, paired samples t-test). (k-o) Netrin-1 phosphorylates CaMKII to potentiate synaptic responses in the adult hippocampus. (k) Representative Western blot (left) and group data (right) from isolated hippocampal slices showing increased levels of phosphorylated CaMKIIα (Thr286) following bath application of netrin-1 (200 ng/ml) (control: 1.00±0.06, netrin-1: 1.19±0.02; t6=2.60, p=0.04, independent samples t-test, n=4 slices per mouse, N=4 mice). Effect of netrin-1 (blue bar) on averaged (l) and representative (m) evoked synaptic responses in CA1 pyramidal neurons in the presence of KN62 (5 µM, n =7 cells from 5 mice, green bar). Group data show no significant increase in AMPAR-mediated current (n; 95±7% of baseline, t6=0.73, p=0.49, paired samples t-test) or AMPAR-to-NMDAR ratio (o, 3.2±0.6 in baseline vs. 3.5±0.6 following netrin-1, t6=0.56, p=0.59, paired samples t-test) following netrin-1 (200 ng/ml, 5 min, blue bar).
Netrin-1 binding to DCC mediates axonal chemoattraction during development 10, 32. To determine if netrin-1 potentiation requires DCC, we deleted a floxed dcc allele from glutamatergic neurons in the adult forebrain by generating T-29-CaMKIIα∷Cre/DCCfl/fl mice (DCC cKO) 15. Bath perfusion of netrin-1 potentiated eEPSCs in acute slices from control littermates and young 4-6 week old DCC cKO mice that still express high levels of residual DCC, but failed to potentiate eEPSCs in CA1 pyramidal neurons from 6-month-old adult DCC cKO mice (Figure 6d-e). Additionally, while bath application of netrin-1 increased AMPAR-mediated current and AMPAR-to-NMDAR ratios in control littermates and 4-6 week old DCC cKO mice, no increase was detected in aged DCC cKO (Figure 6f). We conclude that netrin-1 potentiation of Schaffer collateral inputs requires DCC.
Both NMDAR and DCC activation result in Ca2+ influx and increased intracellular Ca2+ concentration, which in turn can promote changes in AMPAR trafficking via Ca2+/calmodulin kinase II (CaMKII) activation 33-35. To assess the contribution of increased intracellular Ca2+ to netrin-1 potentiation, we included the Ca2+ chelator 1,2-bis-(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid (BAPTA; 20 mM) in the intracellular recording pipette. BAPTA completely blocked netrin-1 potentiation of AMPAR-mediated synaptic responses and the increase in AMPAR-to-NMDAR ratio (Figure 6g-j), indicating that intracellular Ca2+ is critical for netrin-1 potentiation.
CaMKII activation and stargazin phosphorylation are required to trap and accumulate GluA1-containing AMPARs at synapses 33. Bath application of netrin-1 resulted in increased CaMKII phosphorylation in hippocampal slice homogenates (Figure 6l), suggesting that netrin-1-mediated activation of CaMKII may potentiate synaptic responses. Consistent with this, bath application of the CaMKII inhibitor KN62 (5 µM) completely blocked netrin-1-induced increases in the amplitude of evoked AMPAR-mediated currents and AMPAR-to-NMDAR ratio in acute hippocampal slices (Figure 6l-o). These results support the conclusion that netrin-1 activates CaMKII, and that CaMKII activation is required for netrin-1 potentiation of evoked responses.
Netrin-1 potentiation is mediated via postsynaptic accumulation of GluA1-containing receptors
Increases in intracellular Ca2+ can trigger AMPAR recruitment to synapses 34. AMPARs are composed of homo-or heteromeric configurations of four different subunits (GluA1-4), with Q/R-edited GluA2-containing receptors lacking Ca2+ permeability 36, 37. Early stages of LTP require interactions mediated by the “TGL” PDZ domain binding motif at the carboxyl terminus of the GluA1 AMPAR subunit 34. However, recent studies suggest that multiple forms of AMPARs contribute to this type of synaptic plasticity 38. We assessed the subunit composition of AMPARs inserted into the plasma membrane 20 min following application of netrin-1 (200 ng/ml, 5 min) using cell-surface biotinylation of isolated acute adult hippocampal slices. Bath application of netrin-1 significantly increased the amount of cell surface GluA1 compared to control slices from the same animal that were not exposed to netrin-1 (Figure 7a). In contrast, levels of biotinylated GluA2 were not different between conditions (Figure 7b). These findings indicate that netrin-1 selectively increases the amount of GluA1-containing, GluA2-lacking AMPARs at the neuronal surface.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Synaptic recruitment of Ca2+-permeable GluA1-containing AMPA receptors by netrin-1. (a-b) Western blots (left) and group data (right) of GluA1 (a; 306±73% of baseline, t2=2.81, p=0.04, independent samples t-test) and GluA2 (b; 145±49% of baseline, t2=0.45, p=0.44, independent samples t-test) surface membrane distribution in acute hippocampal slices from adult mice with (blue) or without (black) 5 min bath application of netrin-1 (200 ng/ml; n=4 slices per mouse, N = 3 mice per condition). (c-e) Representative traces of synaptic rectification experiments during baseline (black) and following netrin-1 (200 ng/ml; blue). Effect of netrin-1 on I-V curve of AMPAR-mediated synaptic responses (d) and rectification index (e; 2.7±0.7 in baseline vs. 7.7±1.9 following