

Abstract
Mobilization of transposable elements (TEs) in plants has been recognized as a driving force of evolution and adaptation, in particular by providing genes with regulatory modules that impact their transcription. In this study, we employed an ATCOPIA93 Long terminal repeats (LTR) promoter-GUS fusion to show that this retrotransposon behaves like an immune-responsive gene during plant defense in Arabidopsis. We also showed that the reactivation of the endogenous ATCOPIA93 copy EVD, in the presence of bacterial stress, is not only negatively regulated by DNA methylation but also by Polycomb-mediated silencing — a mode of repression typically found at protein-coding and microRNA genes. Interestingly, one of the ATCOPIA93-derived soloLTRs is located upstream of the disease resistance gene RPP4 and is devoid of either DNA methylation or H3K27m3 marks. Through loss-of-function experiments, we demonstrated that this soloLTR is required for proper expression of RPP4 during plant defense, thus linking the responsiveness of ATCOPIA93 to biotic stress and the co-option of its LTR for plant immunity.
Introduction
TEs are repeated sequences that can potentially move and multiply in the genome. Their mobilization has been recognized as a driving force of evolution and adaptation in various organisms, in particular by providing genes with regulatory modules that can create or impact transcriptional programs (Chuong et al, 2016). The study of TE regulation is thus important in order to understand both the conditions for their transposition but also their influence, as full-length or truncated elements, on nearby gene regulation. This role in cis has been demonstrated in plants by artificially inducing insertions that confer gene regulation, e.g., the rice TE mPing (Naito et al, 2009) or the Arabidopsis TE ONSEN (Ito et al, 2011). However, the causal link between cis-regulatory properties of TEs and established expression patterns of nearby genes, requires loss-of-function experiments and has rarely been demonstrated (Chuong et al, 2016).
TE cis-regulatory effects can either be genetic in nature, such as when the TE contains regulatory motifs, or epigenetic through recruitment of dimethylation of histone 3 lysine 9 (H3K9m2) and cytosine DNA methylation, which are hallmarks of transposon control. DNA methylation in Arabidopsis is carried out by four pathways. While METHYLTRANSFERASE1 (MET1) (Kankel et al, 2003) maintains CG methylation, CHROMOMETHYLASE2 and 3 (CMT2 and CMT3) maintain CHG methylation (where H is any base pair but not a G) (Zemach et al, 2013; Stroud et al, 2014). The maintenance of CHH methylation requires either CMT2 for long heterochromatic repeats or RNA-directed DNA methylation (RdDM) mediated by DOMAINS REARRANGED METHYLASE 2 (DRM2) and accompanying small RNA machinery (Cao & Jacobsen, 2002; Chan, 2004; Deleris et al, 2016). In addition, the SNF2 family chromatin remodeler DECREASED DNA METHYLATION 1 (DDM1) is necessary for heterochromatic DNA methylation in all cytosine sequence contexts (Jeddeloh et al, 1999; Zemach et al, 2013). Furthermore, DNA methylation and H3K9m2 are mechanistically interconnected and, as a result, are largely co-localized throughout the genome (Du et al, 2015). Importantly, this histone and cytosine marking can also impact the nearby genes which then become epigenetically controlled because of the inhibitory effect of DNA and H3K9 methylation on promoter activity (Lippman et al, 2004; Liu et al, 2004; Huettel et al, 2006; Gehring et al, 2009). In addition, in both plants (Mathieu et al, 2005; Deleris et al, 2012; Weinhofer et al, 2010) and animals (Reddington et al, 2013; Saksouk et al, 2014; Basenko et al, 2015; Walter et al, 2016), the removal of DNA methylation at some TEs leads to H3K27 trimethylation (H3K27m3), an epigenetic mark deposited and interpreted by Polycomb Group (PcG) proteins, which normally target and silence protein coding genes that are often developmentally important (Förderer et al, 2016). Thus, there exists a potential for this alternative repression system to mediate silencing of TEs, but it has not been fully explored in plants, except in the endosperm, a nutritive and terminal seed tissue that is naturally DNA hypomethylated (Weinhofer et al, 2010; Moreno-Romero et al, 2016).
In accordance with the epigenetic control mediated by DNA and histone H3K9 methylation, mutations in DNA methylation pathway genes lead to reactivation of various subsets of TEs; however, these defects in chromatin regulation are not always sufficient for TE expression, and the activation of specific signaling pathways is sometimes needed. This has been exemplified by the Arabidopsis Long-Terminal Repeats (LTR)-retrotransposon ONSEN, which was shown to be reactivated after heat-stress, in wild-type plants and independently from a loss of DNA methylation in this context (Ito et al, 2011; Cavrak et al, 2014). In addition, ONSEN was not expressed in unstressed RdDM-defective mutants (nor in ddm1), but its induction was enhanced in RdDM-defective mutants subjected to heat stress (Ito et al, 2011). To our knowledge, ONSEN is the only described example of a TE which expression is modulated by DNA methylation during stress response. Thus, it is important to characterize other TEs that exploit plant signaling, in order to gain a deeper understanding of the connection between biotic/abiotic stresses and transposon activation, and how epigenetic silencing pathways exert their influence on this relationship.
In this study, we unravel the responsiveness of another family of Arabidopsis retroelements, ATCOPIA93 (Mirouze et al, 2009; Marí-Ordóñez et al, 2013), during PAMP-triggered immunity (PTI). PTI is defined as the first layer of active defense against pathogens and relies on the perception of evolutionary conserved Microbe- or Pathogen-associated molecular patterns (MAMPs or PAMPs) by surface receptors (Boutrot & Zipfel, 2017). ATCOPIA93 is a low-copy, evolutionary young family of LTR-retroelements, which is tightly controlled by DNA methylation, in particular CG methylation (Mirouze et al, 2009). The family representative EVD (AT5G17125) was found to transpose in ddm1 after eight generations of inbreeding (Tsukahara et al, 2009) as well as in genetically wild-type epigenetic recombinant lines (epiRILs) derived from crosses between wild-type and met1 (Mirouze et al, 2009, Marí-Ordóñez et al, 2013) or ddm1 (Marí-Ordóñez et al, 2013). EVD is 99.5% identical in sequence to the pericentromeric ATR (AT1G34967), which is predicted to encode a polyprotein but does not seem to be active in the latter conditions (Mirouze et al, 2009). Here, we first took advantage of an unmethylated ATCOPIA93 LTR-GUS fusion that we used as a reporter of promoter activity, since LTRs of retroelements contain cis-regulatory sequences that can recruit RNA Pol II (Chuong et al, 2016). We showed that this LTR exhibits the hallmarks of a promoter of an immune-responsive gene in the absence of epigenetic control. Accordingly, the corresponding methylated endogenous ATCOPIA93 retroelements, EVD and ATR, were only significantly reactivated after PAMP-elicitation in a DNA hypomethylated background, in met1 or ddm1 mutants. Interestingly, we demonstrated for the first time in wild-type plant vegetative tissues, a second layer of control of TE expression mediated by Polycomb silencing. Importantly, we showed that H3K27m3 co-exists with DNA methylation at EVD sequences but not at ATR, leading to a differential negative control between these two copies during immunity. Furthermore, we were able to test the implications of these findings for the regulation of the immune response. We identified an ATCOPIA93-derived soloLTR, unmethylated and not marked by H3K27m3, upstream of the RPP4 disease resistance gene. By measuring the impact of the genetic loss of this soloLTR, we could show that it has been co-opted for the proper expression of RPP4 during PAMP-triggered plant basal defense and thus that it plays a role as regulatory “enhancer” element. Thus, we established a link between the responsiveness of a TE to biotic stresses and the co-option of its derived soloLTR for plant immunity, where the repressive epigenetic modifications controlling the full-length active elements are absent on the derived regulatory sequence.
