

ABSTRACT
Multi-subunit tethering complexes control membrane fusion events in eukaryotic cells. CORVET and HOPS are two such multi-subunit tethering complexes, both containing the Sec1/Munc18 protein subunit VPS33A. Metazoans additionally possess VPS33B, which has considerable sequence similarity to VPS33A but does not integrate into CORVET or HOPS complexes and instead stably interacts with VIPAR. It has been recently suggested that VPS33B and VIPAR comprise two subunits of a novel multi-subunit tethering complex (named ‘CHEVI’), analogous in configuration to CORVET and HOPS. We utilised the BioID proximity biotinylation assay to compare and contrast the interactomes of VPS33A and VPS33B. Overall, few proteins were identified as associating with both VPS33A and VPS33B, suggesting these proteins have distinct sub-cellular localisations. Consistent with previous reports, we observed that VPS33A was co-localised with many components of class III phosphatidylinositol 3-kinase (PI3KC3) complexes: PIK3C3, PIK3R4, NRBF2, UVRAG and RUBICON. Although in this assay VPS33A clearly co-localised with several subunits of CORVET and HOPS, no proteins with the canonical CORVET/HOPS domain architecture were found to co-localise with VPS33B. Instead, we identified two novel VPS33B-interacting proteins, VPS53 and CCDC22. CCDC22 co-immunoprecipitated with VPS33B and VIPAR in over-expression conditions and interacts directly with the VPS33B-VIPAR complex in vitro. However, CCDC22 does not appear to co-fractionate with VPS33B and VIPAR in gel filtration of human cell lysates. We also observed that the protein complex in HEK293T cells which contained VPS33B and VIPAR was considerably smaller than CORVET/HOPS, suggesting that, unlike VPS33A, VPS33B does not assemble into a large stable multi-subunit tethering complex.
- Abbreviations
- ARC
- Arthrogryposis, Renal dysfunction, and Cholestasis syndrome
- ARKID
- Autosomal recessive Keratoderma-Ichthyosis-Deafness syndrome
- CHEVI
- class C Homologs in Endosome-Vesicle Interaction
- CORVET
- class C core vacuole/endosome tethering
- EARP
- endosome-associated recycling protein
- GARP
- Golgi associated retrograde protein
- EGFP
- GFP (enhanced) green fluorescent protein
- HEK293T
- human embryonic kidney 293T
- HOPS
- homotypic fusion and vacuole protein sorting
- MS
- mass spectrometry
- PI3KC3
- class III phosphatidylinositol 3-kinase
- XLID
- X-linked intellectual disability
INTRODUCTION
Membrane trafficking in eukaryotic cells is tightly controlled by a range of proteins, including markers of membrane identity (such as small GTPases) and multi-subunit tethering complexes. Tethering complexes, containing Sec1/Munc18 protein subunits, are soluble cytoplasmic proteins that work together with SNAREs to fuse membranes (1). One such Sec1/Munc18 protein is Vps33, found in all eukaryotes as a subunit of class C core vacuole/endosome tethering (CORVET) and homotypic fusion and vacuole protein sorting (HOPS) complexes (2). The mammalian HOPS and CORVET complexes share four common components, VPS33A, VPS16, VPS18 and VPS11, and have two distinct components, VPS8 and TRAP-1 (CORVET), or VPS39 and VPS41 (HOPS) (3–5). CORVET is active on Rab5-positive membranes (early endosomes) (3,6) and HOPS on Rab7-positive membranes (late endosomes, autophagosomes, lysosomes) (7–9).
In metazoans there are two Vps33 homologs (10), with the functions of yeast Vps33 being carried out by VPS33A. Metazoan VPS33B shares 30% amino acid sequence identity with VPS33A, but cannot integrate into CORVET or HOPS complexes (4,8,11). Instead, VPS33B has been observed on Rab10- and Rab25-positive membranes, functioning in post-Golgi trafficking (12), and on Rab11A-positive membranes acting in an apical recycling pathway (13).
Mutations in human VPS33A and VPS33B produce distinct phenotypes. Recently, a small population of patients have been identified with a single mutation in VPS33A (R498W) and a mucopolysaccharidosis-like phenotype (14,15). Although the mechanism is as yet unclear, this mutation results in over-acidification of lysosomes and an inability to catabolize glycosaminoglycans. This phenotype is quite different to that of patients with VPS33B mutations. VPS33B forms a complex with VIPAR (also known as SPE-39, VIPAS39 or VPS16B), and mutations in either of these proteins can cause Arthrogryposis, Renal dysfunction, and Cholestasis (ARC) syndrome (13). ARC syndrome is a multi-system disorder, with some symptoms attributable to the known functions of VPS33B and VIPAR in apical recycling pathways (13), post-Golgi collagen processing (12), and α-granule formation in megakaryocytes (16,17). Further, mutations in VPS33B that affect its interactions with Rab proteins cause Autosomal recessive Keratoderma-Ichthyosis-Deafness (ARKID) syndrome (18). The differences in phenotype conferred by mutations in VPS33A and VPS33B confirm that these proteins act in distinct cellular pathways.