netrin-1, t4=3.13, p=0.03, paired samples t-test). n = 5 cells from 3 mice. (f-i) Effect of netrin-1 (200 ng/ml; blue) on Schaffer collateral evoked synaptic response in CA1 pyramidal neurons from acute adult hippocampal slices during continuous bath application of the CP-AMPAR antagonist, IEM1460 (f; 50 µM, purple bar; n = 7 cells from 4 mice). Representative evoked EPSCs (g) and group data show AMPAR-mediated current (h; 112±22% of baseline, t6=0.01, p=0.99, paired samples t-test) and AMPAR-to-NMDAR ratio (i; 3.5±0.9 in baseline vs. 3.3±0.9 following netrin-1, t6=0.16, p = 0.87, paired samples t-test) in the presence of IEM1460 following bath application of netrin-1. (j-l) Inhibition of GluA1 insertion with intracellular Pep1-TGL (100 µM, yellow bar, n=8 cells from 4 mice) blocks netrin-1 potentiation of synaptic responses (j). Representative evoked EPSCs (k) and group data (l) show mean AMPAR-mediated current during baseline and following netrin-1 in neurons loaded with Pep1-TGL (i; 93±9% of baseline, t7=0.13, p=0.89, paired samples t-test). (m) Sample images of dendritic segments expressing both SEP-GluA1 (left, green on right) and Homer1C-DsRed (middle, red on right) during baseline (top) and after netrin-1 (200 ng/ml, bottom) in dissociated rat hippocampal neurons. Overlay (right) shows SEP-GluA1 insertion at Homer1C-DsRed sites (white arrows). Scale = 5 µm. (n) Average SEP-GluA1 fluorescence variation at Homer1C-DsRed puncta for each neuron (control: ΔFmedian=-0.0364, n=10 cells vs. netrin-1: ΔFmedian=0.0226, n=12 cells). Wilcoxon test for paired observations, *: p<0.05. (o) Circular plots show proportion of synaptic states (blue: “down”, black: “stay”, red: “up”; see Methods) after netrin-1 compared to control sister cultures.
GluA2-lacking AMPARs composed of hetero- or homomeric combinations of GluA1, GluA3, and GluA4 subunits are permeable to Ca2+, and characterized by pronounced inward rec-tification due to blockade by endogenous polyamines 39, 40. Bath application of netrin-1 in the presence of GABAA and NMDAR antagonists significantly enhanced rectification of AMPAR-mediated currents at depolarized voltages (Figure 7c-e). To determine if netrin-1-induced potentiation is mediated by Ca2+-permeable AMPARs (CP-AMPARs), netrin-1 was co-applied with the CP-AMPAR blocker, IEM1460 (50 µM) 37. Blockade of CP-AMPARs eliminated the netrin-1 induced potentiation of synaptic responses, and blocked the increases in AMPAR-mediated current and AMPAR-to-NMDAR ratio (Figure 7f-i). These findings are consistent with GluA2-lacking CP-AMPARs underlying the netrin-1-induced synaptic potentiation, likely engaging the incorporation of hetero- or homotetramers of GluA1. To test this, we then blocked the incorporation of GluA1-containing AMPARs by including the competitive peptide Pep1-TGL (100 µM) in the in-tracellular solution 34, 41. Pep1-TGL, which prevents SAP97 binding to the “TGL” PDZ binding motif at the GluA1 C-terminus, completely blocked netrin-1 potentiation of AMPAR-mediated current (Figure 7j-l). To determine if netrin-1 promotes GluA1 synaptic localization, we co-expressed super ecliptic pHluorin-tagged GluA1 (SEP-GluA1) and the post-synaptic marker Homer1c-DsRed in cultured rat hippocampal neurons (14 DIV). Bath application of netrin-1 (200 ng/ml) triggered a rapid increase in SEP-GluA1 fluorescence at a subset of synapses compared to cultures not treated with netrin-1 (Figure 7m-n, Supplemental movie 2-3). Moreover, bath application of netrin-1 resulted in an increase in the number of SEP-GluA1 positive synapses (“up” puncta), while decreasing the number of SEP-GluA1 negative synapses (“down” puncta) compared to control cultures without netrin-1 application (Figure 7o). Together, these findings indicate that netrin-1 promotes the insertion of GluA1-containing AMPARs at a subset of synaptic sites in hippocampal pyramidal neurons.
Discussion
Netrin-1 is essential for normal embryonic neural development, directing cell migration, axon guidance, and synaptogenesis. Neuronal expression and enrichment at synapses in the adult CNS suggested that netrin-1 might also influence the plasticity of mature neural circuits. Here, we demonstrate that netrin-1 expressed by neurons plays a critical role in the modification of glutamatergic transmission at Schaffer collateral synapses between CA3 and CA1 pyramidal neurons in the adult mammalian hippocampus. Our findings reveal that netrin-1 is secreted by neurons in response to depolarization and NMDAR activation. We demonstrate that neuronal expression of netrin-1 in principal forebrain neurons is critical for activity-dependent LTP, that brief application of exogenous netrin-1 triggers long-lasting synaptic potentiation, and that netrin-1-induced synaptic potentiation occludes LTP induced by high-frequency stimulation. Further, we show that netrin-1-induced potentiation enhances glutamatergic synaptic transmission through DCC-mediated recruitment of GluA1-containing AMPARs to hippocampal synapses in a CaMKII-dependent manner. These findings reveal an essential role for neuronal secretion of netrin-1 in glutamatergic synaptic plasticity in the adult hippocampus that is critical for classic activity-dependent LTP.