Results
ATCOPIA93-LTR::GUS transcriptional fusion behaves as a canonical immune-responsive gene
An ATCOPIA93-LTR::GUS construct –comprising the full EVD/ATR LTR upstream of a sequence encoding a GUS protein (schema Fig 1A, EV1A) – was transformed into the Arabidopsis wild type reference accession Columbia (Col-0), initially to serve as a reporter of DNA methylation levels. Unexpectedly, the LTR::GUS transgenes were not methylated in any of the transgenic lines obtained (Fig 1A, Fig EV1B). Instead, we generated a reporter of ATCOPIA93 promoter activity that we could exploit to assess ATCOPIA93 responsiveness during PTI in the absence of DNA methylation-mediated control. Plants containing the LTR::GUS transcriptional fusion were further elicited with either flg22 (a synthetic peptide corresponding to the conserved N-terminal region of bacterial flagellin that is often used as a PAMP surrogate) (Zipfel et al, 2004; Boutrot & Zipfel, 2017), or PtoΔ28E (a non-pathogenic Pseudomonas syringae pv. tomato DC3000 (Pto DC3000) in which 28 out of 36 effectors are deleted (Cunnac et al, 2011)), and the accumulation of GUS mRNA and protein was monitored over a 24 hour time course. In water-treated plants, at 24 hours post-infiltration (hpi), there was barely any GUS staining, indicating that the activity of the ATCOPIA93 LTR promoter is weak in this condition. By contrast, in both flg22 and PtoΔ28E treatments, an intense GUS staining was observed at 24 hpi (Fig 1B, top panel). This was associated with progressive GUS protein accumulation over the time-course, until it reaches a plateau (Fig 1B, bottom panel). At all the time points analyzed, the GUS expression was generally stronger in response to PtoΔ28E than flg22 and thus we focused on the former bacterial elicitor for the rest of the study. By analyzing GUS mRNA levels, we could then show that the GUS induction was transient, similarly as an immune-responsive gene induced rapidly during PTI such as WRKY29 (Asai et al., 2002) (Fig 1C, Fig EV1C). In addition, treatment with the virulent wild-type Pto DC3000 strain, which can inject type III effectors into the host cell, resulted in a partially compromised induction of GUS protein accumulation compared to a similar inoculum of PtoΔ28E (Fig 1D), suggesting that some bacterial effectors suppress the LTR responsiveness during PTI. This transcriptional behavior is reminiscent of typical PTI-induced genes whose induction is impaired by bacterial effectors that have evolved to suppress different steps of PTI to enable disease (Asai & Shirasu, 2015). Together, these data show that the ATCOPIA93 LTR::GUS transgene behaves like a canonical immune-responsive gene, which is transcriptionally reactivated during PTI and whose induction is suppressed by bacterial effectors. In support of this, we noticed in the LTR sequence the presence of two putative W-box elements, i.e. DNA sequences with the C/TTGACC/T (A/GGTCAA/G) motif, which are the cognate binding sites for WRKY transcription factors that are known to orchestrate transcriptional reprogramming during PTI (Rushton et al, 2010) (Fig EV1A). Importantly, we found that these two putative W-box elements are functional as the PtoΔ28E-mediated transcriptional induction of GUS was lost in part or entirely when W-box 1 and W-box 2 were mutated, respectively (Fig 1E).

A. Alignment between the LTRs of ATCOPIA93-EVD and ATCOPIA93-ATR showing that they are identical in sequence. The two W-boxes tested in Fig 1 are highlighted in blue; the beginning and end of the LTRs of the ATCOPIA93 family are highlighted in green and orange respectively; the GGGCC sequence in red is the site recognized by Sau96I methylation-sensitive restriction enzyme and underlined is the CG site analyzed by Chop-qPCR in Fig EV1B and Fig 3D. The five CG sites of the LTR, unmethylated in the transgene, are highlighted with pink boxes.
B. Methylation status of the DNA (CG site) at the LTR in LTR::GUS transgenic plants by Sau96I Chop-qPCR. DNA from a pool of leaves of single primary transformants and pools of T3 homozygous plants was digested with the methylation sensitive restriction endonuclease Sau96I which recognizes GGNCC sites- here GGGCCG- and is sensitive to methylation of the second C. Digested DNA was quantified by using qPCR with primers spanning a Sau96I restriction site in the LTR. On the left, primers were specific for the transgenic LTR (one primer in the vector): lack of amplification shows unmethylation of the CG site in the transgenic LTR. On the right, as a control, the same DNA was analyzed with primers specific for the endogenous EVD LTR (one primer upstream of EVD): amplification shows methylation of the CG site in the endogenous 5’ LTR sequence. The signal was normalized to an undigested control. The assay principle is schematized under the graph.
C. Additional independent experiment for the time-course analysis of GUS mRNA (plain lines) and PTI-marker WRKY29 mRNA (dashed lines) by RT-qPCR. The experiment was performed as in Fig 1C.

A. Cytosine methylation analyzed by bisulfite-sequencing at the LTR::GUS transgene. Genomic DNA of a pool of LTR::GUS transgenic plants (one T3 line, four plants, two rosette leaves each) was treated with sodium bisulfite, amplified with primers specific for the LTR contained in the LTR::GUS construct and cloned for sequencing (19 clones). The endogenous LTR of ATCOPIA93 EVD was sequenced as a positive control (17 clones). The percentage of methylated cytosines is indicated by vertical bars. This result was also reproduced in various T3 and T1 lines by a Sau96I methylation-sensitive assay analyzing the first CG site (red asterisk)(Fig EV1B).
B. Accumulation of GUS protein detected in response to bacterial elicitors of basal immunity. Upper panel: representative pictures of leaves infiltrated with water (mock), Pto DC3000 deleted of 28 effectors (PtoΔ28E) at 2.108 colony-forming unit per ml (cfu/ml) or 1 μM of flg22, and incubated with GUS substrate 24 hours post-infiltration (24hpi). The number of leaves showing this representative phenotype is shown into brackets. Lower panel: Western blot analysis over a 24h time-course; RbC: Rubisco. Three to four plants (two leaves per plant) were infiltrated for each condition and time point, and leaves pooled by condition and time-point before extracting the proteins. Samples derived from the same experiment, and gels and blots were processed in parallel. This experiment was repeated twice with similar results.
C. Time-course analysis of GUS mRNA (plain lines) and PTI-marker WRKY29 mRNA (dashed lines) by RT-qPCR. Leaves were infiltrated with water (mock), or effectorless bacteria Pto DC3000 (PtoΔ28E) at 2.108 cfu/ml; two similar leaves of three to four plants were pooled by condition and by time-point after infiltration (as in B) before extracting the RNA subjected to RT-qPCR. Values are relative to the expression of the UBIQUITIN gene (At2g36060). This experiment was repeated twice independently and another independent experiment is shown in Fig EV1C.
D. Accumulation of GUS protein detected in response to virulent bacteria Pto DC3000 versus PtoΔ28E. Upper panel: representative pictures of leaves infiltrated with water (mock), effectorless (PtoΔ28E) and virulent (Pto) bacteria Pto DC3000, both at 1.107 cfu/ml, and incubated with GUS substrate at 24hpi. The number of leaves showing this representative phenotype is shown into brackets. Lower panel: Western blot analysis of the GUS protein accumulated at 24hpi; RbC: Rubisco. Two similar leaves of three to four plants were pooled by condition and time-point before extracting the proteins.
