

ABSTRACT
Intestinal barrier immaturity, or “leaky gut,” is the proximate cause of susceptibility to necrotizing enterocolitis in preterm neonates. However, the impact of intestinal microbiota development on intestinal mucosal barrier maturation has not been evaluated in this population. In this study, we investigated a longitudinally sampled cohort of 38 preterm infants monitored for intestinal permeability (IP) and fecal microbiota during the first two weeks of life. Rapid decrease in IP indicating intestinal barrier function maturation correlated with significant increase in community diversity. In particular, members of the Clostridiales and Bifidobacterium were highly transcriptionally active, and progressively increasing abundance in Clostridiales was significantly associated with decreased gut permeability. Further, neonatal factors previously identified to promote intestinal barrier maturation, including early exclusive breastmilk feeding and low antibiotic exposure, favor the early colonization of the gut microbiota by members of the Clostridiales, which altogether are associated with improved intestinal barrier function in preterm infants.
Introduction
The intestinal mucosa paracellular trafficking of macromolecules is controlled by a competent epithelial barrier 1. The intestinal barrier constitutes a protective shield to the diffusion of pathogens and other elements with pro-inflammatory and tissue injury properties, and regulates absorption and secretion of essential nutrients 2. A functional intestinal barrier is driven by a complex structure that includes physical barrier with coinciding chemical, immunological and microbiological components 3. The colonization with microorganisms starts at birth and undergoes rapid shifts in composition and structure as the host matures over time 4. These microorganisms perform essential functions mechanistically linked to intestinal barrier competency, including epithelial metabolism, proliferation and survival, mucin and antimicrobial compound production, and cell-cell communication signaling molecule secretion 3. The microbial community in general is considered to play critical roles in the early development of the intestinal epithelium, the immune system, nutrient acquisition and energy regulation, and opportunistic pathogens suppression 3,5.
Disrupting intestinal microbiota, on the other hand, leads to dysbiosis, a state of ecological imbalance where the community loses diversity, key bacterial species, and more critically metabolic capacity with reduced colonization resistance to opportunistic pathogens 6. Early life gut dysbiosis is associated with disease susceptibility along with short-term and lifelong health issues, such as necrotizing enterocolitis (NEC) 7, sepsis 7, asthma and allergies 8, type 1 diabetes 9, celiac disease 10, inflammatory bowel disease 11 and obesity 12, among others. NEC is a life-threatening, gastrointestinal emergency affecting approximately 7-10% of preterm neonates with mortality as high as 30-50% 13. In this condition, bacteria across the intestinal wall leading to local and systemic infection and inflammation, and bowel wall necrosis and perforation. Intestinal barrier immaturity, characterized as elevated intestinal permeability (IP), or “leaky gut”, is the proximate cause of susceptibility to NEC in preterm neonates 14,15. It is critical to characterize the preterm infant intestinal microbiota to identify dysbiotic states associated with increased intestinal leakiness, as well as beneficial bacteria associated with improved intestinal barrier function, for subsequent stratification of early diagnosis, early intervention and primary prevention of leaky gut and its sequelae.
Despite the critical role of the microbial community in intestinal barrier function, its effect on newborn IP is unknown. In particular, the microbiota of preterm neonates with measured elevated IP, a high-risk population for NEC, has not been studied previously. We hypothesize that the intestinal microbiota plays a pivotal role in modulating IP and that the presence of “beneficial” bacteria will be associated with improved intestinal barrier function in preterm infants. In this study, we studied a cohort of 38 preterm infants born prior to 33 weeks of gestation. IP was measured by urinary detection of orally administered sugar probes lactulose and rhamnose using high pressure liquid chromatography 16 with coinciding measures of the composition and function of the fecal microbial communities were investigated. We sampled three time points, study day 1, 8, and 15, during the first two weeks of life, which is a critical period corresponding to the initiation of the intestinal microbiota development process 16-18. A rapid decrease in IP was observed to correlate with increased fecal microbiota biodiversity, indicating intestinal barrier function maturation over the first two weeks of life with a shift in the composition and structure in intestinal microbial community. We subsequently revealed an association between decreased IP and the abundance of Clostridiales, which was highly transcriptionally active along with members of the Bifidobacterium. Our study highlights the multifactorial processes involved in intestinal barrier maturation, and the importance to consider microbiological and neonatal factors for diagnosing, monitoring, and modulating IP in preterm infants.