Netrin-1 contributes to activity-dependent LTP
Long-lasting changes in synaptic strength in the adult hippocampus depend on activation of postsynaptic NMDARs 42. Here, we report that netrin-1 is enriched in dendrites (Figure 2) and that NMDAR activation can result in netrin-1 exocytosis (Figure 1), suggesting a role in activity-dependent synaptic plasticity in the hippocampus. Consistent with this, conditional loss of netrin-1 from excitatory hippocampal neurons results in attenuated expression of LTP with no change in short-term plasticity following HFS (Figure 2). Short-term facilitation of synaptic responses following HFS is mediated through changes in presynaptic Ca2+ that enhance transmitter exocytosis 43. In contrast, changes in the distribution of post-synaptic receptors can be detected within 5-10 min after HFS, consistent with the time course observed following exogenous application of netrin-1. Moreover, our findings indicate that following LTP-inducing stimulation, the NMDAR-mediated Ca2+ influx mobilizes the secretion of netrin-1 from dendrites at or near synaptic sites (Figure 1). We propose that netrin-1 then activates postsynaptic signaling mechanisms such as CaMKII, independent of NMDAR activation, that locally recruit GluA1-containing CP-AMPARs to excitatory synapses, resulting in postsynaptic potentiation of synaptic transmission.
Activity-dependent secretion of netrin-1 enhances synaptic transmission through maturation of immature synapses
Intracellular vesicles isolated from adult rat brain synaptosomes contain readily detectable netrin-1 and DCC. Patches of DCC immunoreactivity decorate dendritic spines, and DCC is associated with a detergent-resistant component of postsynaptic density 15. Postsynaptic expression of LTP requires soluble N-ethyl-maleimide-sensitive factor-attachment proteins (SNAPs), which promote vesicle exocytosis in a t-SNARE-dependent manner 44, 45. DCC interacts with the t-SNARE synatxin-1 and v-SNARE TI-VAMP. However, it remains to be demonstrated that netrin-1 activation of DCC in dendritic spines is required for SNARE function during activity-dependent AMPAR recruitment.
Netrin-1 is secreted from Homer1C-positive sites along dendrites in response to plasticity-inducing stimulation (Figure 1). We also observed exocytotic events at sites lacking Homer1C-DsRed. Homer1C is an adaptor for PSD enriched proteins, where it regulates glutamatergic synaptic transmission through interactions with Group I metabotropic glutamate receptors (mGluR1/5) 46, 47. Recent evidence indicates that ∼20% of excitatory synapses in the hippocampus may lack the PSD proteins necessary for synaptic stabilization 48, and that these non-PSD-containing synapses exhibit relatively low AMPAR to NMDAR ratios 49, 50. Bath application of netrin-1 potently increases the AMPAR-to-NMDAR ratio (Figure 3), indicating that netrin-1 secretion promotes insertion of GluA1-containing AMPARs, recruiting these receptors to immature synapses, possibly including those that initially lack PSD proteins. This conclusion is further supported by the demonstration that challenging cultured rat pyramidal neurons with an immobilized netrin-1 coated micro-bead is sufficient to rapidly recruit PSD-95 7, a critical post-synaptic adaptor that drives glutamate receptor clustering and promotes synaptic maturation 51.
Previous work suggests that HFS LTP selectively recruits GluA1-containing CP-AMPARs and increases AMPAR single channel conductance 52, 53. Consistent with increases in immature synapses in the absence of netrin-1, conditional deletion resulted in significantly decreased amplitude of mEPSCs compared to control littermates, suggesting that lack of netrin-1 results in reduced AMPAR density at synapses (Supplemental figure 4). In contrast, following bath application of exogenous netrin-1 to acute hippocampal slices from wild-type mice, we observed ∼2 fold increase in mEPSC frequency with no appreciable change in mEPSC amplitude. These findings suggest that netrin-1 may recruit of CP-AMPARs to immature synapses to serve as “placeholders” for subsequent synaptic stabilization. Indeed, previous work has suggested that LTP can lead to increases in mEPSC frequency 54, 55 by increasing the density of mature synapses 56, 57. Therefore, netrin-1 may facilitate the maturation of immature synapses by promoting the incorporation of transient GluA1-containing AMPARs, which may be replaced with GluA2-containing AMPARs by synaptic local protein synthesis 14, 58, 59.
AMPAR trafficking is a key process for LTP expression in the hippocampus, and is mediated in part through NMDAR-mediated increases in intracellular Ca2+ and CaMKII activation 60,61. Interestingly, LTP results in enlargement of thin-type dendritic spines, which are largely devoid of AMPARs under control conditions 62, 63. Increases in thin-type spine volume is dependent, in part, on activation of CaMKII and RhoGTPases including RhoA and Cdc42 64, 65. Netrin-1 binding to DCC, which is critical for netrin-1-mediated potentiation of AMPAR currents (Figure 6), directs cytoskeletal reorganization in developing neuronal growth cones by regulating Cdc42, Rac1 and RhoA 66-68. However, it remains to be determined how netrin-1 may contribute to AMPAR trafficking and structural modification of dendritic spines associated with LTP in the adult hippocampus.