E. Activation of the GUS expression upon PtoΔ28E elicitation in LTR::GUS plants with mutated W-boxes. Experiments were performed on 28, 22, and 19 primary transformants for the LTR::GUS WT, m1 and m2 constructs respectively; the point-mutations introduced are depicted on the right (W-boxes are the sequences in bold). Mock and PtoΔ28E (at 2.108 cfu) treatments were performed on four similar leaves of each individual transformant and GUS staining performed 24 hours later on two leaves. One representative picture (into brackets is the number of plants showing this phenotype) for one primary transformant is shown for each construct with each treatment. Plants were classified into three categories: normal GUS induction, loss of GUS induction, constitutive GUS expression and percentages of plants belonging to each category over the total number of plants tested were calculated.
AtCOPIA93 reactivation is negatively controlled by DNA methylation during PAMP-triggered immunity
We next analyzed, over the same period of time, the reactivation of the almost identical endogenous copies of AtCOPIA93: EVD and ATR. The induction of their expression upon bacterial challenge was generally weak in wild-type leaves (Fig 2A and 2B, Fig EV2). By contrast, we observed a consistent and transient induction of EVD/ATR at 3, 6 and 9 hpi in a ddm1 hypomethylated background (Stroud et al, 2013, Fig 2A and 2B left panel, Fig EV2), specifically in response to the PtoΔ28E strain. EVD/ATR transcript levels were also enhanced at 6 hpi in a bacteria-challenged met1 mutant (Fig 2B, right panel), which is impaired in CG methylation (Ordonnez et al., 2013). Together, these data indicate a tight negative control of ATCOPIA93 induction which is exerted by DNA methylation and is particularly relevant during bacterial challenge when the LTR is activated. Notably, at this developmental stage, mutations in the components of the DNA methylation pathways were not sufficient to enhance ATCOPIA93 expression in the absence of bacterial stress, in accordance with the transcriptional behavior of the unmethylated LTR::GUS fusion which displays weak promoter activity in water-treated plants (Fig 1).

Biological replicate (independent experiment) for the time course analysis of ATCOPIA93 mRNA by RT-qPCR. The experiment was performed as in Fig 2A.

A. Time course analysis of ATCOPIA93 mRNA by RT-qPCR. Leaves were infiltrated with water (mock), or PtoΔ28E bacteria at 2.108 cfu/ml; two similar leaves of three to four plants were pooled by condition and by time-point after infiltration before extracting the RNA. Values were determined by RT-qPCR and are relative to the expression of the UBIQUITIN gene (At2g36060). This experiment was repeated twice with similar results and another independent experiment is shown in Fig EV2.
B. ATCOPIA93 mRNA analysis in two methylation-defective mutants, ddm1 and met1, at 6 hours post-treatment with either water or PtoΔ28E at 2.108 cfu/ml. Material and data were generated as in A. The data points for three independent experiments are plotted.
ATCOPIA93-EVD is marked by H3K27m3 chromatin modification in addition to DNA methylation
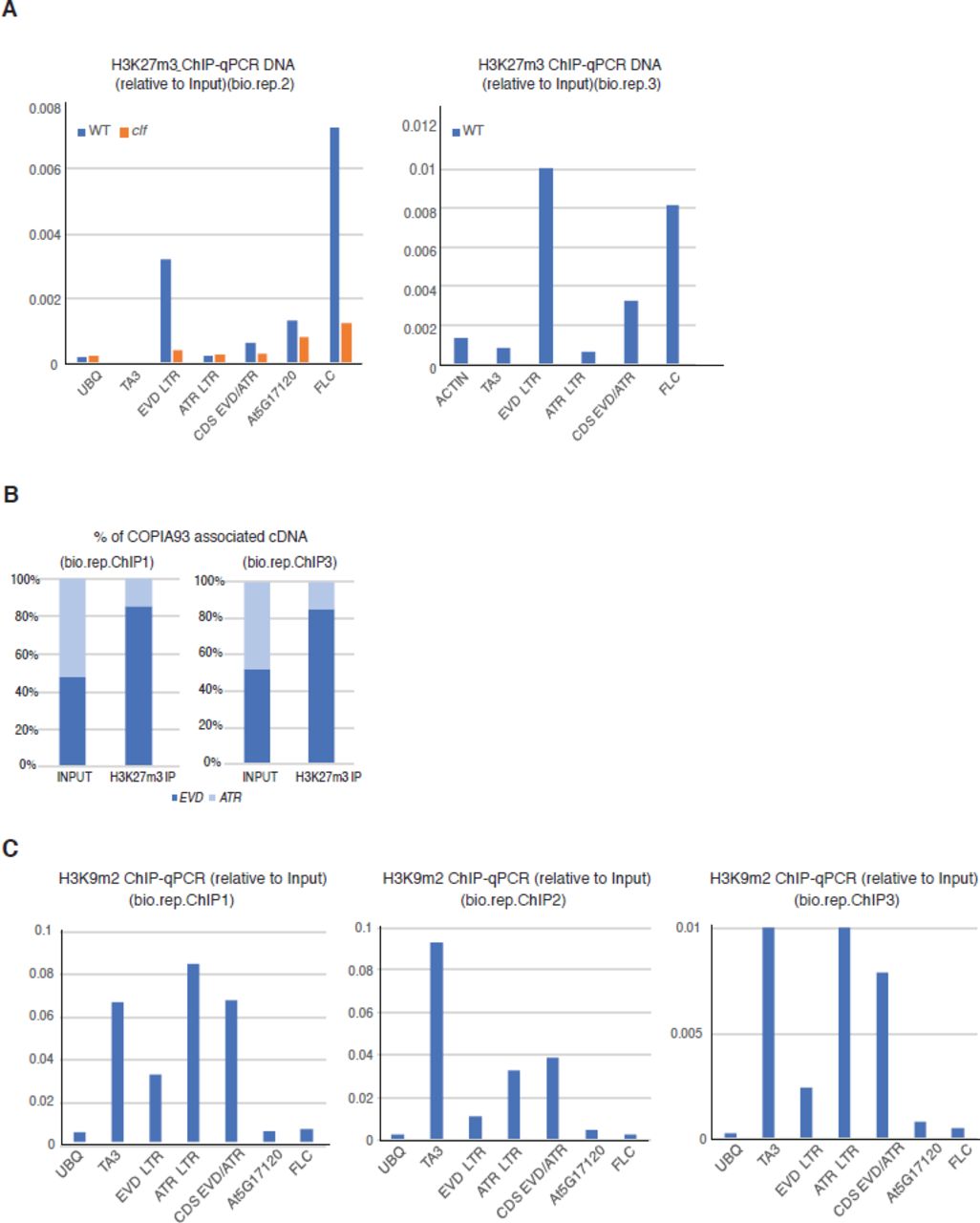
Given previous observations in plants and mammals that some loci gain H3K27m3 marks upon their loss of DNA methylation (Mathieu et al, 2005; Deleris et al, 2012; Weinhofer et al, 2010; Reddington et al, 2013; Saksouk et al, 2014; Basenko et al, 2015), presumably mediating “back-up” transcriptional silencing of hypomethylated sequences, we thought that there could be an increase in H3K27m3 marks at ATCOPIA93-LTR in ddm1 plants. To test this possibility, we inspected publically available ChIP-chip datasets and found that ATCOPIA93 is marked by H3K27m3, not only in an hypomethylated mutant met1, but also unexpectedly in wild-type plants (Fig 3A). However, one limit of ChIP-chip and ChiP-seq datasets is that they do not allow precise determination of the genomic localization of immunoprecipitated repeated sequences, either because of cross-hybridization (ChIP-chip) or the impossibility to accurately map multiple repeated reads (ChIP-seq). To circumvent this problem, we designed specific qPCR primers to discriminate EVD-LTR from ATR-LTR sequences after ChIP by using upstream genomic sequences. In addition, we took advantage of the rare SNPs between EVD and ATR and used pyrosequencing to analyze the immunoprecipitated fragments from the ATCOPIA93 coding sequence (CDS), where specific primer design is impossible. Interestingly, with both these approaches, we found that there was a strong bias towards EVD molecules in the H3K27m3-IPs (Fig 3B, Fig EV3A and Fig 3C, Fig EV3B). This could be possibly due to a positional effect, as the EVD sequence is embedded in a larger domain of H3K27m3 that comprises seven adjacent genes (Fig 3A) (At5g17080 to At5g17140, coding for either cysteine-proteinases or cystatin-domain proteins). Nevertheless, there was less H3K27m3 in the CDS than in the LTR region (Fig 3B, Fig EV3A); this likely reflects the previously observed antagonism of DNA methylation/H3K9m2 and H3K27m3 (Mathieu et al, 2005; Deleris et al, 2012; Weinhofer et al, 2010) since DNA/H3K9 methylation levels are higher in the coding sequence of EVD than in the LTR (Fig EV3C)(Marí-Ordóñez et al, 2013). Finally, we found that H3K27m3 levels were strongly reduced in clf plants mutated for the Polycomb-Repressive Complex 2 (PRC2) H3K27 methylase CURLY LEAF (Förderer et al, 2016)(Fig 3B).