Results
Intestinal barrier maturation correlates with increased microbiota biodiversity over the first two weeks of life
The demographic, obstetric, and neonatal characteristics for all thirty-eight preterm infants enrolled in the study are summarized in Table 1. As previously reported 16, 20 infants (57%) experienced a rapid decrease in intestinal permeability (IP), 5 infants (14%) had a decreased IP during the first week and subsequent substantial increase and 10 infants (29%) showed a delayed IP decrease maintaining high IP throughout the study period. At each time point, infants were assigned to either high or low IP (Supplemental File 3). The microbiota of 64 fecal samples were successfully characterized by high-throughput sequencing of the V3-V4 variable regions of 16S rRNA genes. A total of 422,444 high-quality amplicon sequences was obtained, corresponding to 10,544 (±4,029) sequences per sample with an average length of 428 bp. The top 25 most abundant phylotypes are shown in Supplemental Figure 1A. Taxonomic profiles of all samples were clustered into three distinct groups according to similarities in community composition and structure. Klebsiella spp., Staphylococcus epidermidis, and E. coli dominated cluster I, II, and III, respectively. Both La/Rh ratio and taxonomic profile of each sample are shown in Supplemental File 3. Taxonomic profiling of corresponding metagenomes further resolved Klebsiella spp. to Klebsiella pneumoniae. Not surprisingly, older term infants at 6-24 months old, or phase II/III as defined previously 17-19, clustered together in a different and more diverse cluster (Supplemental Figure 1B). Rapid decrease in IP over the two-week observation period indicates intestinal barrier function maturation (p-value = 0.002), which is correlated with a significant increase in community diversity (p-value = 0.02) (Figure 1A); while delayed increase in community diversity was associated with maintenance of high intestinal permeability (p-value < 0.001) (Figure 1B). The results indicated that preterm infant intestinal barrier maturation correlates with increased fecal microbiota biodiversity and a change in microbiota structure.
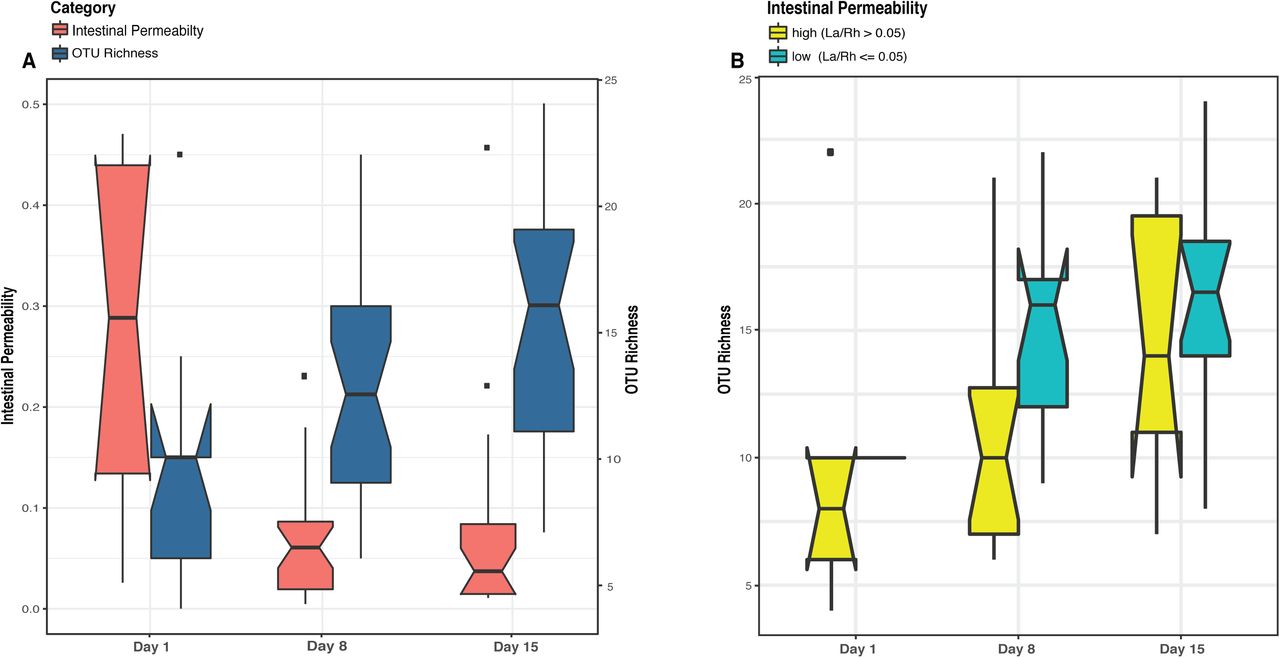
Boxplots comparing levels of intestinal permeability and microbial community diversity at study days 1, 8, and 15 in a cohort of 43 preterm infants (<33 weeks gestational age). Intestinal permeability is measured by non-metabolized sugar probes lactulose (La) (marker of intestinal paracellular transport)/rhamnose (Rh) (marker of intestinal transcellular transport). Microbial community diversity was calculated by OTU (Operational Taxonomic Units) richness. Wilcoxon rank sum test and a false discovery rate of 5% was used in significance test. Median values and interquartile of the values were shown in box. (A) Intestinal permeability (p-value = 0.002) and community diversity at the three study time points (p-value = 0.02). (B) Community diversity (p-value < 0.001) in infants with low and high intestinal permeability defined by a La/Rh >0.05 or <=0.05 respectively (1).
Characteristics of Study Subjects (preterm infants <33 weeks gestational age) (n=38).
Subject variation, PMA and IP explain most of the variation in microbial community composition
We employed multivariate response linear regression on the “balance” of microbial community and evaluated the effect of covariates of demographic, obstetric, and neonatal factors on the microbiome using Gneiss 20. Covariates of antibiotics use, maternal antibiotics use, delivery mode, PPROM, feeding pattern, IP, birthweight, gender, ethnicity, gestational age (GA) and postmenstrual age (PMA) were included in the analysis. Subject, PMA, and IP had the greatest correlation with intestinal microbiota, together they explained 63.4% of the variation of the intestinal microbial community composition observed in the cohort (Supplemental File 4). The plots (Supplemental Figure 2) show the predicted points lie within the same region as the original communities and the residuals have roughly the same variance as the predictions within ±2. Overall our result indicates the microbial differences between subjects are large (R squared difference is 0.23±0.10), and the covariate with strongest effect is PMA (R squared difference is 0.44). IP correlates with the intestinal microbiota (R squared difference is 0.20), and its effect is lower than PMA and similar to the average among-subject difference.
