

Abstract
Sepsis is associated with significant mortality that persists despite advances in the care of critically ill patients. Concomitant development of acute kidney injury (AKI) markedly increases the mortality of sepsis through unclear mechanisms. While electrolyte disturbances and toxic metabolite buildup likely play a crucial role, loss of a protective molecule(s) from the injured kidney could also contribute to the dire outcomes observed in sepsis with AKI. Uromodulin (Tamm-Horsfall protein, THP) is a kidney-derived protein bidirectionally released in the urine and circulation. We previously showed that AKI causes acute systemic THP deficiency. Here, we show that circulating THP increases in experimental murine sepsis without severe AKI through basolateral shifting of its release within the kidney medulla. Concordantly, in asmall cohort of patient with sepsis and preserved kidney function, circulating THP positively correlates with the degree of critical illness, and accumulates in the lungs of a cohort of patients with ARDS. In a knockout mouse model with sepsis, we show that THP deficiency significantly increases mortality. Using single cell RNA-sequencing, we observe that THP expands a macrophage subset enriched with transcripts required for protein translation, migration and phagocytosis. Indeed, treatment of bone marrow-derived macrophages with THP enhances phagocytosis and the loss of THP in vivo causes an increases bacterial burden within organs during sepsis. Finally, treatment of septic THP-/- mice with exogenous THP improves survival. Together, these findings suggest that THP protects from sepsis by enhancing macrophage function and its loss could explain the detrimental outcomes of sepsis with AKI. Our findings also suggest a potential therapeutic role of THP in sepsis.
Introduction
Sepsis is a leading cause of death and disability worldwide (1). This devastating disease is characterized by an aberrant host response to infections, leading to downstream damage and failure of one or more organ systems (2). The kidney is particularly susceptible to sepsis, with acute kidney injury (AKI) occurring in approximately 20-30% of patients with sepsis (3). Sepsis with AKI is approximately twice as deadly as sepsis with preserved kidney function (4, 5). The underlying mechanisms driving this increase are unclear (6). While reduced ability to filter toxins is likely contributing to decreased survival, it is also possible that the kidney produces and releases a protective molecule which is lost in the setting of AKI.
Tamm-Horsfall Protein (THP or Uromodulin) is uniquely produced in the kidney, primarily by the cells of the thick ascending limb (TAL) of the loop of Henle (7, 8). The majority is secreted apically into the tubule and exists primarily as a high molecular weight polymer (9), constituting the major protein component of mammalian urine (10). Urinary THP has multiple functions, including regulation of ion channels and protection from kidney stones and urinary tract infections (11). A smaller portion of THP is secreted basolaterally from the TAL into the interstitium and circulation, existing primarily as a monomer (12). Increased circulating THP is associated with protection against chronic kidney disease (CKD), cardiovascular events and all-cause mortality in patients, which persists even after controlling for overall kidney function (6, 13, 14). Our studies of THP in a knockout mouse (THP-/-) model revealed the importance of circulating THP in protecting against kidney injury (13, 15, 16) and systemic oxidative stress (17) and in regulating the innate immune response within the kidney and systemically (12, 14). Building on that current knowledge, we hypothesize that THP plays a protective role in sepsis. Since severe AKI causes acute systemic THP deficiency in both an animal model and patients (17), we propose that increased mortality in sepsis with AKI could be partially due to the loss of a protective role of circulating THP.
In this work, we studied the role of systemic THP in sepsis. We demonstrated that circulating THP increases in a mouse model of sepsis without severe AKI. We extended these findings to a cohort of patients with sepsis without severe AKI and found that increasing circulating THP positively correlated with sustained critical illness. In a separate cohort with respiratory complications from sepsis, THP was disproportionately enriched, compared to albumin, in the bronchioalveolar lavage of injured lungs. Using a cecal ligation and puncture model of sepsis in THP-/- mice, we showed that THP deficiency significantly reduced survival, and that treatment with a monomeric form of THP increases survival. THP altered transcriptional signaling of macrophages to enhance phagocytosis and bacterial clearance, likely a key mechanism underlying THP protection in sepsis. Our results support that circulating THP is a protective molecule released by the kidney during sepsis, and its deficiency worsens the prognosis. Our findings could have therapeutic implications in the setting of sepsis with AKI.