A recent review suggested that VPS33B and VIPAR were members of a multi-subunit membrane tethering complex with an analogous organisation to CORVET and HOPS (19). This hypothetical complex was called “class C Homologs in Endosome-Vesicle Interaction” (CHEVI). With only two known subunits (VPS33B and VIPAR), there would be four other currently-unidentified subunits in this proposed complex. Here, we have used a quantitative proteomic approach in an attempt to identify members of the proposed CHEVI complex, and to further investigate the differences between the cellular membranes with which VPS33A and VPS33B associate as part of their cognate multi-subunit tethering complexes. We identified known interactors of VPS33A and VPS33B, and novel VPS33B interactors, but did not find evidence for the existence of the CHEVI complex.
RESULTS
It is known that VPS33A must interact with VPS16 in order to be recruited to HOPS and to localise correctly to target membranes (8,11,20). Mutation of two residues of VPS33A (Y438D and I441K) is sufficient to prevent the interaction between VPS33A and VPS16, and therefore inhibit HOPS function (8). We utilised this construct as a negative control for VPS33A localisation in our BioID assays. The VPS33B-VIPAR complex is likely to closely resemble the VPS33A/VPS16 interaction (12). We therefore docked a model of VPS33B generated using I-TASSER (21) onto the structure of human VPS33A in complex with VPS16 (11) to identify point mutations that may disrupt the VPS33B-VIPAR interaction, thus preventing correct VPS33B localisation within the cell (figure 1A).
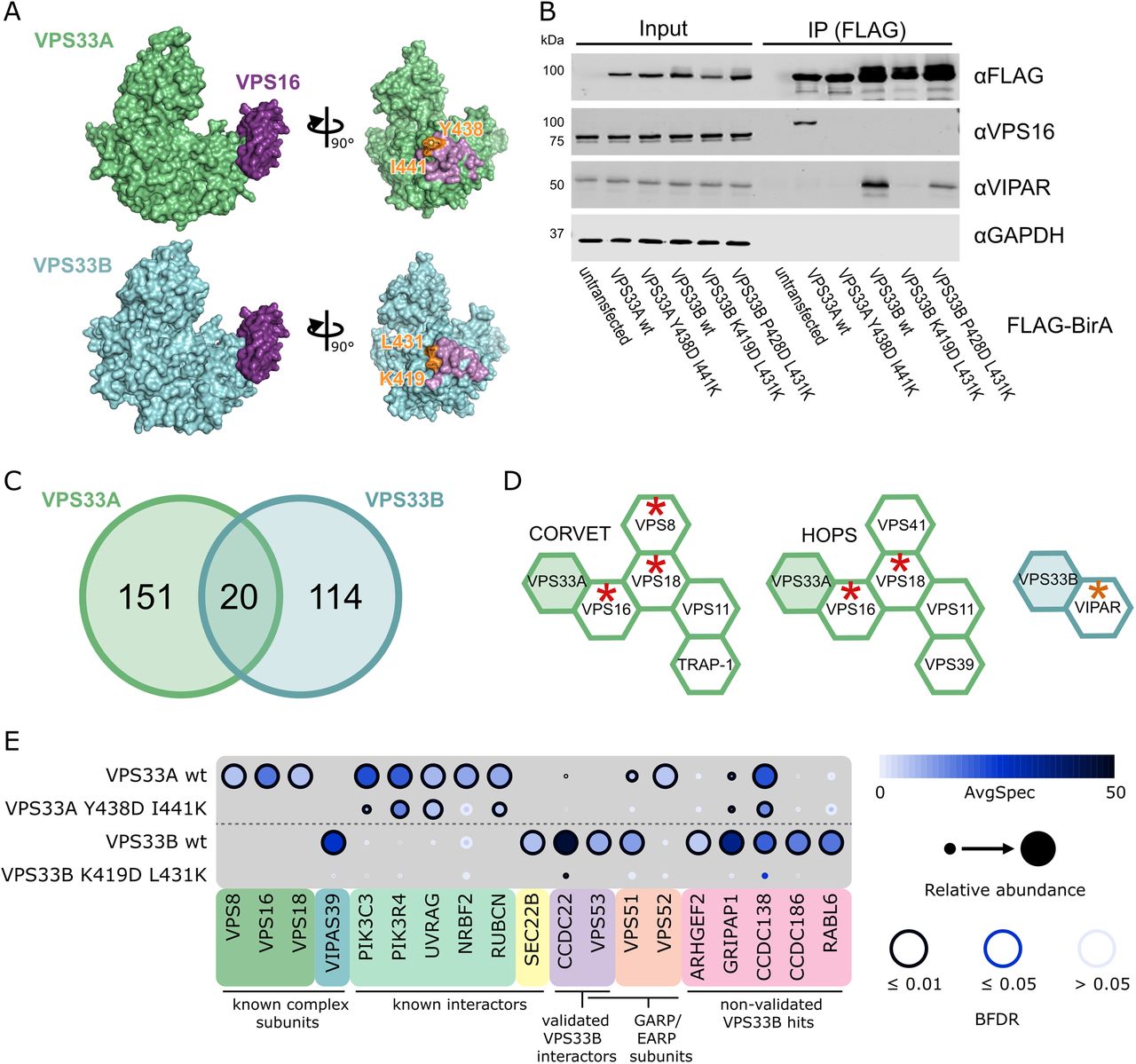
(A) Crystal structure of VPS33A in complex with VPS16 residues 642–736 (top), and a homology model of VPS33B docked onto this complex (bottom). Residues shown in orange are mutated in figure B. Molecular graphics were generated using PyMOL (Schrodinger LLC). (B) HEK293 Flp-In T-REx cells were stably transfected with VPS33A and VPS33B (wildtype and mutant) constructs with C-terminal FLAG-BirA* tags and expression was induced with tetracycline. After immunoprecipitation (IP), samples were immunoblotted for the indicated endogenous protein. (C) Distribution of proteins identified by BioID with a Bayesian False Discovery Rate (BFDR) ≤0.01 and at least two-fold enrichment in the wildtype sample over the negative-control mutant sample. (D) CORVET, HOPS and VPS33B-VIPAR complexes, with subunits identified in BioID results marked with red asterisks (VPS33A as bait) or orange asterisks (VPS33B as bait). (E) Selected BioID results, shown as dot plots (55). The spectral counts for each indicated prey protein are shown as AvgSpec. VIPAS39 = VIPAR, RUBCN = RUBICON. A complete list of the proximal proteins for each bait is available in the supplementary data file.
In order to compare VPS33A and VPS33B, we utilised the proximity-based ligation (BioID) assay (22). This assay utilises an abortive biotin ligase, BirA*, which releases an active intermediate of biotin (biotinoyl-5’-AMP) that can covalently label primary amines on proteins in the vicinity of (but not necessarily in direct contact with) the tagged protein of interest. We anticipated that BirA*-tagged VPS33A and VPS33B would result in biotinylation of stable tethering complex subunits and markers of membrane identity.