A. Additional biological replicates for H3K27m3 analysis at ATCOPIA93 by ChIP-qPCR. The ChIPs were performed with different amounts of starting material for each batch (bio.rep.) which contributes to explain the differences in ChIP efficiency. Left panel: biological replicate for loss of H3K27m3 marks in clf; right panel: additional biological replicate that was used for pyrosequencing of H3K27m3-immunoprecipitated DNA in wild-type plants.
B. Detail of pyrosequencing replicates on H3K27m3 ChIP-DNA at ATCOPIA93 CDS.
C. Analysis of H3K9m2 marks at ATCOPIA93 EVD and ATR by ChIP in rosette leaves, followed by qPCR. Data were normalized to the Input DNA. Loci tested are as in Fig 3B. Because of variability in the ChIP efficiency, the three biological replicates are shown separately.

A. IGB (Integrative Genome Browser) views showing H3K9m2 levels and H3K27m3 levels in WT and met1 rosette leaves, at ATCOPIA93 EVD and ATR (ChIP-chip public data, Deleris et al., 2012). Yellow horizontal bars: protein-coding genes; horizontal blue bars: transposable elements. The LTRs are delineated by pink bars. Vertical light blue bars: H3K9m2 signal relative to H3 (two top lanes) and H3K27m3 signal relative to H3 for each probe.
B. Analysis of H3K27m3 marks at ATCOPIA93 EVD and ATR by ChIP on rosette leaves, followed by qPCR, in wild-type plants and in clf plants mutated for the H3K27 methylase CURLY LEAF. Data were normalized to the input DNA. ATCOPIA93 CDS is a region in ATCOPIA93 GAG common to EVD and ATR. At5g17120 is a region in the protein-coding gene located upstream of EVD. FLC is a region located in the first intron of FLOWERING LOCUS C which shows high levels of H3K27m3 in vegetative tissues and serves as a positive control. TA3 is a transposon and serves as a negative control. Because of technical variability in the ChIP effciency, one ChIP experiment is presented here and two other independent experiments are presented in Figure EV3. ChIPs were performed on a pool of rosette leaves from eight to ten plants/genotype.
C. Genomic distribution of H3K27m3 marks between EVD and ATR loci by ChIP-PCR pyrosequencing. Upper panel: depiction of the pyrosequenced region (in yellow) within the GAG biotinylated qPCR amplicon obtained after H3K27m3 ChIP-qPCR and purification with streptavidin beads. The position interrogated corresponds to the discriminating SNP between EVD (A/T) and ATR (C/G). The % indicated represents the % of T (EVD, in black bar) or G (ATR, grey bar) at that position. The sequencing primer was designed so that other ATCOPIA93-derived sequences (divergent and non functional) such as At4G04410 and AT1G43775 cannot be amplified and so that the allelic ratio between the two active ATCOPIA93 copies EVD and ATR only can be evaluated. To verify this, the qPCR GAG product is also amplified from the Input gDNA as a control where a 50%-50% ratio is expected. For clarity, an average of two experiments performed on two independent Input and ChIPs samples is shown and individual datasets presented in Figure EV3B.
D. Methylation status of the DNA captured with H3K27m3 by Sau96I Chop-qPCR. H3K27m3 ChIP-DNA from two independent ChIPs was digested with the methylation-sensitive restriction endonuclease Sau96I which is sensitive to the methylation of the second C at the GGGCCG site in the LTR (as in Fig EV1) in two independent. Digested DNA was quantified by using qPCR with primers specific for a region of EVD ATCOPIA93 LTR spanning this Sau96I restriction site, and the signal was normalized to an undigested control. The values from two independent experiments are plotted showing that the WT ChIP-DNA had significantly less digestion compared with the ddm1 control, thus there was more methylation.
Next, we assessed whether H3K27m3 and DNA methylation could co-exist on the same molecules or whether the detection of both marks in wild type rosette leaves was only reflecting the contribution of different cell types, some marked by H3K27m3 at EVD and some by DNA methylation. To distinguish between these two possibilities, we analyzed the DNA methylation status of one representative CG site at the LTR region of EVD using a methylation sensitive enzyme assay on H3K27m3-IPed DNA, followed by qPCR (Fig EV1B, bottom panel). We observed amplification of the enzyme-treated DNA, comparable to the total input genomic DNA (Fig 3D). Based on this result we can conclude that the EVD DNA associated with H3K27m3 is methylated and that DNA methylation does not inhibit H3K27m3 deposition in this region.
Polycomb-group proteins and DNA methylation exert differential negative control on ATCOPIA93 induction during PAMP-triggered immunity
The co-existence of DNA methylation and H3K27m3 at EVD-LTR suggests that there is dual control by both PcG- and DNA methylation- mediated silencing on the same molecule, in the same cell type. To test for the functional relevance of PcG silencing at ATCOPIA93-EVD, we challenged wild-type and clf mutant plants with PtoΔ28E and monitored ATCOPIA93 transcript levels by RT-qPCR analyses, using ddm1 mutant plants as a positive control. Results from these analyses revealed a variable but consistent reactivation of ATCOPIA93 expression in bacteria-elicited clf plants, though generally weaker than in elicited ddm1 plants (Fig 4A and 4B). These data indicate that both DNA methylation and PcG negatively regulate the transcriptional activation of ATCOPIA93 during plant defense. Furthermore, by pyrosequencing ATCOPIA93 cDNA in bacteria-elicited plants, we observed that while both EVD and ATR are induced in ddm1, EVD almost exclusively is reactivated in clf-elicited mutant background (Fig 4C, Fig EV4A). This is consistent with EVD exhibiting comparatively stronger H3K27m3 enrichment than ATR in wild-type (Fig 3). The pyrosequencing result also presumably explains, at least partly, the weaker ATCOPIA93 induction observed in clf compared to ddm1 after bacterial challenge (Fig 4A, 4B), as the mRNA is almost only contributed by EVD in clf (Fig 4C). In addition, as anticipated, we observed that in clf mutants, where H3K27m3 is reduced, H3K9m2 marks were retained to levels comparable to WT at EVD (Fig EV4B, top panel); this likely contributes to explain lower accumulation of EVD transcripts in clf than ddm1 (Fig 4D). In ddm1 mutants, where H3K9m2 is reduced, H3K27m3 marks were also retained to levels comparable to WT at EVD (Fig EV4B, lower panel) suggesting that H3K9m2/DNA methylation may exert a stronger repressive effect on EVD than polycomb group proteins.