We have demonstrated that netrin-1 is enriched in the stratum radiatum of adult mice and is detected intracellularly within dendritic shafts and spines of CA1 pyramidal neurons (Figure 2). Our findings suggest that NMDAR-dependent exocytosis of netrin-1 during activity-dependent LTP promotes recruitment of Ca2+-permeable GluA1-containing AMPARs at immature or nascent synapses. Although certain forms of activity-dependent plasticity do not require GluA1 receptor insertion 38, trafficking of GluA1-containing AMPARs is dependent on increases in intracellular Ca2+ and is critical for LTP in the adult hippocampus 52, 69, 70. Application of netrin-1 is sufficient to trigger a local increase in the concentration of intracellular Ca2+ in developing neurons 35, 71-73. We propose that NMDAR-mediated synaptic Ca2+ transients in dendritic spines facilitate netrin-1 release, which in turn locally increases intracellular Ca2+ from internal stores to activate CaMKII, and promote the focal insertion and synaptic trapping of GluA1-containing AMPARs. Together, our findings indicate that NMDAR-mediated increases in intracellular Ca2+ promote the activity-dependent secretion of netrin-1, which functions as a critical effector of long-term synaptic plasticity.
Author contributions
S.D.G., S.L., I.V.B, P.D.K., E.S.R., and T.E.K. conceived the study and designed the experiments. S.D.G. and J. G. performed and analyzed electrophysiological data. S. D. G. and I.V.B. performed and collected molecular biological data. S.L. performed live imaging experiments. S.D.G. and S.A. performed and collected immunohistological data. D.H., S.N.H., R.A.M., and P.S. provided reagents and resources. S.D.G, S.L., P.D.K, E.S.R., and T.E.K. wrote the paper, and all authors read and approved the final manuscript. P.W., P.S., P.D.K., E.S.R., and T.E.K supervised the study.
Methods
Animals
The study was conducted in accordance with the guidelines of the Canadian Council for Animal Care and approved by the McGill University and Université Laval Animal Care Committees. All animals were housed in group housing, and provided ad libitum access to food and water. For experiments using wild-type animals, C57/B6 mice (8-12 weeks) were used (Charles River, St Constant, Canada). Thy1-GFP (line 15) mice were used in immunohistochemical experiments, and express low levels of membrane-targeted EGFP (mGFP)-positive cells within the CA1 region of the hippocampus under the control of Thy1.2 promoter 74. Floxed alleles of ntn1 (NTN1fl/fl) and dcc (DCCfl/fl) were generated in mice and maintained on a C57BL/6 genetic background as described 15, 25. T29-1 CaMKIIα∷Cre mice (JAX 005359) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and maintained on a C57BL/6 background. Both male and female T-29-CaMKIIα∷Cre/netrin-1fl/fl (T-29-CaMKIIα∷Cre/NTN1fl/fl > 3 months old) and T-29-CaMKIIα∷Cre/DCCfl/fl (T-29-CaMKIIα∷Cre/DCCfl/fl > 6 months old) mice were used for molecular biological and electrophysiology experiments. We observed no statistically significant differences between sexes, and therefore all data were pooled for analysis. Control experiments were performed using littermates that were negative for Cre and homozygous for floxed alleles of ntn1 and dcc.
Hippocampal neuronal culture
For cell biological assays, hippocampi from E18 or P1 Sprague Dawley rats were isolated, dissociated, plated and cultured as described 7, 15, 75.
Immunohistochemistry
Immunohistochemistry was performed on free-floating sections derived from Thy1-GFP (line 15). Briefly, animals were deeply anaesthetized by intraperitoneal injection of a mixture of 2,2,2 – tribromoethyl alcohol and tert-amyl alcohol diluted at 2.5% in PBS, and transcardially perfused with cold PBS (pH: 7.4) followed by 4% paraformaldehyde (PFA). Brains were removed and post-fixed for 24 h in 4% PFA, followed by 24 h cryoprotection in 30% sucrose in PBS. Thirty µm sections were cut using a cryostat (Leica CM1850), and stored in PBS at 4°C.
Antigen retrieval was performed by boiling sections in 0.1M citrate buffer for 10 min, and allowing to cool to RT. Sections were then washed three times in PBS-T (0.3% Triton X-100), and blocked for 1.5 h in PBS-T containing 3% BSA. Brain sections were incubated sequentially over-night with rabbit monoclonal anti-netrin-1 (1:500, Abcam, RRID:AB_11131145) and chicken polyclonal anti-GFP (1:1000, Abcam, RRID:AB_300798), washed 3 ×; 10 min in blocking solution, and coupled to corresponding secondary antibodies (donkey anti-rabbit Alexa 555, 1:1000, RRID:AB_2536182; and goat anti-chicken Alexa 488, 1:1000, RRID:AB_2636803) for 1h at RT. Sections were then successively washed twice for 10 min with PBS-T and PBS, mounted on gelatin-coated glass microscope slides, and coverslipped using DAPI-Fluromount-G (Southern Biotech).