Results
Circulating THP increases in the CLP mouse model of sepsis through a shift towards basolateral release in TAL cells within the kidney outer medulla
We used the murine cecal ligation and puncture (CLP) model, which mimics sepsis pathophysiology and does not cause early severe AKI in young mice ((18) and our data below). Circulating THP increased by 6 hours after CLP surgery (5.99 ± 1.16 ng/ml to 9.54 ± 2.49 ng/ml, p = 0.0019, Figure 1A) and remained elevated in surviving animals (14.18 ± 8.35 ng/ml, p = 0.0113, Figure 1A). To identify the source of the increased circulating THP, we measured levels of mRNA message and protein within the kidney 6 hours following surgery. When compared to sham-operated animals, CLP-operated animals did not show a significant difference in levels of the UMOD transcript which encodes the THP protein (1.06 ± 0.12 in sham versus 1.08 ± 0.16 relative transcript level in CLP animals, p = 0.8564, Figure 1B) or the protein itself (1.05 ± 0.85 in sham versus 0.95 ± 0.35 relative protein level in CLP animals, normalized to levels of actin and expressed as fold change compared to the mean, p = 0.8360, Figure 1C). Since there was no increase in total amount of THP, we then investigated whether more THP is being trafficked to the basolateral side of the TALs, which is expected to supply THP to the serum, rather than the apical side of the TAL, which supplies THP to the urine. In the inner stripe of the outer medulla, a renal compartment with the highest density of TAL tubules, we found an increase in the amount of basolaterally vs. apically located THP (0.26 ± 0.08 in sham versus 0.33 ± 0.11 basolateral THP:apical THP in CLP animals, p < 0.0001, Figure 1D). Together, these results suggest that the increased levels of circulating THP are due to a shift in trafficking of THP towards the basolateral side of the TAL within the outer medulla.

(A) Circulating THP levels increase following a mice model of sepsis (CLP) at both 6 and 48 hours after surgery