A. Detail of the pyrosequencing biological replicates for ATCOPIA93 cDNA analysis. Each color corresponds to each biological replicate shown in Fig 4A.
B. Analysis of H3K9m2 and H3K27m3 marks in clf and ddm1 at ATCOPIA93 EVD and ATR by ChIP in rosette leaves, followed by qPCR. Data were normalized to the Input DNA. Two biological replicates are presented. ChIPs were performed in parallel in WT, ddm1 and clf samples. The analysis of the WT ChIP DNA is already presented in Fig 3B and Fig EV3C (bio.rep.1 and 2) for comparisons between LTR and CDS, and between EVD and ATR. Here, the ddm1 and clf data were included to show that H3K9m2 marks persist in clf mutant (and, as expected, are almost absent in the ddm1 negative control) and H3K27m3 marks persist in ddm1 (and are strongly reduced in clf as already shown in Fig 3B).

A. ATCOPIA93 mRNA levels in pools of rosette leaves of DNA methylation mutant ddm1 and PRC2 mutant clf (three to four plants per condition), 6 hours post-infiltration with either water or PtoΔ28E bacteria at 2.108 cfu/ml. Values were determined by RT-qPCR and are relative to the expression of the UBIQUITIN (At2g36060) gene. Three independent experiments were performed and the three corresponding biological replicates are shown and represented by black, blue and green symbols respectively. The average is represented by a black horizontal bar.
B. ATCOPIA93 mRNA levels in seedlings of ddm1 and clf mutants. About thirty to forty of three week-old seedlings (grown in plates then transferred to liquid medium) were vacuum-infiltrated with either water or a suspension of PtoΔ28E bacteria at 2.108 cfu/ml and collected two hours later (this time-point was determined on the basis of LTR::GUS expression in a pilote experiment). Less variability is observed at this stage as shown by the two biological replicates (black and grey symbols).
C. Qualitative analysis by pyrosequencing of the RT-qPCR products quantified in 4A. The pyrosequenced region and SNP interrogated are the same as in Fig 3. For clarity, the average of three experiments on the three biological replicates (4A) is shown; the independent replicates are shown individually in Figure EV4 with the corresponding color code.
D. Determination of EVD and ATR transcripts levels 6hpi with PtoΔ28E by integrating ATCOPIA93 total transcript levels (4A) with pyrosequencing data. Calculations were made by applying the average respective ratios of EVD and ATR (4C) to the average RNA values (relative to UBIQUITIN) of the three biological replicates shown in 4A. Pyrosequencing could not be performed in wild type because of too low amount of ATCOPIA93 transcript thus the EVD/ATR ratio could not be determined and an intermediate grey color is used.
Cis-regulation of the RPP4 disease resistance gene by a ATCOPIA93-derived, unmethylated soloLTR
The corollary of our findings on ATCOPIA93 regulation is that the presence of a ATCOPIA93 LTR in the genome, if deprived of DNA methylation and H3K27m3, can potentially lead to the transcription of downstream sequences, thus potentially affecting the transcription of adjacent genes. We found through a BLAST search, three new ATCOPIA93-derived sequences in the genome on the chromosome 3, in addition to the ones that were previously annotated on chromosomes 1, 3 and 4. Interestingly, apart from two copies on chromosomes 1 and 4, all other five sequences are present in the form of a soloLTR, which is the product of unequal recombination between the LTRs at the ends of a single retroelement (Fig EV5A). The functional W-box 1 was conserved in all of them making them potentially regulatory units responsive to PAMP-triggered immunity (Fig EV5). Interestingly, detection of transcription was detected in response to various bacterial challenges downstream of soloLTR-1, soloLTR-2 and soloLTR-5 (Fig EV5A). While soloLTR-1 and -2 are located upstream of a pseudogene and an intergenic region respectively, both of unknown function, the soloLTR-5 is embedded in the predicted promoter of the RECOGNITION OF PERONOSPORA PARASITICA 4 (RPP4) gene, less than 500 bp upstream of the annotated transcriptional start site. RPP4 is a canonical and functional disease resistance gene that belongs to the RPP5 cluster on chromosome 4, which is composed of 7 other Toll Interleukin-1 Receptor (TIR) domain-Nucleotide binding site (NBS) and Leucine-rich repeat (LRR) domain (TIR-NBS-LRR genes) (Noel, 1999). Among these genes, RPP4 was previously shown to confer race-specific resistance against the oomycete Hyaloperonospora arabidopsidis isolates Emwa1 and Emoy2 (Van Der Biezen et al, 2002). In addition, we observed that RPP4 was generally induced during PTI, either triggered by PtoΔ28E (Fig EV5B) or by various bacterial and oomycete PAMPs tested at 4 hpi (https://bar.utoronto.ca/eplant/AT4G16860, Tissue and Experiment eFP viewers, Biotic Stress Elicitors, Waese et al., 2017). Interestingly, the soloLTR-5 is completely DNA unmethylated and not marked by H3K27m3 nor H3K9m2 (Fig 5A, Ordonnez et al., 2013).

A. Summary of the characteristics of the ATCOPIA93-derived soloLTRs in Col-0 wild-type plants. chr: chromosome; (+): plus strand; (-): minus strand; mC: methylated cytosines. Methylation status at cytosines was inferred from inspection of public data at unique reads (http://neomorph.salk.edu/arabidopsis_methylomes/stressed_ath_methylomes.html); trimethylation status of H3K27 was inferred from inspection of both ChIP-chip (Deleris et al., 2012) and ChIP-seq data for unique reads (Wang et al., 2016). Transcription downstream of the soloLTRs was inspected in plants treated with various bacteria (http://neomorph.salk.edu/arabidopsis_methylomes/stressed_ath_methylomes.html)(Dowen et al., 2012); in addition, RPP4 induction was observed in PtoΔ28E-treated plants (Fig EV5B) and in response to various bacterial and oomycete elicitors (https://bar.utoronto.ca/eplant).
B. RPP4 mRNA levels in wild-type plants, at 6hpi with either water or PtoΔ28E bacteria. Two similar rosette leaves of three plants were used per condition. Values were determined by RT-qPCR and are relative to the expression of the UBIQUITIN (At2g36060) gene. Three independent biological replicates are shown.
C. Alignment between the soloLTR-5 (upstream of RPP4) and the EVD/ATR soloLTR. The W-box 1 (GGTCAA), which is conserved in both, is depicted in blue.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A. Absence of H3K27m3 and H3K9m2 marks at the soloLTR-5 (2 primers sets (1) and (2)), upstream of RPP4. qPCRs were performed on ChIP-DNA previously analysed in Fig 3B and Fig EV3C (bio.rep.1) to further validate the epigenetic status inferred from both H3K27m3 and H3K9m2 ChIP-chip (Deleris et al., 2012) and H3K27m3 ChIP-seq data for unique reads (Wang et al., 2016). UBQ: negative control for both H3K27m3 and H3K9m2 ChIPs; FLC: positive control for H3K27m3; TA3: positive control for H3K9m2.
B. Depiction of the constructs used to transform the rrp4 null mutant to assess the impact of LTR mutations on RPP4 expression. Blue large bars: exons, blue medium bars: transcribed and untranslated regions (UTRs), purple bar: soloLTR-5.