Wildtype and mutant VPS33A and VPS33B were C-terminally tagged with FLAG-BirA*, and stably integrated into the tetracycline-inducible HEK293 Flp-In T-REx cell line. As expected, when expressed in this cell line, wildtype VPS33A-FLAG-BirA* was able to co-immunoprecipitate endogenous VPS16, while the VPS33A Y438D I441K mutant could not (figure 1B). Similarly, wildtype VPS33B-FLAG-BirA* was able to co-immunoprecipitate endogenous VIPAR (figure 1B). The VPS33B K419D L431K mutant entirely disrupted binding to endogenously-expressed VIPAR, while the VPS33B P428D L431K mutant retained some binding (figure 1B). We thus proceeded to use VPS33A Y438D I441K and VPS33B K419D L431K mutants as negative controls for their respective wildtype constructs, thereby facilitating the identification of interactions formed only when VPS33A and VPS33B were correctly assembled into their cognate multi-subunit tethering complexes: CORVET and HOPS for VPS33A, and with VIPAR (potentially as part of CHEVI) for VPS33B.
High-confidence proximity interactions (preys) in BioID assays were considered as having a Bayesian false discovery rate (BFDR) of ≤0.01 (as determined by SAINT analysis (23)) and at least two-fold enrichment of the protein in the wildtype compared to the negative control mutant. Of these high-confidence preys, very few proteins (20 of 285, 7%) were captured by both VPS33A and VPS33B (figure 1C). The majority of preys were found exclusively with either VPS33A or VPS33B, providing strong evidence that VPS33A and VPS33B occupy distinct subcellular locations.
Proteins identified for VPS33A included two of the common subunits of CORVET and HOPS, VPS16 and VPS18, demonstrating that the placement of the BirA* tag did allow for biotinylation of central complex subunits (figure 1D). We also identified VPS8, a subunit specifically found in CORVET. Most of the subunits of CORVET and HOPS (excluding VPS33A) follow a similar architecture: an N-terminal β-propeller, an α-solenoid, and (often) a C-terminal zinc-finger domain (2,24). Although the mass spectrometry (MS) results for VPS33B included VIPAR (gene name VIPAS39), we did not find any other proteins with a domain structure reminiscent of that of CORVET or HOPS subunits (a β-propeller followed by an α-solenoid) as high-confidence preys.
In our BioID results for VPS33A, we identified two out of three of the core subunits of class III phosphatidylinositol 3-kinase (PI3KC3) complexes - PIK3C3 (a.k.a. Vps34) and PIK3R4 (a.k.a. Vps15). PI3KC3 complexes are found on Rab5- (25) and Rab7-positive membranes (26), including early endosomes and late endosomes or autophagosomes (reviewed in (27)) where CORVET and HOPS function, respectively. We also identified NRBF2, a regulator of the PI3KC3-C1 complex (28–30). Furthermore, we identified UVRAG, which is a member of the PI3KC3-C2 complex and is a known interactor of HOPS during autophagosome-lysosome fusion (31). RUBICON (gene name RUBCN), an inhibitor of PI3KC3-C2 complexes, was also identified as a VPS33A prey. RUBICON reportedly binds directly to UVRAG and prevents UVRAG from binding to the HOPS complex (32,33). Our observation that VPS33A can capture RUBICON indicates that UVRAG may not be required for HOPS recruitment to these membranes.