(B) UMOD relative transcript levels are not significantly different in CLP versus sham operated mice 6 hours after surgery
(C) THP relative protein levels (normalized to actin) are not significantly different in CLP versus sham operated mice 6 hours after surgery. Western Blot, left, quantitation, right.
(D) Confocal microscopy of THP localization and intensity in CLP and Sham-operated kidneys 6 hours after surgery, left. Arrowheads indicate areas of high basolateral THP staining in the inner stripe of CLP-operated animals. The inner stripe of CLP-operated animals has increased basolateral THP:apical THP mean fluorescence intensity as compared to sham-operated animals. This phenomenon is restricted to the inner stripe, right.
CLP – cecal ligation and puncture
Circulating THP increases in clinical sepsis without severe AKI and concentrates in injured organs
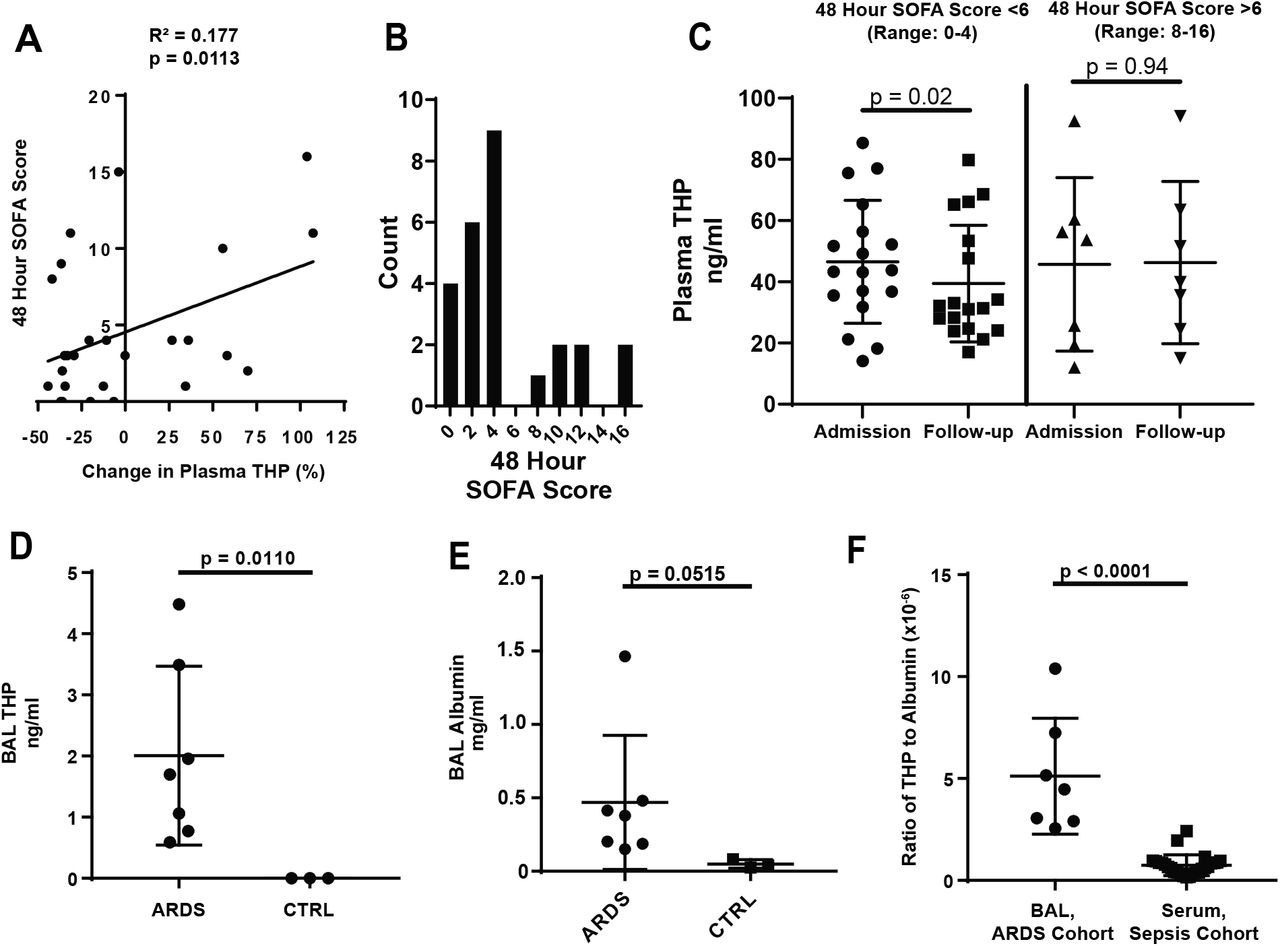
To extend these findings to the clinical setting, we prospectively enrolled a cohort of 26 patients from the Methodist Hospital Intensive Care Unit (Indianapolis) who received a diagnosis of sepsis (Table 1). Plasma and clinical parameters, used to calculate the Sequential Organ Failure Assessment (SOFA) score (19), were collected at admission and 48-hour follow-up. Circulating THP was measured in plasma samples from these time points. The percent change in THP was positively correlated with follow-up (48 hour) SOFA score (R2 = 0.177, p = 0.0113, Figure 2A). Patients were also stratified by follow-up SOFA score (Figure 2B). Those with SOFA score less than 6 had decreased plasma THP between admission and follow-up (49.3 ± 22.9 ng/ml versus 39.4 ± 19.0 ng/ml, p = 0.0200, Figure 2C). In patients with elevated (>6) follow-up scores, plasma THP remained high (45.7 ± 28.3 ng/ml versus 46.3 ± 26.5 ng/ml, p = 0.9453, Figure 2C), suggesting that when renal function is preserved (serum creatinine remained stable/improved between admission and 48-hour follow up, Table 1) the kidney reacts to sepsis by circulating high levels of THP.