Embryonic hippocampal neurons infected with AAV8-hSyn-HA-hM3D(Gq)-IRES-mCitrine were fixed with 4% PFA, washed 3 ×; 10 min with 0.1% PBS-T, blocked in 0.1% PBS-T containing 3% BSA for 1h, incubated overnight in rabbit anti-HA (1:1000, Abcam, RRID:AB_307019), washed 3 ×; 10 min with 3% BSA in 0.1% PBS-T, and coupled with corre-sponding secondary anti-rabbit Alexa 488 (1:1000, ThermoFisher Scientific, RRID: AB_143165) and Hoescht stain (1:1000, ThermoFisher Scientific, RRID:AB_2307445). Coverslips were washed 2 ×; 5 min with PBS-T followed by 2 ×; 5 min in PBS and mounted on glass microscope slides.
Immunofluorescence images were obtained using either a Zeiss Axiovert (S100TV) with epifluorescence illumination at 40x (0.75 N.A.), or a Zeiss LSM 880 laser-scanning confocal microscope equipped with an Airyscan detection unit using a high NA oil immersion alpha Plan-Apochromat 100X/1.46 Oil DIC M27 (Zeiss) with a zoom adjustment to 1.8. All imaging was performed using Immersol 518 F immersion media (ne = 1.518 (23 °C); Carl Zeiss). Detector gain set to 700 and pixel dwell times were adjusted for each dataset keeping them at their lowest values in order to avoid saturation and bleaching effect. The pinhole was adjusted to have an AU of ∼1.25 considering the axial position in tissue. The different fluorophores were acquired sequentially with appropriate filter conditions. The information is listed in the following as “fluorophore – excitation wavelength – dichroic mirror – detection filter”. Alexa Fluor 488 – 488 nm (Argon) – MBS488 – BP 495-550 + LP 570; CF 568 – 561 nm (DPSS 561-10) - MBS 458/561 – BP 495-550 + LP 570; Alexa Fluor 647 – 633 nm (HeNe633) – MBS 488/561/633 – BP 570-620 + LP 645. For processing Airyscan images, Zen Black 2.3 software was used to enhance the spatial resolution 1.7-fold of the diffraction limit by processing each of the 32 Airy detector channels separately with filtering, deconvolution and pixel reassignment. The visualization and rendering was processed with Imaris 8.1.2 (Bitplane AG, Zurich CH).
For images in Supplemental figure 2c-e, cultured hippocampal neurons were fixed for 10 min in 4% PFA solution (0.1 M phosphate buffer, 4% sucrose, and 2 mM EGTA, pH 7.4). Neurons were then washed once with PBS and twice with 0.1 M PBS/glycine and were permeabilized for 30 min in blocking solution (PBS, 2% normal goat serum, and 0.1% Triton X-100). Primary antibody against netrin-1 (1:500, Abcam, RRID:AB_11131145) was diluted in blocking solution and incubated for 2 h at room temperature. After washes in PBS, secondary antibodies (goat anti-rabbit STAR488, Abberior, 1:1000) was diluted in blocking solution and incubated for 45 min at room temperature. Coverslips were mounted in Prolong Gold Antifade mounting media (Thermofisher). Images were acquired on a confocal system (LSM700; Carl Zeiss) using a 63x 1.4 NA oil immersion objective.
Protein media concentration
For protein media concentration experiments, rat hippocampal neurons from E18 Sprague Dawley rats were plated at 2.5 ×; 106 cells and used at 14 DIV. For hM3D/CNO experiments, cells were infected at 6 DIV with pAAV8-hSyn-HA-hM3D(Gq)-IRES-mCitrine (MOI: 1:10000; Neurophotonics Molecular Tools Platform, Université Laval, Québec, QC, Canada). At 14 DIV, cells were washed 3x with warm (37°C) control ACSF containing (in mM): 135 NaCl, 3.5 KCl, 1.2 MgCl2, 2 CaCl2, 10 HEPES, and 20 D-Glucose (pH: 7.4, 300 mOsm). Cells were then incubated at 37°C in ACSF with or without clozapine-N-oxide (CNO, 10 µM) for 1 h. For KCl experiments (Supplemental figure 1), cells were washed 3x with serum-free warmed NeuroBasal media (NBM, ThermoFisher Scientific). Cells were then incubated in NBM supplemented with high KCl (20 mM) for 20 min at 37°C. Protease inhibitors (in 1 mg/ml aprotonin, 1 mg/ml leupeptin, 100 mM PMSF, and 0.5 M EDTA) were then added to the media, and cells and media were rapidly cooled to 4° C. Equal volumes of supernatant were collected into a 4 ml Amicon Ultra Centrifugal tube with Ultracel 10-membrane (EMD Millipore), and goat anti-chicken Alexa 633-conjugated secondary antibody (40 ng per tube) was added to ensure equal concentration. Control and experimental media tubes were then centrifuged at 5,000 x g for 60 min. The resultant supernatant was concentrated at 200x, lysed in Laemmli sample buffer, and boiled for 5 min. Cells were harvested and processed as described above.