Clostridiales is associated with low intestinal permeability in preterm infants
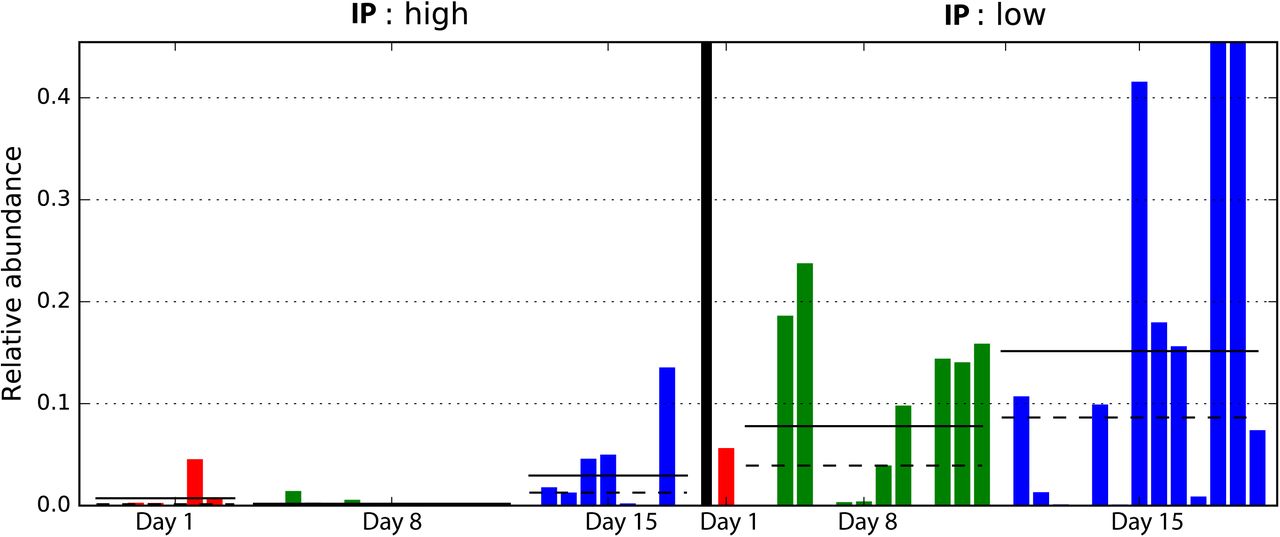
Comparative analysis of fecal microbiota with high and low IP showed that Clostridia, the class containing the order Clostridiales in this cohort, was significantly more abundant in samples with low IP compared to those with high IP (p-value = 0.01) (Figure 2, Supplemental Figure 3). In particular, a progressive and significant increase in members of Clostridiales over the first two week after birth significantly associated with low IP (p-value = 0.0002) (Figure 3). Based on Bayesian nonparametric adaptive smoothing models and subject-specific changes in relative abundance of Clostridiales at each of study day 1, 8, and 15, the results demonstrated: (1) at baseline study day 1, the abundance of Clostridiales was low in subjects with either high or low IP; (2) However, in samples measures with low IP but not high IP, a significant increase in Clostridiales was observed that reached ~8% median and >20% maximal relative abundance from study day 1 to day 8, and ~16% median and ~45% maximal relative abundance again from study day 8 to 15; (3) on the other hand, in samples measured in high IP at study day 1 that are also high at the follow up days, members of Clostridiales was almost completely absent on study day 8 and no increase was observed from study day 1 to 8, and the increase from study 8 to 15 was small at ~3% median and ~10% maximal relative abundance; (4) in infants 6-24 months old, Clostridiales is the most abundant taxonomic groups with >50% median and >85% maximal relative abundance (Supplemental Figure 4). Together, our results suggest preterm infants at birth have low abundance of Clostridiales, which became progressively and significantly more abundant only in the group with rapid progression of intestinal barrier maturation, while remained low in those with persistent high IP over the first two weeks of life.

Cumulative relative abundance of bacterial groups in high and low IP infants. (A) cumulative abundance between phase II/III subjects (6-24 months of age) and phase I infants (within first two weeks of life) with high and low IP; (B) Cumulative abundance at different study day at day 1, 8, and 15 for phase I infants with high and low IP. The most outstanding difference between high and low IP in preterm infants is in the Clostridiales (p-value = 0.01), which is the most abundant bacterial group in phase II/III infants.

Comparison of samples with different intestinal permeability on the relative abundance of members of Clostridiales. Bars represent the relative abundance of Clostridiales in each sample. Dotted line represents mean, solid line represents median relative abundance. The alpha value for the non-parametric factorial Kruskal-Wallis sum-rank test was 0.05 and the threshold for the logarithmic LDA model (3) score for discriminative features was set at 2.0. Low IP: La/Rh < 0.05; high IP: La/Rh >= 0.05.