Sepsis cohort characteristics at admission and follow-up
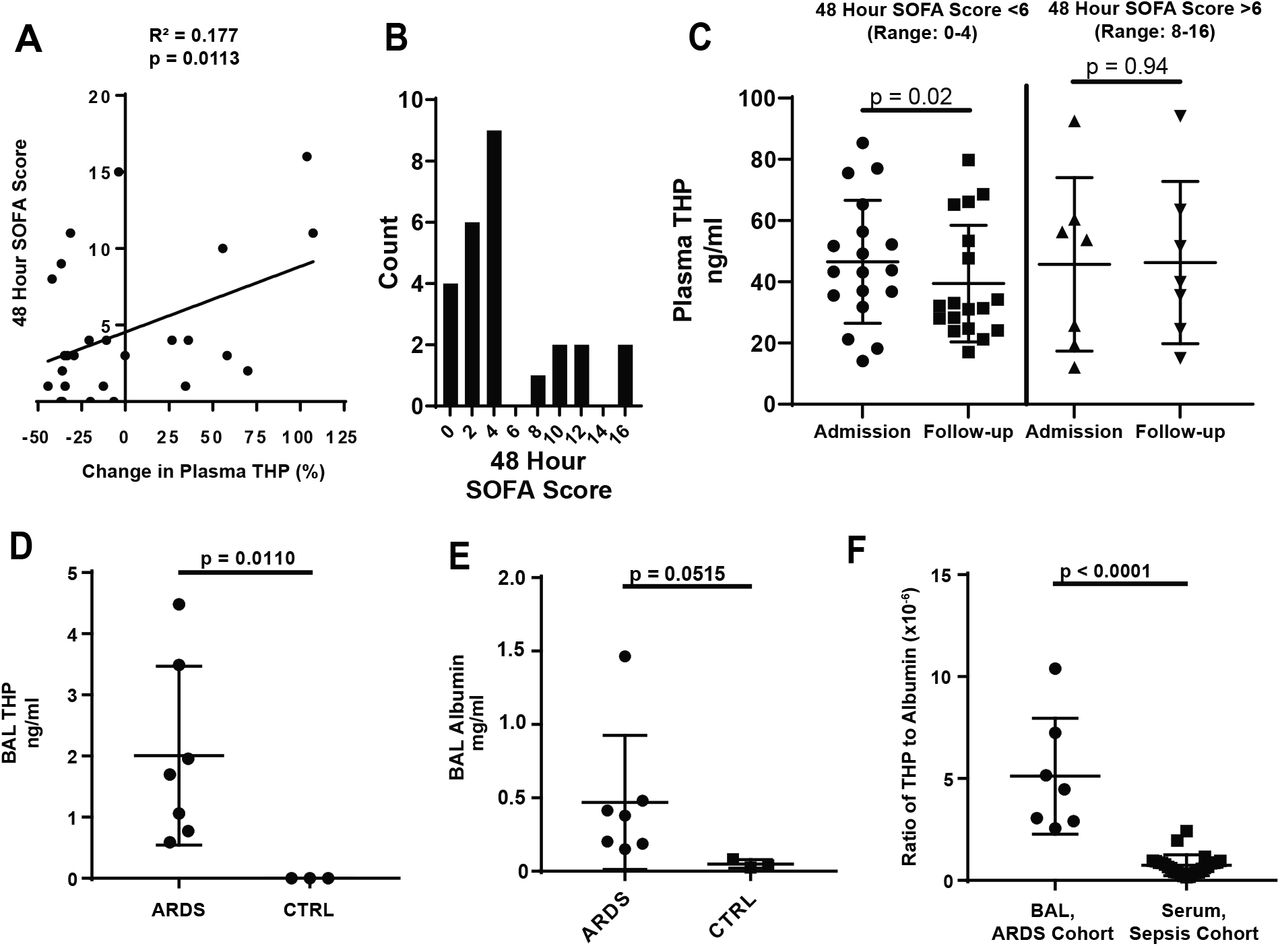
(A) In a cohort of patients admitted to the ICU with a diagnosis of sepsis, the percent increase in circulating THP from admission to 48-hour follow-up is positively correlated with 48-hour SOFA score (R2 = 0.177, p = 0.0323)
(B) Histogram of 48 hour SOFA scores shows a break between patients with low organ dysfunction (0-4) or sustained organ dysfunction (8-16)
(C) Plasma THP in patients with 48 hour SOFA score less than 6 drops significantly between admission and 48 hour follow-up, while patients with scores above 6 maintain levels of plasma THP
(D) THP is found in the BAL fluid of patients with ARDS, but not in fluid from healthy controls (CTRL)
(E) Albumin is elevated in patients with ARDS as compared to healthy controls
(F) The ratio of THP:albumin is increased in the BAL fluid of the ARDS cohort as compared to the plasma of the sepsis cohort
ICU – Intensive Care Unit, SOFA – Sequential Organ Failure Assessment, BAL – bronchoalveolar lavage, ARDS – acute respiratory distress syndrome
Since circulating THP has immunomodulatory and anti-inflammatory functions (11), we tested whether THP localized to distant injured organs in sepsis and critical illness. Acute Respiratory Distress Syndrome (ARDS) is a rapidly progressing form of lung injury that can occur secondary to sepsis. THP was assayed in bronchoalveolar lavage (BAL) samples from ARDS patients (Table 2) and healthy controls. THP was detected in BAL fluid of patients with ARDS (2.00 ± 1.46 ng/ml, Figure 2D, Table 2), but not healthy controls (p = 0.0110). In ARDS, disruption of alveolar barriers leads to interstitial edema with a protein-rich fluid (20), providing a potential mechanism for THP entry. To test this, we measured levels of human serum albumin (HSA) in these samples. As previously described (21), levels of HSA in the BAL fluid of ARDS patients increased compared to healthy controls (0.39 ± 0.40 mg/ml versus 0.05 ± 0.03 mg/ml, p = 0.0267, Figure 2E). THP and HSA levels in ARDS BAL fluid were positively correlated (Figure S1A, R2 = 0.6779, p = 0.0034), supporting that THP probably enters the lungs from the circulation following barrier dysfunction. In contrast, THP and HSA in plasma samples from the sepsis cohort were not significantly correlated (Figure S1B, R2 = 0.005668, p = 0.7147). To test whether THP disproportionately accumulates in the lungs compared to albumin, we generated a ratio of THP to HSA concentrations for the BAL samples (in the ARDS cohort) and plasma samples (in the sepsis cohort). We found the ratio of THP:HSA is higher in the BAL samples (4.56 x 10−6 ± 2.92 x 10−6) compared to plasma samples (0.75 x 10−6 ± 0.51 x 10−6), suggesting that THP is likely enriched in the lungs of ARDS patients (Figure 2F). These results support that the clinical response to sepsis without significant kidney injury is to increase levels of THP in the circulation and possibly in damaged organs.