Cell surface biotinylation and phosphorylation assays in acute brain slice
Cell surface biotinylation and phosphorylation assays were performed as described 76, 77. For surface biotinylation in brain slices, acute hippocampal slices from 2-3 month old C57/B6 mice were made using the same protocol as for electrophysiological experiments. Slices were then transferred to a recording chamber and continuously perfused with control ACSF. Exogenous netrin-1 (200 ng/ml) or vehicle was bath applied for 5 min, followed by a 20 min wash in ACSF (ACSF). Individual hippocampi were isolated by microdissection in ice-cold ACSF, and washed 3x in cold ACSF. For biotinylation assays, 3-4 hippocampi from the same animal and condition were transferred to chilled ACSF containing Ez-link Sulfo-NHS-LC-biotin (1 mg/ml; ThermoFisher Scientific) for 45 min. The biotin reaction was then quenched by washing the slices twice for 25 min at 4° C in 10 mM glycine in ACSF, followed by 3x in cold ACSF (5 min). Slices were harvested in RIPA buffer (containing 150 mM NaCl, 20 mM Tris, pH 8.0, 1%, NP-40 (USB Corp., Cleveland, OH, USA), 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA) supplemented with protease inhibitors (1mg/ml aprotonin, 1mg/ml leupeptin, 100 mM PMSF, and 0.5M EDTA). Biotinylated proteins were precipitated using streptavidin-agarose beads (ThermoFisher Scientific) at 4°C for 2h, centrifuged at 13,800 x g for 15 min, and washed twice with cold PBS. Proteins were then eluted from beads using Laemmli sample buffer, and boiled for 5 min.
For phosphorylation assays, hippocampi were dissected in ACSF and harvested in RIPA buffer supplemented with protease and phosphatase inhibitors (1mM Na3V04 and 1mM NaF), homogenized, and centrifuged for 10 min at 13,800 x g. Supernatant was then collected, lysed in Laemmli sample buffer, and boiled for 5 min.
Western blot analysis
Netrin-1 expression was assessed from hippocampal and cerebellar homogenates derived from 2-3 month old C57/B6 or 3 month old T-29-CaMKIIα∷Cre/NTN1fl/fl mice. Briefly, mice were deeply anaesthetized, and the brain was quickly extracted in ice-cold Ringer solution. Cerebellum, cortex, and hippocampi were dissected and isolated in cold DMEM (Life Technologies) supplemented with protease and phosphatase inhibitors, and homogenized in cold RIPA buffer containing (10 mM phosphate buffer [pH 7.2], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) with protease and phosphatase inhibitors (aprotinin 2 mg/ml, leupeptin 5 mg/ml, EDTA 2 mM, sodium orthovanadate 1 mM, sodium fluoride 1 mM, and PMSF 1 mM).
Extracts from protein media concentration supernatant, cell lysates, whole hippocampal homogenates, or hippocampal slice homogenates were assessed using Bradford protein assay (Pierce BCA kit, ThermoFisher Scientific). Equal protein levels were loaded and subjected to 10% SDS-page gel electrophoresis and transferred to nitrocellulose membranes, as described 15. Western blots were analyzed using mouse beta-III tubulin (1:1000; Sigma Aldrich, RRID:AB_477590), rabbit anti-active caspase 3 (1:1000; Abcam, RRID:AB_302962), rabbit anti-CaMKII (1:1000; ThermoFisher, RRID:AB_2533032), mouse anti-DCC (1:1000; BD Pharmingen, RRID:AB_395314), chicken anti-GAPDH (1:5,000; Millipore, RRID:AB_11211911), rabbit anti-GluA1 (1:10,000; Abcam, RRID:AB_10860361), rabbit anti-GluA2 (1:10,000; Abcam, RRID:AB_2620181), rabbit anti-netrin-1 (1:10,000; Abcam, RRID:AB_11131145), rabbit anti-phospho-Thr286(1:1000; PhosphoSolutions, RRID: AB_2492051). Blots were developed using Immobilon Western Chemiluminescent HRP Substrate (Millipore). Densitometry and quanti?cation of relative protein levels were performed on scanned images of immunoblots using Fiji image software 78.
Optical imaging of NTN1-SEP
NTN1-SEP was generated by subcloning the SEP coding sequence (GenBank: AY533296.1) 79 lacking the initiating methionine in frame at the C terminus of the sequence encoding netrin-1 9 in plasmid eGFP-N3 (Clontech). For optical imaging of NTN1-SEP, rat hippocampal neurons were plated at a density of 75 cells / mm2on glass coverslips coated with poly-D-lysine and maintained in Neurobasal/B27 (Thermo Fisher Scientific). Neurons were transfected with plasmids encoding NTN1-SEP and Homer-DsRed using Lipofectamine 2000 (Thermo Fisher Scientific) at 12 DIV for NTN1-SEP imaging, as described 80. Neurons (13-14 DIV) were imaged at 32-35 °C in an open perfusion chamber (0.2-0.5 ml/min) (Warner Instruments) mounted on an Olympus IX-71 inverted microscope equipped with a 100X objective (N. A. = 1.49) with Toptica Chrome MLE laser sources, and a backlit thinned CCD (Princeton ProEm 512B-FT). Signals for NTN1-SEP and Homer1C-DsRed were discriminated using the following laser lines and Semrock filters: SEP, excitation 488nm, emission Brightline 520/35; Homer1C-DsRed, excitation at 560 nm, emission with BrightLine 617/73.