We further calculated the predictive power of microbiota composition in classifying IP using random forest supervised machine learning scheme. The top 15 phylotypes with the highest mean decrease gini index importance measure (Supplemental Figure 5) were used to fit a random effect logistic regression model of IP, 4 of which resulted significantly associated with low IP (Supplemental File 5), including three members of the order Clostridiales, Coprococcus (p-value = 0.004), Lachnospiraceae (p-value = 0.007), Veillonella dispar (p-value = 0.01), and Bifidobacterium (p-value = 0.01) from the order of Bifidobacteriales. Interestingly, Bifidobacteriales was the second most abundant taxonomic groups in infants 6-24 months old, only lower than Clostridiales (Supplemental Figure 4).
Clostridiales and Bifidobacterium are highly active members of the intestinal microbiome
The level of bacterial transcriptional activities was characterized by studying the suite of genes present and expressed in preterm infant intestinal microbiota. A total of 869 million metagenomic sequence reads (average of ~31.0 million sequence reads per sample) and 694 million metatranscriptomic sequence reads (average of ~53.4 million sequence reads per sample) were obtained after quality assessment. Figure 4 shows that Bifidobacterium breve (Actinobacteria), Veillonella and Clostridiales Family XI incerteae Sedis (Clostridiales) are the most transcriptionally active bacteria with high ratio of transcript abundances over gene abundances in all samples. Further, the levels of transcriptional activities of Bifidobacterium breve and Clostridiales Family XI incerteae Sedis are correlated with a spearman correlation of 0.89, suggesting these two taxonomic groups are either functionally dependent or co-regulated (Supplemental Figure 6). We observed increased abundance of both Clostridiales and Bifidobacteriales through the transition from the first two weeks (phase I) to later age of 6-24 months (phase II/III) as further supporting their active contribution to the function of the GI microbiota after birth. Interestingly, Clostridiales and Bifidobacteriales are also the most abundant taxonomic groups in the intestinal microbiota of 6-24 months old infants (Supplemental Figure 4, Supplemental File 3). Specifically, members of the family Clostridiales have an average abundance of 50±3% in phase II and III infants, compared to 0.1±0.4% in phase I infants. Bifidobacteriales have an abundance of 26±5% in phase II and III as opposed to 0.1±0.3% in phase I infants. Together with the previous observation that Coprococcus (Clostridiales), Lachnospiraceae (Clostridiales), Veillonella dispar (Clostridiales), and Bifidobacterium (Bifidobacteriales) are significantly associated with low IP, our results suggest the presence and more importantly the activity of bacterial members of Clostridiales and Bifidobacteriales are associated with improved intestinal barrier function.

Bacterial species transcriptional activity in preterm infant stools. Fecal samples are represented in columns and taxonomic composition quantified using MetaPhlAn (55) version 2 are shown in rows, both are organized by hierarchical clustering. Normalization using Witten-Bell smoothing was performed, and the relative expression of a gene in a sample was calculated by normalizing the smoothed value of the expression level in the metatranscriptome by the smoothed value of the corresponding gene abundance in the metagenome (56, 57). Color scheme indicates an approximate measure of the species’ clade-specific transcriptional activity (56). The colored branches show the clustering of bacterial species that are consistently transcriptionally active (yellow) or consistently transcriptionally inactive (blue) across samples.
Conversely, the two Enterobacteriaceae species, Klebsiella pneumoniae and Escherichia coli, had low transcriptional activity despite their high relative abundance in the infant GI microbiota, questioning their functional contribution to the infant stool microbiota. Interestingly, Enterobacteriaceae and Staphylococcus are the most abundant bacterial taxa present in phase I infants but are rarely observed in phase II and III, suggesting their presence in the infant stools is temporary and might not contribute greatly to the functions provided by the GI microbiota.
Early breast milk feeding and low antibiotic exposure positively correlates with Clostridiales abundance and activities
The associations between intestinal microbiota and demographic, obstetric, and neonatal factors were also evaluated. Gneiss analysis suggests delivery mode, PPROM, gender, ethnicity, birthweight, maternal antibiotics use are not contributing covariates to the intestinal microbial community variance. Further, no bacterial phylotype was identified to significantly associate with these factors. However, breast milk feeding pattern and antibiotic exposure were significantly associated with increased abundance of Clostridiales. More specifically, early full exclusive breast milk feeding by study day 10 (p-value = 0.0001) (Supplemental Figure 7) and antibiotic exposure limited to no more than 4 days (p-value = 0.05), were associated with the family Lachnospiraceae in the Clostridiales (p-value = 0.004) (Supplemental Figure 8). On the other hand, Enterobacteriaceae, particularly Klebsiella pneumoniae (as identified by metagenomics sequencing), was significantly associated with full breast milk feeding achieved after study day 10 (p-value = 0.01) (Supplemental Figure 7). These results strongly suggest members of the Clostridiales are significantly associated with low intestinal permeability, early full breast milk feeding, as well as shorter duration of antibiotic use.