C. Box plot representing the mRNA levels of RPP4 in the presence/absence of the soloLTR-5. 33 primary transformants were analyzed for the «pRPP4::RPP4+ mock» and «pRPP4::RPP4+ PtoΔ28E» datasets and 18 primary transformants were analyzed for «pRPP4ΔLTR::RPP4+ mock» and «pRPP4ΔLTR::RPP4+ PtoΔ28E» datasets. Mock and PtoΔ28E (2.108 cfu/ml) infiltrations were performed on two leaves of each individual transformant that were collected at 6 hours post infiltration (hpi). RNA was extracted for each transformant individually, for each treatment, and analyzed by RT-qPCR to determine the RPP4 mRNA levels relative to UBIQUITIN (At2g36060) expression. The values obtained for each primary transformant were plotted. The horizontal line in the box represents the median; the edges of the box represent the 25th and 75th percentiles, the whiskers stretch out to the 10-90 percentile above and below the edges of the box; the symbols (dots) represent the outliers. Two tailed p-values were calculated by unpaired T-test with Welch’s correction.
D. Box plot representing the mRNA levels of RPP4 in the presence of the W-box1 or the mutated W-box1 (according to Fig 1E) in the soloLTR-5. 23 primary transformants were analyzed for the «pRPP4::RPP4+ mock» and «pRPP4::RPP4+ PtoΔ28E» datasets and 24 primary transformants were analyzed for «pRPP4w1::RPP4+ mock» and «pRPP4w1::RPP4+ PtoΔ28E» datasets. Analyses were as in C.
E. The oomycete PAMP NLP20 induces the same molecular responses as PtoΔ28E. Left: Representative pictures of leaves infiltrated with water (mock), 1 μM of NLP20 or effectorless bacteria PtoΔ28E at 2.108 cfu/ml as a positive control, and incubated with GUS substrate 24hpi (two leaves from three plants per treatment). This result was repeated three times. Right: GUS mRNA levels at 3hpi with NLP20 or Ptoô28E. Analyses were performed as in Fig 1.
To test whether the presence of this presumably PAMP-responsive ATCOPIA93-soloLTR has a bona fide impact on the expression of RPP4 during PTI and could be co-opted for regulatory functions, we took a loss-of-function approach. We transformed rrp4 knock-out (KO) mutants with transgenes consisting of the entire RPP4 genomic region under the control of its native promoter (~3kb upstream of the TSS, comprising the whole upstream gene unit) or under the control of the same promoter sequence with a deletion for soloLTR-5 (Fig 5B, “WT” and “ΔLTR” constructs). We further analyzed the primary transformants for RPP4 expression in response to either water or PtoΔ28E treatments. In the absence of the soloLTR-5, RPP4 expression was reduced in both mock-inoculated (Fig 5C, significant decrease between the blue boxplots) and PtoΔ28E-elicited plants (Fig 5C, significant decrease between the red boxplots); in addition, the induction of RPP4 expression upon bacterial elicitation was no longer significant in the absence of the soloLTR-5 (Fig 5C). These results provide evidence that the ATCOPIA93-derived LTR is required for both the basal expression and bacterial-stress induction of RPP4. In addition, to assess whether soloLTR-5 contributes to RPP4 induction during PtoΔ28E elicitation through the conserved W-box1 (Fig EV5C), which had a partial effect on the induction of the LTR::GUS fusion (Fig 1E), we transformed the rpp4 KO mutant plants with a construct comprised of the RPP4 gene under the control of its promoter mutated in the W-box 1 element (Fig 5B, “w1” construct). We found that RPP4 expression levels in the absence of PtoΔ28E-challenge were globally unchanged when the W-box element was mutated (Fig 5D, not significant between the blue boxplots). However, the induction of RPP4 expression was significantly reduced in transgenic plants containing the “w1” mutant version versus plants containing the “WT” transgene upon bacterial challenge (Fig 5D, significant decrease between the red boxplots) and induction no longer occurred in these “w1” transgenic plants (not significant between mock-treated and bacteria-treated “w1” plants). These results demonstrate that the LTR responsiveness to bacterial PAMPs contributes to proper induction of RPP4 and is mediated at least in part by a functional W-box element.
Finally, to assess the relevance of this layer of regulation of RPP4 during immune responses against H. arabidopsidis (to which RPP4 confers race-specific resistance), we treated LTR::GUS plants with NLP20, the active peptide of the oomycete PAMP NPP1 that was previously shown to be non-cytotoxic in planta (Oome et al, 2014). We found that the LTR::GUS fusion was similarly responsive to this oomycete PAMP (Fig 5E), showing that NLP20 induces the same ATCOPIA93-LTR regulation as PtoΔ28E, in accordance with the fact that the PTI responses induced by unrelated PAMPs largely overlap (Katagiri, 2004; Schwessinger & Zipfel, 2008; Zipfel et al, 2006).
Together, these results show that a ATCOPIA93-derived soloLTR has been co-opted during evolution to cis-regulate RPP4 expression during basal immunity.
Discussion
ATCOPIA93 has been a widely used model to study plant transposon biology and epigenetics over the last years (Tsukahara et al, 2009; Mirouze et al, 2009; Tsukahara et al, 2012; Marí-Ordóñez et al, 2013; Reinders et al, 2013; Rigal et al, 2016; Oberlin et al, 2017). However, with the exception of DNA methylation-defective mutants, the conditions required for the activation of this family, have not been fully explored. Here, we show that the ATCOPIA93 LTR, in the absence of negative epigenetic control, has the hallmarks of an immune-responsive gene promoter: i) responsiveness to unrelated PAMPs, ii) transient activation upon PAMP elicitation (like early PAMPs-induced genes), iii) suppression of transcriptional activation by bacterial effectors, iv) full dependence on biotic stress-response elements for activation (W-box cis-regulatory elements). While EVD transcripts had been so far only observed in discrete cell types in the absence of DNA methylation (Marí-Ordóñez et al, 2013), we show here that in the presence of the adequate signaling and transcription factors, it can be expressed in other tissues, such as adult leaves.
The connection between TE activation and stress response was particularly well-addressed in studies of the Tnt1 family of transposons in tobacco, which was found to be responsive to various biotic and abiotic stresses (Pouteau et al, 1991, 1994; Moreau-Mhiri et al, 1996; Mhiri et al, 1997; Grandbastien et al, 1997). Additionally, different Tnt families were induced by distinct biotic challenges and functional analyses further proved that the structural motifs present in the LTR sequences of Tnt1 families provided specific transcriptional reactivation to specific stresses (Beguiristain et al, 2001). Here, we show that one single element can be induced by PAMPs from bacterial and oomycetes pathogens; thus, in the future it will be important to determine whether the two functional W-boxes in EVD/ATR LTR are differentially involved in the induction by different pathogens, which would indicate the binding of different transcription factors to the same ATCOPIA93 LTR. Our findings in Arabidopsis, the primary model plant species for epigenetic analyses, should allow for the investigation of the poorly-understood phenomenon of permissiveness of transposon expression to certain stresses but not to others, and test whether this differential permissiveness could be epigenetically regulated, as was previously proposed for Tnt1 (Grandbastien et al, 2005).