To image netrin-1 exocytosis, a segment of dendrite expressing NTN1-SEP and Homer1C-DsRed was selected, and subsequently photobleached to 30-40% of the initial fluorescence by exposing the dendrite to approximately 40s of high power laser (488nm) light. Movies of 1000 images, at 100 ms per image, were then acquired at lower laser power, with neurons in baseline extracellular solution (in mM: 102 NaCl, 5 KCl, 10 Na HEPES, 1 MgCl2, 1.2 CaCl2, 10 Glucose, pH 7.3-7.4, 225-230 mOsm) followed by cLTP solution (same as baseline, except lacking Mg2+, and with 200 µM glycine and 30 µM bicuculline).
Image analysis was performed using a custom-made Matlab (Mathworks Inc., Natick, USA) script allowing semi-automatic detection of exocytotic events. Based on any single exocytotic event occurring faster than the image integration time, we computed maps of the events by subtracting each image from the previous frame. Each map was then segmented to identify regions exhibiting a fluorescence increase, which were extracted to calculate the ΔF/F on 500 ms preceding the event and 1500 ms following. We then computed the area under the ΔF/F curve, using a detection threshold of 2.
Brain slice in vitro electrophysiology
Acute horizontal brain slices containing the hippocampus were obtained from adult C57/B6 mice (8 to 12 weeks), and CaMKII∷Cre/NTN1fl/fl (3-6 months old) and age-matched control littermates. Mice were deeply anaesthetized by intraperitoneal injection of a mixture of 2,2,2 – tribromoethyl alcohol and tert-amyl alcohol diluted at 2.5% in PBS, and transcardially perfused with ice-cold choline chloride-based solution containing (in mM): 110 choline-Cl, 1.25 NaH2PO4, 25 NaHCO3, 7 MgCl2, 0.5 CaCl2, 2.5 KCl, 7 glucose, 3 pyruvic acid, and 1.3 ascorbic acid, bubbled with carbogen (O2 95%, CO2 5%). The brain was rapidly removed, thick horizontal brain slices (300 µm) containing the hippocampus were cut using a vibrating microtome (VT1000s, Leica), and allowed to recover for 1h in artificial cerebrospinal fluid (ACSF) containing, in mM: 124 NaCl, 5 KCl, 1.25 NaH2PO4, 2 MgSO4, 26 NaHCO3, 2 CaCl, and 10 Glucose saturated with 95% O2 and 5% CO2 (pH ∼7.3, 300 mOsm) at room temperature (22°-24° C).
Individual brain slices were placed in a custom-built recording chamber, and continuously perfused with warmed (30 ± 2° C) ACSF (TC324B, Warner Instruments). Voltage-recordings of CA1 hippocampal pyramidal neurons were performed on an upright microscope (Nikon Eclipse or Scientifica SliceScope 2000) equipped with a micromanipulator (Sutter MP-225 or Scientifica Patchstar), a 40x or 60x water immersion objective (0.8 or 1.0 N.A., respectively), differential interference contrast optics, and coupled to a near-infrared charge-coupled device camera (NC70, MTI or SciCam, Scientifica). Borosilicate glass pipettes (Sutter Instruments) were pulled with resistances of 4–8MΩ. The intracellular solution for voltage-clamp recordings was (in mM): 120 CsMeSO3, 20 CsCl, 10 HEPES, 7 di-tris phosphocreatine, 2 MgCl2, 0.2 EGTA, 4 Na2ATP, 0.3 Tris-GTP, and in some experiments, 5 N-ethyllidocaine chloride (QX-314) (pH 7.2-7.26, 280-290 mOsm). AMPAR-mediated and NMDAR-mediated synaptic responses were evoked using a bipolar platinum/iridium electrode (FHC, CE2C275) placed in the Schaffer collaterals, ∼200 µm from the recorded cell. For synaptic experiments, a pair of 0.1 ms bipolar current pulses (50 ms ISI) was delivered via a stimulus isolation unit (Isoflex, AMPI), and stimulus intensity was adjusted to evoke a response 65-75% of the maximal response at 200 µA. After 10-20 min of stable baseline recording in the presence of picrotoxin (100 µM, PTX), netrin-1 (200 ng/ml) was bath applied for 5 min, and synaptic responses monitored for 40 min post-application. AMPAR- and NMDAR-mediated currents were evoked at −70 mV and +40 mV, respectively, during baseline conditions and 20 min post-bath application of netrin-1. Input-output tests were conducted using increasing stimulus intensity from 0-200 µA in 25 µA increments. Access and input resistances were continually monitored throughout the recording through a 50 ms, 5 mV voltage step 150 ms prior to synaptic stimulation, and data were discarded if series resistance changed >20%.
To assess HFS LTP in both NTN1 cKO and control littermates (Figure 2), as well as in the netrin-1 occlusion experiments (Figure 5), the recording mode of the cell was changed from voltage-clamp to current-clamp after a 5-10 min baseline period. Potentiation was induced by a single episode of a 1-s 100 Hz stimulation in current-clamp mode, which can elicit potentiation of evoked EPSCs at ∼150-200% of baseline values. Following stimulation, the cell was returned to voltage-clamp recording mode and held at −70 mV to record evoked responses.