We further characterized the genotypic variation of E. coli through reconstructing MLST loci-sequences from metagenomes 21, and compared them to 25 recently characterized E. coli MLST genotypes associated with NEC 22,23. Five E. coli genotypes were only observed in samples with high IP, two of which, sequence type 73 and 131, were previously identified as uropathogenic E. coli (UPEC) strains associated with NEC and infant mortality 23 (Supplemental File 6). One E. coli genotype 697 which was not recognized as a UPEC E. coli strain nor was observed in NEC 23, was observed in both high and low IP samples. Two new MLST genotypes of E. coli were also observed. A minimum spanning tree on the sequence types is shown in Supplemental Figure 9 and was used to demonstrate the relationship among genotypes of E. coli.
Clostridiales are highly prevalent in the GI microbiota of preterm infants
The most abundant bacterial species included K. pneumoniae, Staphylococcus epidermidis, E. coli, and Enteroccocus faecalis were found with mean abundance of ~10-35% (S.D. ~15%-30%) and ~85-95% prevalence in these samples. In comparison, many species such as Streptococcus agalactiae, B. breve, B. longum, Clostridium perfringens, Propionibacterium acnes, Bacteroides fragilis, Veillonella parvula, and Streptococcus thermophiles were present in 15-70% of all samples and had a much lower level of abundance ranging from ~0.0001% to 1% (S.D. ~0.0001%-6%). Many of the members of Clostridiales were not resolved at the species or genus-level, while those taxonomically identified Clostridiales included Coprococcus, Blautia, SMB53, Ruminococcus gnavus, Clostridium spp., Faecalibacterium prausnitzii, Dorea, Ruminococcus bromii, Roseburia, Pseudoramibacter and Butyricicoccus pullicaecorum were detected in low or extremely low abundance yet high prevalence (Supplemental File 3). A previous study revealed that stool bacterial load varies greatly in the first few days of life but then reached and persisted in most infants in the range of 109 to 1010 bacteria per gram of stool after one week of life 24. Given our average sequencing depth is ~104-105 it is likely that some bacterial taxonomic groups with low relative abundance (<0.001%) are below our detection limit, and their prevalence is underestimated. It is expected that the prevalence of the members of Clostridiales can be higher than the currently observed 15-70% among samples in the GI microbiota of preterm infants. The marked discrepancy between bacterial abundance and prevalence suggests that bacterial species present in the intestinal microbiome of preterm infants can selectively colonize and grow under nutritional or antibiotic pressures.
Discussion
Preterm infants are at elevated risk for leaky gut, feeding intolerance, NEC and sepsis, and other short-term and long-terms morbidities 19. The pathophysiology of these disorders is likely multifactorial, involving a combination of intestinal mucosa barrier immaturity, imbalance in microvascular tone, aberrant microbial colonization and altered immune responses 19,25,26. Previously, our group and others demonstrated that neonatal factors such as gestational age, antibiotic exposure, and exclusive breastmilk feeding affect intestinal mucosa barrier permeability in preterm infants 16,27. With the rapid development of high-throughput sequencing technology, recent studies have evaluated the significant association between the composition of intestinal microbiota, neonatal intestinal health and development 3,5,24. However, the relationships between intestinal microbiota and IP have not yet been evaluated in a high-risk preterm population. In this study, we investigated the early development of the intestinal microbiota and its association with IP in a cohort of 38 preterm infants sampled during the first two weeks of life. We observed that neonatal factors known to be associated with low IP, including early exclusive breast milk feeding and low antibiotic exposure, favored the early colonization of the gut microbiota by members of Clostridiales. The associations between neonatal factors, intestinal microbiota and intestinal barrier function further substantiate the multifactorial processes involved in gut barrier maturation, thus highlighting the impact of neonatal care practices and the potential for therapies such as rationally designed live biotherapeutics strategies to rapidly lower IP after birth in preterm infants.
A critical value of understanding the driver of IP, including associated microbiological biomarkers, is in its clinical significance in NEC risk diagnostics and disease prevention. The etiology of NEC involves the interaction between immature intestinal barrier and the developing intestinal microbial community that leads to an excessive inflammatory response 25,26,28,29. Though IP is high at birth in preterm infants, it rapidly decreases over the first few days, which is associated with diminished risks for adverse outcomes 16,30. Aberrant intestinal barrier function manifests by persistently high and/or late decrease in IP and is likely due to the physiological immaturity of the GI tract barrier function and altered levels of the normal microbial communities 14,15, resulting in microbial invasion of the intestinal wall and gut lamina propria triggering a cascading inflammatory response and ultimately intestinal necrosis and severe infection 2. Multiple studies have revealed microbial community dysbiosis is involved in stimulating a hyperinflammatory response that leads to NEC 25,26,28,29. This community dysbiosis has been characterized by the presence of members of the family Enterobacteriaceae such as E. coli, K. pneumonia, as well as Enterobacter cloacae 23,26,31. However, a generalized bacterial dysbiosis alone does not adequately explain NEC. Many preterm infants that are colonized by high abundance of Enterobacteriaceae do not develop NEC 32, and many NEC cases lacked intestinal colonization of Enterobacteriaceae 33. In this study, Enterobacteriaceae was significantly associated with both elevated IP and later attainment of full exclusive breastmilk feeding (>10 days), while other beneficial bacteria such as members of Clostridiales and Bifidobacterium were significantly associated with improved IP and earlier breastmilk feeding attainment. These results emphasize the importance of a holistic understanding of the etiology of NEC, including the mechanistic characterization of the functional synergy and/or competition among different bacterial groups, as well as nutritional factors, drug uses and host genetics. Further, the links established by previous microbiota association studies could not elucidate the causalities between gut microbiota and NEC development. Our study prospectively associates maturation of gut barrier function with specific microbial community composition and structure for the first time, prior to the onset of NEC. Research on neonatal IP will not only further our understanding of NEC etiology but will help identify the “window of opportunity” for intervention prior to the onset of NEC. Early prediction and prevention of NEC will ultimately improve overall infant survival rates.