For VPS33B, we observed one previously-known interacting protein as a high confidence prey – SEC22B. SEC22B is a SNARE protein with functions in delivering ER-resident proteins to phagosomes in dendritic cells (34) and in plasma membrane expansion (35). It has recently been described as interacting with VPS33B during the formation of α-granules in megakaryocytes (36).
As the BioID results for VPS33A included many interactors that have already been characterised, we focussed on validating our high confidence hits from VPS33B, most of which had not been described previously. Seven proteins were selected for further investigation, based on those appearing with high abundance in our MS results and having a known involvement in membrane trafficking processes. N-terminal myc-tags were added to the shortlisted proteins ARHGEF2, CCDC22, CCDC138, CCDC186, GRIPAP1, RABL6, and VPS53. These were each co-transfected with VPS33B-GFP and FLAG-VIPAR into HEK293T cells, and then a GFP immunoprecipitation was performed (supplementary figure 1). Two of the shortlisted proteins were found to co-immunoprecipitate with VPS33B-GFP under these conditions: myc-CCDC22 and myc-VPS53 (figure S1A). Further, over-expression and co-immunoprecipitation of either of these proteins did not affect the co-immunoprecipitation of FLAG-VIPAR, suggesting that this interaction with VPS33B was not competing with VIPAR binding.
CCDC22 is a subunit of the CCC complex (also called the ‘Commander’ complex), along with CCDC93 and COMMD proteins (37,38). The CCC complex has been found to interact with other multi-subunit complexes: WASH, Retromer and Retriever (37,39-41). VPS53 is a subunit of both the Golgi associated retrograde protein (GARP) and endosome-associated recycling protein (EARP) complexes. Both GARP and EARP have four known subunits, sharing three common subunits (VPS51, VPS52 and VPS53) and each containing one unique subunit (VPS54 in GARP, VPS50 in EARP) (42). Interestingly, VPS51 was also a lower-confidence result in our BioID dataset for VPS33B, and VPS52 was a lower-confidence result for VPS33A.
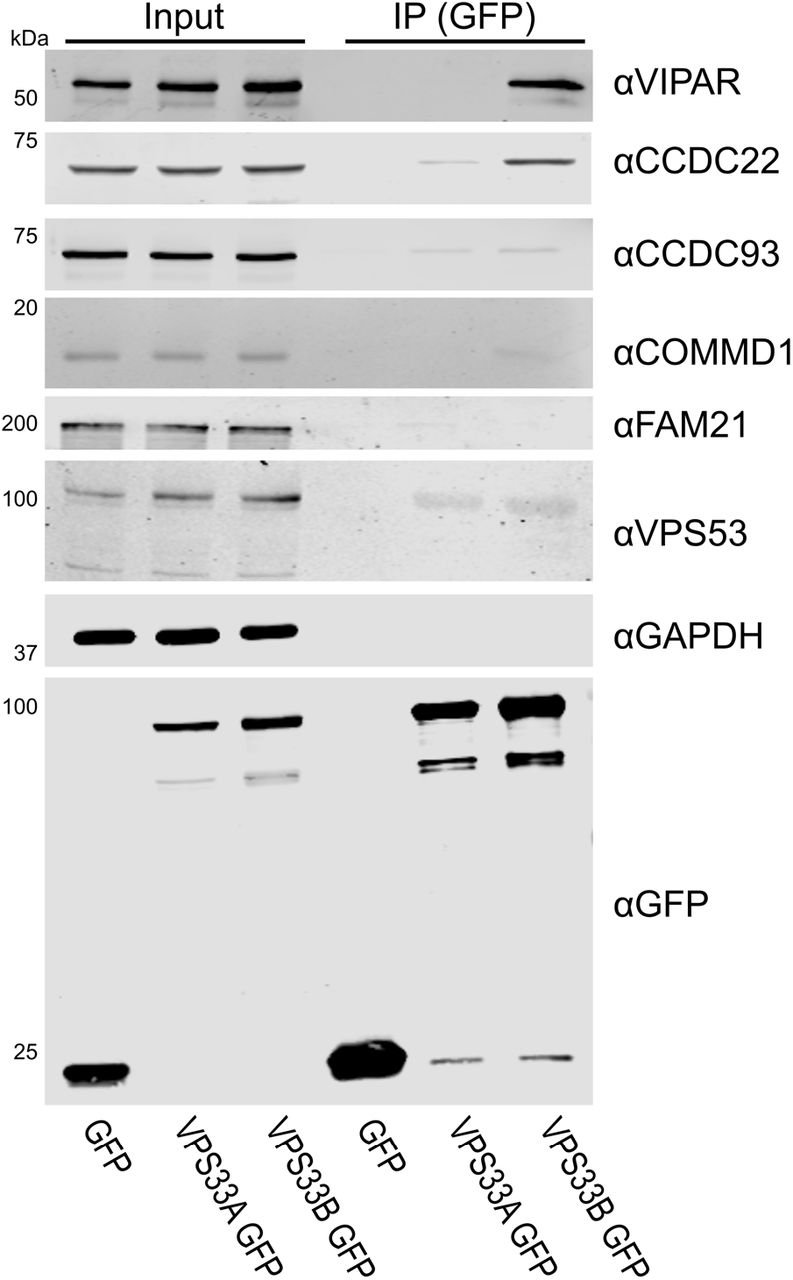
To probe whether GFP-tagged VPS33B was competent to bind endogenous CCDC22 or VPS53, an immunoprecipitation was performed using only transfected VPS33B-GFP (figure 2). Endogenous CCDC22 was efficiently co-immunoprecipitated by VPS33B-GFP. The endogenous CCC complex proteins CCDC93 and COMMD1 were not efficiently captured, nor was the WASH complex protein FAM21. Additionally, endogenous VPS53 did not appear to co-immunoprecipitate with VPS33B-GFP. However, it should be noted that the antibody against endogenous VPS53 indicates that these HEK293T cells expressed VPS53 isoform 4 (94 kDa), while our over-expression constructs are isoform 1 (80 kDa).