The conditional induction of EVD is reminiscent of the heat-stress responsive element ONSEN (Ito et al, 2011; Cavrak et al, 2014) and expands the repertoire of Arabidopsis model TEs that can potentially highjack the transcriptional host machinery during stress responses. Thus, ONSEN is not a unique case, and many Arabidopsis TEs could exhibit this restricted reactivation pattern, where lack of DNA methylation will result in transposition if compounded by specific stress signaling. This would imply that the “mobilome"–the fraction of TEs with transposition activity– observed in ddm1 unstressed mutants (Tsukahara et al, 2009) is likely to be underestimated. Here, we focused on the somatic regulation of ATCOPIA93 in leaf tissues–where transpositions would not be mitotically inherited thus are difficult to detect–with the aim to test the impact of this regulation on defense gene regulation. However, future studies should address the exciting question of enhanced germinal transposition when wild type and ddm1 flowers will be subjected to PAMP elicitation or infected with pathogens.
One major difference between ATCOPIA93 and ONSEN is that ATCOPIA93 stress-induced expression in the wild type is more tightly controlled by epigenetic regulation for ATCOPIA93 than for ONSEN. Importantly, we revealed that ATCOPIA93-EVD, in addition to be subjected to DNA/H3K9 methylation, is targeted by an additional layer of epigenetic control through PcG silencing, which is generally associated with negative regulation of protein-coding genes and miRNA genes in vegetative tissues (Förderer et al, 2016). While DNA methylation and H3K27m3 marks have been described to be largely mutually exclusive, with DNA methylation generally preventing K27m3 deposition (Mathieu et al, 2005; Deleris et al, 2012; Weinhofer et al, 2010; Reddington et al, 2013; Saksouk et al, 2014), we found co-existence of these two epigenetic marks at EVD-LTR at the molecular level. This had already been observed in the endosperm at the pericentromeric GYPSY elements (Moreno-Romero et al, 2016) as well as in mammals where lower densities of CG methylation were found to allow H3K27m3 deposition (Statham et al, 2012; Brinkman et al, 2012). Accordingly, EVD LTR has only five methylated CGs (Fig 1, Marí-Ordóñez et al, 2013), which is low relative to the size of the LTR (roughly 400 bp). Thus, in Arabidopsis vegetative tissues, the co-existence of CG methylation and H3K27m3 can occur, likely constrained by CG density. As for the differential marking between EVD and ATR, it is presumably due to, or at least favored by, spreading from the neighboring genes marked by H3K27m3. This idea is supported by the almost complete loss of H3K27m3 at EVD-LTR in clf, a mutant for the PRC2 component CLF, which was recently shown to be involved in the spreading phase of H3K27m3 at the Flowering Locus C (FLC) (Yang et al, 2017). Future studies should identify and delineate TEs that can potentially recruit PRC2 in cis from the ones that become H3K27m3-marked due to an insertional effect, and the ones that exhibit both characteristics. Importantly, we showed that PcG silencing is functional at EVD ATCOPIA93, the modest effect of H3K27m3 loss on total ATCOPIA93 expression presumably due to the specific EVD control by PcG and the functional redundancy of H3K27m3 with DNA methylation, which co-exists at this site. The role of PcG in TE silencing is actually supported by the recent evidence of negative regulators of PcG, such as ALP1, which is encoded by a domesticated transposase (Liang et al, 2015) and which is likely evolved to protect TEs from Polycomb-mediated silencing. Interestingly, this negative control of EVD by PcG adds an additional epigenetic layer of restriction specifically at the functional ATCOPIA93 member (which is also less DNA methylated than its pericentromeric counterpart ATR), and presumably limits its somatic transposition while the corresponding soloLTR in the RPP4 promoter is activated for proper regulation during immune response. Notably, this double and differential mC/H3K27m3 marking could allow for unique members of one TE family to be differentially regulated, in particular at discrete stages of development since DNA methylation and PcG are not equivalent in their lability; this might provide the family members with different and discrete windows of opportunities to be expressed and transpose in some restricted tissues, and account for a not-yet appreciated strategy of TEs to adapt to their host.
Interestingly, the ATCOPIA93-LTR::GUS fusion was consistently found to be unmethylated in various transgenic lines in the wild-type Col-0 background, concordant with LTR::GUS reactivation in response to PtoΔ28E. This lack of de novo methylation upon transformation may be explained by weak LTR transcriptional activity in untreated plants thus preventing expression-dependent RNA-directed DNA Methylation (Fultz & Slotkin, 2017) and/or by low levels of CHH methylation/siRNAs at ATCOPIA93 (Fig 1A) (Mirouze et al, 2009; Marí-Ordóñez et al, 2013)), thus preventing identity-based silencing in trans (Fultz & Slotkin, 2017). Similarly, in wild type plants, the ATCOPIA93-soloLTRs appear usually to be unmethylated (Fig EV5A), in particular the soloLTR-5 described in detail here. This absence of methylation allowed us to test for a role of the ATCOPIA93 LTR as a “fully competent” transcriptional module in immunity, i.e., not masked by DNA methylation. This is a different role from the one previously described as an epigenetic module, interfering negatively with downstream expression, when the LTR was artificially methylated in trans by siRNAs produced by EVD after a burst of transposition in specific epiRIL lines (Marí-Ordóñez et al, 2013). The latter results may provide an explanation for the peculiar epigenetic control of EVD, which is almost exclusively controlled by CG methylation (Fig1A, Mirouze et al, 2009), although it belongs to an evolutionary young family of TEs: the preferred targets of POLYMERASE V, siRNAs and the RNA-directed DNA methylation (RdDM) pathway (Zhong et al, 2012). We propose that the low levels of EVD LTR siRNAs, which could methylate the soloLTR-5 in trans if present in larger quantities, could be the result of evolutionarily selection, so that soloLTR-5 remains unmethylated and proper immune response can be properly activated.
Transposable elements have been proposed to contribute not only to the diversification of disease resistance genes, which are among the fastest evolving genes, but also, following their diversification, to the evolution of their cis-regulation, as part of their maturation process (Lai & Eulgem, 2017). Compelling evidence exists for the latter role (Hayashi & Yoshida, 2009; Tsuchiya & Eulgem, 2013; Deng et al, 2017; Lai & Eulgem, 2017). In the present study, we have brought another demonstration for the cis-regulatory role of TEs, and for the first time we have linked the co-option of a soloLTR for proper expression of a functional disease resistance gene (RPP4) and the responsiveness of the corresponding full-length retroelement (COPIA93 EVD/ATR), through its LTR, during basal immunity. TEs have been long thought to be a motor of adaptive genetic changes in response to stress (McClintock, 1984). The link we established between responsiveness of a retroelement to biotic stress and its co-option for regulation of immunity provides experimental support to McClintock’s early model that TEs play a role in the genome response to environmental cues. Future studies should address the extent of the TE repertoire with such restricted expression to adapt to their host, in the light of recent findings showing that not only do disease resistance gene clusters contain many TEs, but also they are among the most frequent transposition targets observed (Quadrana et al, 2016).
Materials&Methods
Plant material and growth condition
Plants were grown at 22°C with an 8h light/16h dark photoperiod (short days) and experiments were generally performed on 4.5 to 5-week-old rosette leaves. Apart from Figure 3, where plants were analyzed in the absence of treatment, plants were infiltrated with a syringe with either water (“mock”), synthetic flg22 (Genescript) at 1µM concentration or a suspension of bacteria as described below, always at the same time in the morning (between 10 and 11.30am, depending on the number of plants to infiltrate). Plants were then covered with a clear plastic dome until tissue harvest to allow high humidity (Xin et al, 2016). For Figure 4B, 3.5-week-old seedlings grown on MS plates were transferred to liquid MS for at least 24h then infiltrated with either water or a suspension of bacteria and put back to light for 2 hours.
Mutant lines
We used the met1-3 allele (Saze et al, 2003), the ddm1-2 allele (Vongs et al, 1993) and the clf-29 allele (Bouveret, 2006). For Figures 1 and 2, first generation homozygous met1-3 and ddm1-2 mutants were genotyped and used for analysis; for Figure 4, second generation ddm1-2 homozygous mutants were used.
Generation of transgenic lines
- LTR::GUS transgenic lines
EVD-LTR was cloned into a pENTR/D-TOPO vector, then in a pBGWFS7 binary vector, upstream of the GUS sequence. LTR::GUS constructs were transformed in the Col0 accession by standard Agrotransformation protocol (Clough & Bent, 1998). Primary transformants were selected with Basta herbicide. Four lines were selected on the basis of 3:1 segregation of the transgene (single insertion) and brought to T3 generation (#2, #12, #4, #6) where the transgene was in a homozygous state and all four lines behave similarly as for GUS expression. Most experiments were performed on stable T3 lines (#12) homozygotes for the LTR::GUS transgene, and some in their progeny (T4) after checking that the absence of DNA methylation persisted. The mutations in W-boxes 1 and 2 were introduced in pENTR vector by overlapping PCR and the mutated LTRs cloned in pBGWFS7. Experiments were performed on individual primary transformants for WT and mutated constructs. Transgenic plants were sequenced to verify the presence of the mutations at the LTR transgene.
- pRPP4::RPP4 transgenic lines
For the “WT” construct, a 3kb sequence upstream of RPP4 predicted TSS was cloned in pENTRD-TOPO; for the “ΔLTR” construct, the same 3kb sequence minus the soloLTR-5 was synthesized and cloned in the same vector; for the “w1” construct, site-directed mutagenesis was used on the “WT” pENTRD-TOPO. A fragment corresponding to the RPP4 gDNA (with introns and UTRs) was then amplified from wild-type plants and cloned after the RPP4 promoter in the three different “WT”, “ΔLTR” and “w1” pENTRD-TOPO vectors using restriction enzymes. The resulting vectors were recombined with pH7WG. Primary transformants were selected on hygromycin and analyzed individually.
Bacterial strains and preparation of inocula
The bacterial strains used are Pseudomonas syringae pv tomato Pto DC3000 (“Pto”) and a non-pathogenic derivative of Pseudomonas syringae pv tomato Pto DC3000 in which 28 out of 36 effectors are deleted (Cunnac et al, 2011), referred here as “PtoΔ28E”. Bacteria were first grown on standard NYGA solid medium at 28°C with appropriate selection, then overnight on standard NYGB liquid medium. Bacteria were pelleted and washed with water twice. Suspensions of 2.108cfu/ml were used for PtoΔ28E except for Figure 1C where suspensions of 1.107cfu/ml were used to compare to a same inoculum of Pto DC3000.
Histochemical GUS staining
GUS staining was performed as in (Yu et al, 2013). Briefly, leaves were placed in microplates containing a GUS staining buffer, vacuum infiltrated three times during 15 min, and incubated overnight at 37 °C. Leaves were subsequently washed several times in 70% ethanol.
SDS-PAGE and Western blotting
Leaf total protein extracts were obtained by using the Tanaka method, quantified by standard BCA assay and 100 µg were resolved on SDS/PAGE. After electroblotting the proteins on a Polyvinylidene difluoride (PVDF) membrane, GUS protein analysis was performed using an antibody against the GFP since pBGWFS7 contains a GUS-GFP fusion and the anti-GFP antibody (Clontech #632380) was more specific, and stained with a standard coomassie solution to control for equal loading.
DNA extraction and bisulfite conversion
DNA extraction and bisulfite conversion was performed as in Yu et al, 2013 except that the DNA was not sonicated before bisulfite conversion and 17 to 22 clones were analyzed per experiment.
RNA extraction and qRT-PCR analyses
Total RNA was extracted using RNeasy Plant Mini kit (Qiagen or Macherey-Nagel). One µg of DNA-free RNA was reverse transcribed using qScript cDNA Supermix (Quanta Biosciences) and either oligo(dT) and random hexamers mix or a transcript-specific primer for GUS mRNA analysis. cDNA was then amplified in RT q-PCR reactions using Takyon SYBR Green Supermix (Eurogentec) and transcript-specific primers on a Roche Light Cycler 480 thermocycler. For each biological replicate, two or three technical replicates were averaged when the qPCR corresponding values were within 0.5 cycles. Expression was normalized to UBIQUITIN (At2g36060) expression. In addition, for Figure 4A, two reverse-transcription reactions were performed for each biological replicate – in particular, in order to obtain enough cDNA for pyrosequencing- and qPCRs technical replicates averaged. The PCR parameters are: 1 cycle of 10 minutes at 95°C, 45 cycles of 10 s at 95°C, 40 s at 60°C.
Chromatin immunoprecipitation and ChIP-qPCR analyses
ChIPs were performed as in (Bernatavichute et al, 2008), starting with 0.3 g to 1 g (per ChIP) of adult leaves that were previously crosslinked by vacuum-infiltration of a 1% formaldehyde solution. Antibodies against H3K27m3 and H3K9m2 are from MILLIPORE (07-449) and ABCAM (ab1220) respectively. 2 µl of a 1:10 dilution of the IP was used for qPCR. The PCR parameters are: 1 cycle of 10 minutes at 95°C, 45 cycles of 10 s at 95°C, 40 s at 60°C.
Methylation-sensitive enzyme assay (“Chop-assay”)
Two hundred ng of gDNA (Fig EV1) or 10 to 20 ng of ChIP-DNA (10 ng for Input DNA) (Fig 3) was digested overnight at 37C with 1 µL or 0,5 µl respectively of Sau96I enzyme (Thermoscientist FD0194). As Sau96I cannot be heat-inactivated, DNA was then purified with a clean-up column (Macherey Nagel nucleospin column) (Fig 1A) or, when the amount of material was limited (Fig 3) by standard phenol-chloroform extraction using glycogen to precipitate the DNA. DNA was eluted in 20 µl of water or pellets were resuspended in 20 µl of water; qPCR were performed using 0,3ul and primers that amplify an amplicon spanning the Sau96I site. The same amount of the corresponding non-digested DNA was used for qPCR as a control and to normalize the data.
Pyrosequencing
ATCOPIA93 DNA (ChIP-DNA, cDNA or gDNA as a control) was amplified with a biotinylated (forward) primer in the same region where RNA levels were analyzed and containing a SNP between EVD and ATR; the biotinylated PCR product (40 µl reaction) was pulled down with streptavidin beads (sigma GE17-5113-01) and the sense biotinylated strand sequenced with a Pyromark Q24 (Qiagen) on the sequencing mode. Input DNA was used as a control for equal contribution of each SNP. Analysis and quality check of the peaks were done with the Pyromark Q24 companion software which delivers pyrograms indicating the % of each nucleotide at the interrogated SNP. These percentages were directly plotted for each biological replicate in the Extended Views and averaged for clarity of presentation in the main figures.
Authors contributions
A.D., J.Z., A.Y., J.W. performed the experiments, A.D. and J.Z. analyzed the data; J.D. performed bioinformatic searches; A.D, J.Z. and L.N. designed the experiments; A.D. wrote the manuscript.
Acknowledgments
We thank the members of the Navarro Lab for their input and discussions as well as the Bourc’his and Felix Labs for their help with pyrosequencing, D. Bouyer and L. Quadrana for discussions, H. Keller for valuable comments, M. Greenberg and M. Boccara for critical reading of the manuscript and discussions.
Funding source: This work was funded by an ANR-retour post doc (ANR-11-PDOC-0007-01) granted to A.D. and a Human Frontier Scientific Program Career Development Award (HFSPCDA-00018/2014) granted to A.D.
All authors have seen and approved the manuscript.
References