Multiple intrinsic and extrinsic factors affect newborn intestinal microbiota, such as maternal diet, delivery mode, breast milk feeding, antibiotic exposure, and other early life environmental exposures 7,34. In this study, early exclusive breast milk feeding and low antibiotic exposure was associated with the presence of members of Clostridiales in the stool microbiota of preterm infants. We have previously observed these two factors are associated with improved IP in preterm infants 16, which has been shown to be critically protective against NEC 35. This observation emphasizes the importance of factors such as clinical administration of nutritional supplement and limiting exposure to antibiotic in neonatal care units. Interestingly, Clostridiales strains were recently shown to be sensitive to many antibiotics, including ampicillin and amoxicillin 36, both commonly used for the neonate clinical management. Further understanding of the selective nutritional requirement that favor the growth of these bacteria would afford the development of novel nutritional supplemental strategies to limit the incidence of NEC and improve clinical outcomes in preterm infants.
Current therapies for NEC are mostly ineffective, and involve antibiotic treatment and surgical interventions, including drain placement or bowel resection. These procedures are associated with poor prognosis and a mortality rate of ~50% due to the rapid progression of the disease 37. Live biotherapeutics products (LBP) are being considered, but selecting the appropriate one remains a major challenge. LBP therapies are promising, low-cost, and constitute a likely safe preventive measure to improve intestinal barrier maturation and reduce NEC incidence in at-risk preterm infants 38. In an experimental mouse model of NEC, the administration of Bifidobacterium infantis prevented an increase in IP, stabilized tight junction proteins, and reduced NEC incidence 28. Translating these findings in human has been challenging. There have been at least 11 randomized controlled trials and a recent meta-analysis of LBP supplementation to prevent NEC in preterm neonates 39. Although there was a 30% reduction in NEC incidence in these trials, various formulations, doses, and duration of therapy were used, infants <1000 g BW with the highest NEC incidence were under-represented, and no Food and Drug Administration-approved products are available to assure quality and safety under good manufacturing practices.
Clostridiales offer a new opportunity to develop a LBP for the prevention of NEC, in combination with strains of Lactobacillus and Bifidobacterium already available. Members of the family Clostridiales often have anti-inflammatory properties associated with their fermentative metabolism of carbohydrates and amino acids 40. Because of the difficulties to culture Clostridiales, it has been largely overlooked. A few species belonging to this family are known for their pathogenicity and include C. botulinum, C. perfringens, C. tetani, and C. difficile 41, however these are opportunistic pathogens and not commensal of the intestinal microbiota. The application of culture-independent high-throughput sequencing identified many formerly unculturable Clostridiales species, and the group is now thought to be one of the predominant groups of microbes inhabiting the GI tract, comprising ~30-40% abundance of the adult intestinal microbiota 42. These species form the basis of the microbiome therapeutics product, SER109, for the treatment of C. difficile infection in adults 43. Clostridiales are heterogeneous in terms of their enzymatic, and metabolic properties, and produce beneficial short-chain fatty acid (SCFA) such as acetate, propionate, and butyrate 44. Further, Clostridiales have been shown to stimulate the production of intestinal epithelial cytokines that have been associated with the improvement of intestinal dysbiosis, and marked reduction in inflammation 36,45,46. The recent characterization of 46 strains of newly isolated Clostridiales revealed their ability to induce regulatory T cells and a protection against colitis and allergic responses 45. Seventeen strains of human-derived Clostridiales species were rationally selected using gnotobiotic mice and the cocktail shown to have prophylactic effect in mouse colitis 36,46. In addition, the administration of Clostridiales protects the host from pathogen infection and abrogated intestinal pathology 47. In term infants, the presence of Clostridiales in the intestinal microbiota was demonstrated to prevent colonization by bacterial pathogens such as S. Typhimurium 48. Unfortunately, the current standard application of 16S rRNA V4 or V3-V4 amplicon sequencing is not capable to resolve the species of Clostridiales present in a sample 49. Future taxonomic and functional characterization of Clostridiales species will greatly improve our capability to develop novel diagnostic and treatment strategies, and potentially prevent microbial community-mediated intestinal dysbiosis in preterm infants to optimize intestinal maturation and limit the burden of prematurity 23.