(A) HEK293T cells were transfected with VPS33A-GFP or VPS33B-GFP. After immunoprecipitation (IP), samples were immunoblotted for endogenous proteins using the antibodies indicated.
In order to perform in vitro binding experiments, VPS33B and GST-VIPAR were co-expressed in Escherichia coli and purified by GSH affinity and size-exclusion chromatography (supplementary figure 2). The VPS33B/GST-VIPAR complex was used as bait in pulldown experiments, with myc-CCDC22 and myc-VPS53 expressed by cell-free in vitro transcription/translation in wheat germ lysate. Myc-CCDC22 was very efficiently pulled down by VPS33B/GST-VIPAR, whereas myc-VPS53 was not pulled down (figure 3A). This suggests that the interaction between VPS53 and the VPS33B-VIPAR complex is either indirect (i.e. other proteins contribute to the interaction) or requires a post-translational modification not conferred in the plant cell-free expression system. To test the former, we repeated the pulldown experiment with each of the other subunits of the GARP and EARP complexes (VPS50, VPS51, VPS52, VPS54), each with an N-terminal myc tag. However, none of these singly-expressed subunits interacted with VPS33B/GST-VIPAR (figure 3A). We also attempted co-expressing all the subunits of GARP or EARP simultaneously, in the hope of reconstituting the complexes, but this expression strategy was inefficient and did not result in binding to VPS33B/GST-VIPAR (data not shown).

Myc-tagged CCDC22 or GARP and EARP subunits were produced by in vitro transcription/translation and then subjected to GST pulldown (PD) using VPS33B/GST-VIPAR or GST alone. Samples were analysed by immunoblotting with anti-myc. (A) CCDC22 is efficiently pulled down by VPS33B/GST-VIPAR, but no GARP or EARP subunits are pulled down. (B) While full length (1–627) CCDC22 is pulled down by VPS33B/GST-VIPAR, no CCDC22 truncations retained binding. (C) Point mutations of CCDC22 demonstrate reduced binding to VPS33B/GST-VIPAR compared to the wildtype (wt) protein. (D) Immunoblot band intensities were quantified and normalised (intensity of PD band divided by input band, normalised to the PD efficiency of the wildtype construct in each of three independent experiments), and analysed by one-way ANOVA with Dunnett’s multiple comparisons test (*p<0.05, **p<0.01). (E) Schematic of CCDC22, showing predicted N-terminal calponin homology (CH)-like (NN-CH) and C-terminal coiled-coil domains (59), regions required for binding to COMMD1 and CCDC93 (37), and constructs used in this paper.
To further understand how VPS33B-VIPAR may interact with CCDC22, we attempted to refine the region of CCDC22 that interacts with VPS33B/GST-VIPAR by generating a series of truncated forms of CCDC22. However, none of these CCDC22 truncations were able to bind to VPS33B/GST-VIPAR. We hypothesise that truncated forms of CCDC22 are unstable and unable to fold correctly in this assay system.
Multiple mutations have been reported in CCDC22, found in a cohort of X-linked intellectual disability (XLID) patients (43). Several of these mutations have been previously used to study the interaction between CCDC22 and COMMD proteins (found in the CCC complex) (44). Interestingly these mutations cluster in the N-terminal half of CCDC22. Four of these CCDC22 point mutants were assayed in the pulldown experiment described above, and each demonstrated reduced binding to VPS33B/GST-VIPAR. (The CCDC22 T17A mutant was not included because it also affects splicing (43).) For the two most N-terminal CCDC22 mutants, T30A and R128Q, we found statistically-significant disruption of binding to VPS33B/GST-VIPAR. This might suggest that the N terminus of CCDC22 is critical for binding to VPS33B/GST-VIPAR. However we cannot discount the possibility that these point mutations cause CCDC22 to be poorly folded in our assay system.
In order to determine whether VPS33B and VIPAR form a stable complex with CCDC22 in untransfected cells, we performed a cell fractionation experiment. In brief, whole cell lysates of HEK293T cells were separated by size exclusion chromatography. A series of elution fractions were collected from the column, the protein content of alternate fractions was concentrated, and the concentrated fractions were analysed by immunoblotting. In order from largest to smallest, we observed elution peaks for WASH, CORVET/HOPS, CCC, and then VPS33B-VIPAR complexes during cell lysate fractionation (figure 3A, 3B). It is thus evident that VPS33B and VIPAR do not co-fractionate with the majority of CCDC22 under these conditions. CCDC22 instead elutes in the same fractions as other members of the CCC complex (CCDC93 and COMMD1), proteins that are not efficiently co-immunoprecipitated by over-expressed VPS33B-GFP (figure 2). Further, the complex containing VPS33B and VIPAR elutes later than (and is thus considerably smaller than) the CORVET and HOPS complexes, indicating that VPS33B and VIPAR did not elute from the gel filtration column as part of a larger multi-subunit tethering complex.
DISCUSSION
In this study we performed a proteomic comparison of the cellular interaction partners of VPS33A and VPS33B using proximity-based ligation (BioID). VPS33A was found to associate with members of the PI3K3C complexes, confirming previous observations (31–33). We also corroborated a recent publication that found VPS33B to co-localise with SEC22B (36). This confirms that our BioID-tagged constructs were able to localise to the correct intracellular membranes and were competent to label bona fide endogenous binding partners. Overall, the majority of proteins identified in our assay co-localised exclusively with either VPS33A or VPS33B, indicating that these proteins are found on different subcellular membranes.
VPS33A is a subunit of both the CORVET and HOPS complexes, the subunits of which share a very similar architecture (2,24). While VPS33B-FLAG-BirA* did label VIPAR, we did not find any other proteins proximal to VPS33B that shared the canonical architecture of CORVET and HOPS components, as would have been predicted in a VPS33B- and VIPAR-containing CHEVI complex. Further, our results from the whole cell fractionation experiment provide compelling evidence that VPS33B and VIPAR are found in a complex which is considerably smaller than CORVET and HOPS (figure 4). Given that CORVET/HOPS components co-eluted during this experiment, confirming that such complexes remain assembled under these assay conditions, we conclude that the hypothesised large, multi-subunit CHEVI complex does not exist in HEK293T cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) HEK293T cell lysates were injected onto a Superose 6 10/300 GL gel filtration column and eluted fractions were analysed by immunoblotting. (B) Immunoblot band intensities were quantified, normalised to the band of maximum intensity, and fitted to a Gaussian distribution (for all except FAM21, which could not be reliably fitted to a single Gaussian distribution).
VPS33B and VIPAR have previously been described as acting in recycling pathways (12,13), as has VPS53 as part of GARP and EARP complexes (42,45), and CCDC22 as part of the CCC complex (37,39-41). Both VPS53 and CCDC22 associated with VPS33B and VIPAR when co-overexpressed (supplementary figure 1). Endogenous VPS53 was not co-immunoprecipitated by overexpressed VPS33B-GFP (figure 2), nor could the interaction with the VPS33B-VIPAR complex be observed in vitro (figure 3A), suggesting that this interaction is either transient or is tightly regulated by the cell. GFP-tagged VPS33B was able to co-immunoprecipitate endogenous CCDC22 and this interaction could be recapitulated using purified recombinant components (figures 2 and 3A), suggesting that it is a direct physical interaction. Mutations in CCDC22 that cause XLID reduce the interaction of this protein with the VPS33B-VIPAR complex (figure 3C–D). In agreement with our results, a recent large-scale proteomic study also identified CCDC22 as interacting with VPS33B-VIPAR (46). We do not observe co-immunoprecipitation of VPS33B-GFP with other members of the CCC complex (figure 2). Further, we do not observe efficient co-fractionation of endogenous VPS33B-VIPAR with CCDC22 from HEK293T cell lysates, with CCDC22 instead co-fractionating with other CCC complex components (figure 4). Notably, the CCC complex also did not co-fractionate with the WASH complex, a known interactor. This suggests that the interaction between VPS33B-VIPAR and CCDC22, either alone or as part of the CCC complex, may be weak or transient in cultured HEK293T cells. We hypothesise that this interaction may be inducible upon addition of an external stimulus, for example during bacterial infection, when VPS33B/VIPAR activity has been demonstrated to be important for a robust immunological response (47–49).
In summary, we propose that VPS33B-VIPAR is not assembled into a large multi-subunit tethering complex. Rather, VPS33B and VIPAR form a discrete bimolecular complex that can transiently interact with members of other multi-subunit trafficking complexes such as the CCC complex component CCDC22.
EXPERIMENTAL PROCEDURES
Expression constructs/cloning
Human VPS33A and VPS33B were cloned into pDONR223 (Invitrogen) and then pDEST-pcDNA5-BirA-FLAG (50) using Gateway cloning. VPS33A Y438D I441K, VPS33B K419D L431K and VPS33B P428D L431K were created by site-directed mutagenesis of wildtype constructs in pDONR223, then transferred into pDEST-pcDNA5-BirA-FLAG.
For transient transfection, wildtype VPS33A and VPS33B were cloned into pEGFP-N1 (Clontech), in order to add a C-terminal EGFP tag. An N-terminal FLAG-tag was added to VIPAR and cloned into pF5K CMV-neo (Promega). ARHGEF2 (isoform 1), CCDC22, CCDC138 (isoform 1), CCDC186, GRIPAP1 (isoform 1), RABL6 (isoform 1), and VPS53 (isoform 1) were cloned with N-terminal myc tags into pF5K CMV-neo.
For co-expression in E. coli, DNA encoding VPS33B and VIPAR that had been codon-optimised for bacterial expression (GeneArt) was cloned into positions 1 and 2 (respectively) into the polycistronic vector pOPC (51), VIPAR being tagged at the N terminus with GST.
For in vitro transcription/translation, full-length human CCDC22, VPS50, VPS51, VPS52, VPS53 and VPS54 were cloned into pF3A WG (BYDV) (Promega) with an N-terminal myc tag. Truncation and point mutations of CCDC22 were created by inverse PCR or site-directed mutagenesis.
Cell culture and transfection
Untransfected HEK293 Flp-In T-REx cells were grown in Dulbecco’s Modified Eagle Medium with high glucose (Sigma, cat. D6546), supplemented with 10% (v/v) heat-inactivated foetal calf serum, 2 mM L-glutamine, 100 IU/ml penicillin, 100 mg/ml streptomycin, 3 μg/ml blasticidin and 100 μg/ml zeocin, in a humidified 5% CO2 atmosphere at 37°C. Cells were co-transfected with pOG44 (encoding the Flp recombinase) and the VPS33A or VPS33B constructs in pDEST-pcDNA5-BirA-FLAG vectors (described above) using TransIT-LT1 (Mirus), following the manufacturer’s instructions. Stable cell lines were selected by addition of 200 μg/ml hygromycin.
HEK293T cells (ATCC #CRL-3216, mycoplasma-free) were grown in DMEM with high glucose, supplemented with 10% (v/v) heat-inactivated foetal calf serum, 2 mM L-glutamine, 100 IU/ml penicillin and 100 mg/ml streptomycin, in a humidified 5% CO2 atmosphere at 37°C. Cells were transfected with TransIT-LT1 (Mirus), following the manufacturer’s instructions.
BioID and mass spectrometry analysis
BioID and mass spectrometry analysis was performed essentially as described (52). Briefly, stable HEK293 Flp-In T-REx cells were grown on 15 cm plates to approximately 75% confluency. Bait expression and proximity labelling was then induced simultaneously by addition of tetracycline (1 μg/mL) and biotin (50 μM) for 24 hours. Cells were collected in PBS and biotinylated proteins were purified by streptavidin-agarose affinity purification. Proteins were digested on-bead with sequencing-grade trypsin in 50 mM ammonium bicarbonate pH 8.5. Peptides were then acidified by the addition of formic acid (2% (v/v) final concentration) and dried by vacuum centrifugation. Dried peptides were suspended in 5% (v/v) formic acid and analysed on a TripleTOF 5600 mass spectrometer (SCIEX) in-line with a nanoflow electrospray ion source and nano-HPLC system. Raw data were searched and analysed within ProHits LIMS (53) and peptides matched to genes to determine prey spectral counts (54). High confidence proximity interactions (BFDR ≤ 0.1) were determined through SAINT analysis (23) implemented within ProHits. Bait samples (biological duplicates) were compared against 12 independent negative control samples (6 BirA-FLAG only and 6 3xFLAG only expressing cell lines). The specific control samples used in this study were previously published as part of Chapat et al. (52). Dotplots were prepared in ProHits-Viz (55). Mass spectrometry data have been deposited in the MassIVE database (ID MSV000081814; available for FTP download at ftp://MSV000081814@massive.ucsd.edu) and with the ProteomeXchange Consortium (identifier PXD008457) (56). A summary of the results are attached as supplemental data.
Recombinant protein expression and purification
In vitro protein expression was performed using TNT SP6 High Yield Wheat Germ reaction mix (Promega) as per the manufacturer’s instructions.
VPS33B and GST-VIPAR were co-expressed in E. coli B834(DE3). Bacteria were grown in 2x TY medium to an A600 of 0.8–0.9 at 37°C, cooled to 22°C, and protein expression was induced by the addition of 0.2 mM isopropyl β-d-thiogalactopyranoside. After 16–18 hr, cells were harvested by centrifugation at 5000 ×g for 15 min and the pellet was stored at −20°C until required.
Cells were thawed and resuspended in 20 mM Tris (pH 7.5), 300 mM NaCl, 0.5 mM MgCl2, 1.4 mM β-mercaptoethanol, 0.05% TWEEN 20, supplemented with 400 units of bovine DNase I (Sigma–Aldrich) and 200 μl of EDTA-free protease inhibitor mixture (Sigma–Aldrich) per 4–8 L of cell culture. Cells were lysed at 24 kpsi using a TS series cell disruptor (Constant Systems) and lysates were cleared by centrifugation at 40 000 ×g for 30 min at 4°C. Cleared lysate was incubated with glutathione sepharose 4B (GE Healthcare) for 1 h at 4°C, the beads were washed with 20 mM Tris (pH 7.5), 300 mM NaCl, and 1 mM DTT, and bound protein was eluted in wash buffer supplemented with 25 mM reduced glutathione. Protein was injected onto a Superdex 200 10/300 GL column (GE Healthcare) equilibrated in 20 mM Tris (pH 7.5), 200 mM NaCl, and 1 mM DTT. Fractions containing purified proteins were concentrated using 30 kDa nominal molecular mass cut-off centrifugal filter units (Millipore), diluted in glycerol to a final concentration of 50% (v/v) glycerol, and stored at −20°C until required. Purified protein complex identity was verified by peptide mass fingerprinting.
Co-immunoprecipitations and pull-downs
FLAG immunoprecipitations were performed by harvesting cells and lysing in 10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40, and EDTA-free protease inhibitor cocktail (Sigma). Protein concentration in the resulting lysates was quantified by BCA assay (Thermo Scientific), and protein concentration was equalised before immunoprecipitation with anti-FLAG M2 magnetic resin (Sigma). 1 ml of whole cell lysate, containing approximately 1.9 mg of protein, was incubated with 30 μl of anti-FLAG resin for 1 hr at 4°C. Samples were then washed three times in wash buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40), then eluted by incubation with 250 μg/ml FLAG peptide (Sigma, cat. F3290).
GFP immunoprecipitations were performed using GFP-TRAP resin (ChromoTek), as in (57).
GST pulldown experiments were performed as described in (57), using 40-100 pmol purified VPS33B/GST-VIPAR complex as bait (described above).
Antibodies and immunoblotting
The following primary antibodies were used for immunoblotting: polyclonal anti-GFP (Sigma, cat. G1544), monoclonal anti-FLAG M2 (Sigma, cat. 080M6035), monoclonal anti-myc (Millipore, cat. 05-724), monoclonal anti-GAPDH (Life Tech, cat. AM4300), polyclonal anti-VPS33A (as described in (11)), monoclonal anti-VPS33B (Santa Cruz, cat SC-398322), monoclonal anti-VIPAR (a.k.a. anti-SPE39, gift from S.W. L’Hernault, (58)), polyclonal anti-VPS16 (Santa Cruz, cat. sc-86939, lot I1913), polyclonal anti-CCDC22 (Proteintech, cat. 16636-1-AP), polyclonal anti-CCDC93 (Proteintech, cat. 20861-1-AP), polyclonal anti-COMMD1 (gift from E. Burstein, as described in (37)), polyclonal anti-FAM21 (Santa Cruz), polyclonal anti-Strumpellin (Santa Cruz), and polyclonal anti-VPS53 (Sigma cat. HPA024446, lot A106517). IRDye 800CW-conjugated secondary antibodies were supplied by LI-COR: goat anti-mouse (cat. 925-32210), goat anti-rabbit (cat. 925-32211), and donkey anti-rabbit (cat. 925-32213).
All samples were immunoblotted as per (57). Immunoblot band intensity was quantified using Image Studio Lite (version 5.4, LI-COR) and non-linear fitting to a Gaussian distribution was performed using Prism 7 (GraphPad).
Whole cell fractionation
HEK293T cells were harvested and lysed in a buffer similar to that used for immunoprecipitation experiments: 10 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40, EDTA-free protease inhibitor cocktail and Benzonase (Sigma). After centrifugation at 20 000 ×g for 10 min, lysates were injected onto a Superose 6 10/300 column (GE Healthcare) equilibrated in 10 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40. Fractions (0.4 mL) were collected and concentrated using 10 μL of StrataClean resin (Agilent) per 100 μL of fractionated protein. Bound protein was eluted by boiling in SDS-PAGE loading buffer and samples were analysed by SDS-PAGE and immunoblotting.
ACKNOWLEDGEMENTS
We thank S.W. L’Hernault, E. Burstein and M. Seaman for kindly providing reagents. This work was supported by a Sir Henry Dale Fellowship, jointly funded by the Royal Society and the Wellcome Trust, to SCG [098406/Z/12/Z] and an Isaac Newton Trust/Wellcome Trust ISSF/University of Cambridge Joint Research Grant to SCG. GGH was supported by a Parkinson Canada Basic Research Fellowship. ACG is a Tier 1 Canada Research Chair and supported by a CIHR Foundation grant (FDN 143301).
REFERENCES