ARDS cohort characteristics at admission and follow-up
THP protects during sepsis by enhancing macrophage function within the organs
To gain further insight about the role of THP in sepsis and interpret the changes in it systemic levels in the experimental and clinical settings described, we induced a polymicrobial bacterial sepsis in THP-/- mice using the CLP model (22) and observed that THP-/- mice had significantly increased mortality as compared to THP+/+ animals (median survival of 1 day versus 2 days, p = 0.0148, Figure 3). This finding suggests that THP is protective in the setting of sepsis. The early mortality seen in the CLP group (especially in THP-/- mice) is not likely driven by a rapidly progressing kidney injury, since kidney function early in the course of disease (6 hours after CLP) remained stable irrespective of genotype (Figure 3B).

(A) THP-/- mice have increased mortality following CLP as compared to THP+/+ mice
(B) Neither THP+/+ or THP-/- mice have significantly elevated serum creatinine 6 hours post-surgery as compared to sham-operated animals
We previously showed that THP-/- mice have decreased number and function of kidney mononuclear phagocytes (12). Therefore, we hypothesized that THP could regulate the function of macrophages within organs during sepsis, thereby explaining, in part, its protective properties. To gain unbiased insights on the potential role of THP in macrophage activation, we treated mouse bone marrow-derived macrophages (BMDM) with a non-polymerizing truncated form THP (tTHP), followed by single cell RNA sequencing and analysis. tTHP faithfully represents monomeric THP found in the circulation (12). Based on the transcriptomic signatures, BMDM were found to be a heterogenous population which can be unbiasedly sorted into 8 clusters (Figure 4A, left). Cluster 2 expands with tTHP treatment with a log-adjusted fold change of approximately 1.5 (Figure 4A, right). To predict the phenotype associated with macrophages within Cluster 2, we identified the top 15 markers for each cluster (Figure S2). Within Cluster 2, many of the enriched transcripts fell into three functional categories: protein synthesis, cell migration and phagocytosis (sample feature plot, Figure 4B, top, violin plots of expression of selected examples within each cluster in Figure 4B, bottom, (23–28)). Together with our previous findings in THP-/- mice (12), these findings suggested that THP treatment expands a cluster of macrophages that are primed for activation and phagocytosis.