Methods and Materials
Participants and intestinal permeability measurement
The institutional review boards of the University of Maryland and Mercy Medical Center approved the study protocol and informed consent was obtained from parents for participation of their infants in the study. All methods were performed in accordance with the relevant guidelines and regulations. Thirty-eight preterm infants 240/7-326/7 weeks GA were enrolled within 4 days after birth and received 1 ml/kg of the non-metabolized sugar probes lactulose (La) (marker of intestinal paracellular transport)/rhamnose (Rh) (marker of intestinal transcellular transport) (8.6 g La +140 mg Rh/100 mL) enterally on study days 1, 8 ± 2 and 15 ± 2. La/Rh was measured by high-pressure liquid chromatography (HPLC) in urine collected over a 4h period following administration of the sugar probes as previously described 16. High or low intestinal permeability was defined by a La/Rh >0.05 or <=0.05 respectively, as validated and applied previously 16. PMA was calculated as gestational age at birth plus week of life as defined previously 50. Fecal samples (~1g) were collected at the same time, stored immediately in 2 ml of RNAlater (QIAGEN). Urine and fecal samples were archived at −80°C until processed. A standard feeding protocol was used for all preterm participants. To compare microbiota of infants at different growth phases 17,19, 16 samples from older term infants at phase II/III (6-24 months old) from a previous study 51 were included in the comparative analyses.
Stool nucleic acid extraction and sequencing
DNA was extracted from all samples as previously reported 52. Briefly, a 500 μl aliquot of fecal material mixture was homogenized and carefully washed twice in PBS buffer. Enzymatic lysis using mutanolysin, lysostaphin and lysozyme was performed, followed by proteinase K, SDS treatment and bead beating. DNA purification from lysates was done on a QIAsymphony automated platform. PCR amplification of the V3-V4 variable region of 16S rRNA gene was performed using dual-barcoded universal primers 319F and 806R as previously described 53. High-throughput sequencing of the amplicons was performed on an Illumina MiSeq platform using the 300 bp paired-end protocol. Metagenomic sequencing libraries were constructed from the same DNA using Illumina Nextera XT kit according to the manufacturer recommendations.
Total RNA was extracted from 250 μl of stool homogenized in RNALater. Briefly, lysis was performed by bead beating using the FastPrep lysing matrix B protocol (MP Biomedicals), followed with two rounds of protein cleanup using phenol-chloroform in 5PRIME heavy phase lock tubes (QuantaBio) and precipitation of total nucleic acids using isopropanol. Genomic DNA was removed using TURBO DNase (Ambion). Ribosomal RNAs were depleted using the Gram-negative and Human/mouse/rat Ribo-Zero rRNA Removal kits (Epicentre Technologies). The resulting RNA was used for library construction using Illumina TruSeq stranded mRNA library preparation kit according to the manufacturer’s recommendations. Quantification of the constructed RNA libraries was performed on an Agilent Bioanalyzer using the DNA 1000 Nano kit. Both metagenome and metatranscriptome samples were sequenced on an Illumina HiSeq 4000 instrument at the Genomics Resource Center (GRC), Institute for Genome Sciences, University of Maryland School of Medicine using the 150 bp paired-end protocol.
Bioinformatics analysis of intestinal microbiota
Sequencing read quality assessment was performed using strict criteria to ensure high quality and complete sequences of the amplified the V3-V4 regions of the 16S rRNA gene, according to the procedures, programs and citations, and parameters described previously 53. Briefly, a sequence read was trimmed at the beginning of a 4 bp sliding window if the average quality score was less than Q15. The sequence read was then assessed for length and retained if it was at least 75% of its original length. The paired-end reads were assembled to take advantage of the ~90bp overlapping region. These sequences were further de-multiplexed the sequence reads by individual samples. Additional quality filtering was applied that removed sequences with more than one mismatch in the barcode sequence tag or with ambiguous nucleotide. Taxonomic assignments were performed on each sequence using the Ribosomal Database Project trained on the Greengene database (Aug 2013 version), using 0.8 confidence values as cutoff. Clustering taxonomic profiles was performed as previously described 52. The number of clusters was validated using gap statistics implemented in the cluster package in R 54 by calculating the goodness of clustering measure. Within-sample diversity was estimated using both observed OTUs to measure community richness and Shannon diversity index. Linear discriminant analysis (LDA) effect size (LEfSe) analysis55 was used to identify fecal phylotypes that could explain the differences between infants with low or high La/Rh ratio on different sampling days. For LEfSe, the alpha value for the non-parametric factorial Kruskal-Wallis (KW) sum-rank test was set at 0.05 and the threshold for the logarithmic LDA model 56 score for discriminative features was set at 2.0. An all-against-all BLAST search in multi-class analysis was performed.
Balance tree analysis was applied as implemented in Gneiss, and trees were generated using Ward hierarchical clustering of abundance profiles. Balance was computed as the isometric log ratio of mean abundances at each bifurcating node in the tree, to characterize the “flow” of changes in the abundance of a group of correlated bacteria in a microbial community 20. Multivariate response linear regression on the calculated balances was performed, and multiple factors were included as covariates, including antibiotics use, maternal antibiotics use, delivery mode, preterm premature rupture of membranes (PPROM), feeding pattern and source, intestinal permeability, birthweight, gender, ethnicity, GA and PMA. Leave-one-variable-out approach was used to calculate the change in R square to evaluate the effect of a single covariate on the community. Ten-fold cross validation was performed to mitigate the common overfitting issues in statistical modelling.
Statistical Analysis
An adaptive spline logistic regression model implemented in spmrf R package 57 was adapted to determine the associations between intestinal permeability and relative abundance of bacterial phylotypes. This model is a locally adaptive nonparametric fitting method that operates within a Bayesian framework, which uses shrinkage prior Markov random fields to induce sparsity and provides a combination of local adaptation and global control 57. The analysis was performed on the phylotypes present in at least 15% of all samples, and the effect size was defined as the difference between the extreme values of the probability of intestinal permeability index. Given that there were multiple samples collected from each subject, this model takes into consideration of the dependencies among samples within a subject. Bayesian goodness-of-fit p-value implemented in R package rstan 58 was used to access the significance of the association between phylotypes and metadata including antibiotics use, maternal antibiotics use, delivery mode, PPROM, feeding pattern, intestinal permeability, birthweight, gender, ethnicity, gestational age (GA), and postmenstrual age (PMA). R code implementation of the model is provided in Supplemental File 1. We further adapted random forest supervised machine learning scheme implemented in R package randomForest 59 to test the predictability of the phylotypes of microbial community on intestinal permeability. The top 15 phylotypes relative abundance with highest mean decrease gini index importance measure, were fitted to a random effect logistic regression model of intestinal permeability that was defined as a dichotomous variable high (La/Rh >0.05) or low (La/Rh <=0.05). The relative abundances of phylotypes were centered to the mean and scaled by standard deviation to apply to the model to normalize relative abundances. R code implementation of the model is provided in Supplemental File 2.
Intestinal microbiome analyses
Metagenomic and metatranscriptomic sequence data were pre-processed using the following steps: 1) human sequence reads and rRNA LSU/SSU reads were removed using BMTagger v3.101 60 using a standard human genome reference (GRCh37.p5) 61; 2) rRNA sequence reads were removed in silico by aligning all reads using Bowtie v1 62 to the SILVA PARC ribosomal-subunit sequence database 63. Sequence read pairs were removed even if only one of the reads matched to the human genome reference or to rRNA; 3) the Illumina adapter was trimmed using Trimmomatic 64; 4) sequence reads with average quality greater than Q15 over a sliding window of 4 bp were trimmed before the window, assessed for length and removed if less than 75% of the original length; and 5) no ambiguous base pairs were allowed. The taxonomic composition of the microbiomes was established using MetaPhlAn version 2 65. Normalization using Witten-Bell smoothing was performed since metatranscriptomes are a random sampling of all expressed genes and transcripts can be identified that correspond to genes not represented in the metagenome, particularly for low abundance species that were metabolically active 66. The relative expression of a gene in a sample was calculated by normalizing the smoothed value of the expression level in the metatranscriptome by the smoothed value of the corresponding gene abundance in the metagenome, as suggested previously 66,67. Correlation plots were generated using R corrplot package68. Genotypic variation of Escherichia coli was performed through reconstructing MLST loci-sequences from metagenomes using metaMLST program 21. The resulting STs were visualized to show related genotypes of E. coli strains on a minimum spanning tree computed by a goeBURST algorithm 69 implemented in PHYLOViZ 70.
Conclusion
At birth there is low abundance of Clostridiales in preterm infants with progressive, significant increase in abundance in the group with rapid progression toward intestinal barrier maturation, but remained low in those with persistent high IP over the first two weeks of life. We further identified neonatal factors previously identified to promote intestinal barrier maturation, including early exclusive breastmilk feeding and shorter duration antibiotic exposure, favor the early colonization of the gut microbiota by members of the Clostridiales, which altogether are associated with improved intestinal barrier function in preterm infants. This highlights the importance of factors such as clinical administration of nutritional supplement and limiting exposure to antibiotic in the high-risk preterm population. Our study suggests rationally selected and formulated Clostridiales species could constitute a promising LBP candidate for the prevention of NEC, especially when combined with already available strains of Bifidobacterium and Lactobacillus. The rationale for this intervention is supported by our correlative finding between increased Clostridiales abundance and intestinal barrier maturation of preterm neonates at-risk for NEC development. Identification of specific strains of Clostridiales, their functions in mediating intestinal barrier maturation, LBP formulation and manufacturing, dosing, safety and efficacy evaluation will be needed to support their application as oral supplementation to promote intestinal barrier maturation and overall health of preterm neonates. Early prediction and prevention of NEC will ultimately improve overall infant survival rates.
Data Availability
All 16S rRNA sequence data were deposited in SRA SUB3616368 under BioProject PRJNA432222 (release upon acceptance).
Contributions
B.M., A.W., A.F., J.R., and R.V. designed the research. B.M., E.M., H.Y., and M.H. performed the research. B.M. and P.G. analyzed the data. B.M., E.M., J.R., and R.V. wrote the paper.
Competing interest statement
The authors declare no competing financial and non-financial interests.
Acknowledgements
This study was funded by The Gerber Foundation and NCCIH (National Center for Complementary and Integrative Health, AT006945).
This work is dedicated to the memory of our colleague Bushra Saleem, M.B.B.S., who contributed to the design and conduct of the study.
The authors thank Dr. Emmanuel Mongodin and Dr. Lauren Hittle, PhD at the Institute for Genome Sciences - University of Maryland School of Medicine for their helpful assistance in total RNA extraction.





{kind=link}
{kind=link}
{kind=link}
{kind=link}