For rectification experiments following bath application of exogenous netrin-1 (200 ng/ml), AMPAR-mediated currents were evoked in the presence of PTX (100 µM) and D-(-)-2-Amino-5-phosphonopentanoic acid (50 µM, D-APV) at a range of voltages (−110 mV to +50 mV in 20 mV intervals). APV was validated by measuring the lack of NMDAR-mediated currents at +40 mV. QX-314 (5 mM) was also included in the intracellular solution to prevent action potential generation. All responses were normalized to peak AMPAR-mediated current. Rectification index was computed as a ratio of current amplitude of responses evoked at −70 mV and +50 mV.
Miniature excitatory postsynaptic currents (mEPSCs) were recorded in voltage-clamp mode at a holding potential of −70 mV in the presence of PTX (100 µM) to block GABAA-mediated synaptic currents and tetrodotoxin (1 µM, TTX) to block sodium currents. Currents were analyzed using MiniAnalysis (Synaptosoft), and events were detected using a threshold of 7 pA (>3 pA root mean square of baseline noise levels). Cumulative distribution plots were generated using an equal number of events per condition (70 per condition), which were randomly selected from each cell, rank-ordered, and averaged across each condition.
An Axopatch 200B or 700B amplifier (Molecular Devices) were used for recordings, and signals were digitized (Digidata 1322A or 1550A, Molecular Devices). Voltage-clamp recordings were sampled at 10 kHz, filtered at 2 kHz, and acquired using pClamp software (v9.0 or v10.4, Molecular Devices).
Hippocampal culture electrophysiology
Whole cell patch clamp recordings were made from embryonic rat hippocampal neurons (14 DIV) prepared as described above. Neurons were plated on glass coverslips at high density, and individual coverslips transferred to an upright SliceScope 2000 (Scientifica), and perfused with ACSF containing (in mM): 135 NaCl, 3.5 KCl, 1.2 MgCl2, 2 CaCl2, 10 HEPES, and 20 D-Glucose (pH: 7.4, 300 mOsm). Current clamp recordings were performed using pipettes filled with (in mM): 120 K-gluconate, 20 KCl, 10 HEPES, 7 phosphocreatine di-Tris, 2 MgCl2, 0.2 ethylene-glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid (EGTA), 4 Na-ATP, 0.3 Na-GTP (pH: 7.2-7.26, 280-290 mOsm). Access resistance was monitored throughout the recording, and cells were held at resting membrane potential. Current-clamp recordings were sampled at 20 kHz and filtered at 10 kHz using pClamp (v10.4, Molecular Devices).
Pharmacology
Stocks were prepared at 1000x by dissolving tetrodotoxin citrate (T-550, Alomone labs), D-APV (0106, Tocris), BAPTA (ab120449, Abcam), IEM-1460 (ab141507, Abcam), Pep1-TGL (1601, Tocris), and clozapine-N-oxide (ab141704, Abcam) in water, and picrotoxin (P1675, Sigma-Al-drich) and KN62 (ab120421, Abcam) in DMSO. All other salts and drugs were from Sigma-Alrich unless otherwise noted. For certain experiments, pharmacological agents were included in the intracellular patch solution (Figures 6g-j and 7j-l). In these cases, cells were dialyzed with pipette solution for at least 15 min before beginning baseline recordings.
Statistical analysis
Statistical analyses on parametric data were assessed using repeated measures ANOVAs, pairwise comparisons (Tukey), within-samples paired t-tests, and independent t-tests where appropriate. Normality, homoscedasticity, and outlier tests were performed on all datasets. Non-Gaussian distributed data sets were tested by Wilcoxon test for paired observations or Friedman’s repeated measures ANOVA on ranks. For mEPSC cumulative distribution data, comparisons were performed using the Kolmogorov-Smirnov test. Significant interactions and main effects were assessed using pairwise comparisons and compensated using the Tukey method for parametric data. Statistical significance was determined with p ≤ 0.05 using two-tailed tests. All data are presented as mean ± SEM. Data were analyzed using Clampfit 10.3 (Axon Instruments), Matlab (Mathworks), Fiji 78, Photoshop (Adobe), MiniAnalysis (Synaptosoft), Prism 7 (Graphpad), and Sig-maplot 11 (Systat). Plotted data were then formatted in Adobe Illustrator CS6 (Adobe Systems).
Acknowledgments
The authors thank Wayne Sossin, and the members of the Kennedy, Ruthazer, Séguéla, and De Koninck labs for comments on drafts of the manuscript. We also thank Francine Nault, Charleen Salesse, Nathalie Marcal, Yi Jiang, and Hanan Elimam for technical assistance. S.D.G. was supported by postdoctoral fellowships from Fonds de la Recherche Québec – Santé (FRQS) and Canadian Institute for Health Research (CIHR). I.V.B. was supported by a graduate scholarship from Natural Sciences and Engineering Research Council (NSERC) of Canada. J.G. was supported by a postdoctoral fellowship from FRQS. The project was supported by grants from NSERC (P.D.K., P.S., E.S.R.), CIHR (R.A.M., P.D.K., P.S., E.S.R., T.E.K.) and the Alzheimer Society of Canada (T.E.K.). E.S.R. holds a FRQS Research Chair.
References
- 1.↵
- 2.↵
- 3.↵
- 4.
- 5.
- 6.↵
- 7.↵
- 8.
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵