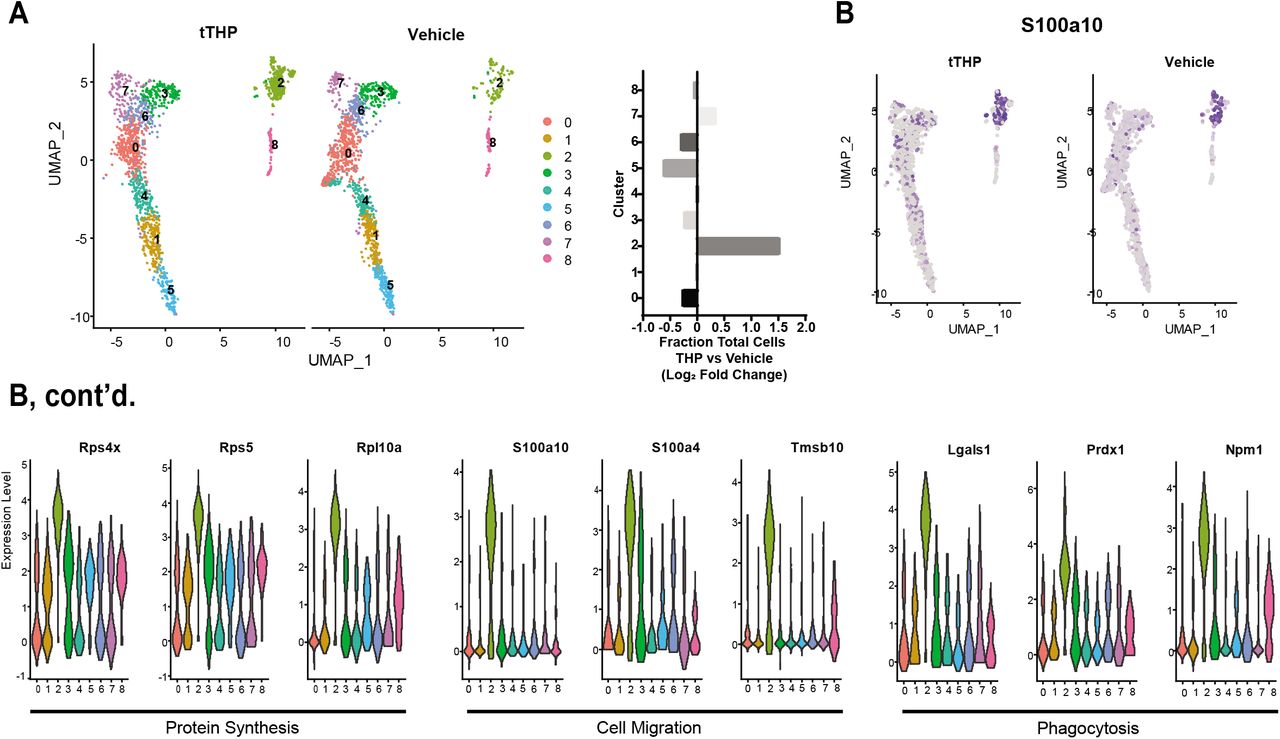
(A) Single cell RNA sequencing of tTHP and vehicle-treated mouse BMDM reveals eight distinct clusters, left. The relative proportion of cluster two increases with THP treatment, right.
(B) Markers of cluster 2 have roles in protein synthesis, bottom left, cell migration, bottom middle, and phagocytosis, bottom right. Violin plots for representative transcripts show relative expression of markers in cluster 2 compared to other clusters, bottom, and a representative feature plot shows expression of a marker gene across all cells.
tTHP = truncated THP
Since macrophages are important effectors of the innate immune response during infections, we then hypothesized that THP enhances pathogen clearance at the organ level in sepsis by stimulating their phagocytic activity. Given the high early mortality in THP-/- animals in CLP sepsis, we expected THP would impact bacterial clearance early during the course of disease. Since CLP induces a peritoneal infection, we examined bacterial burden in the peritoneal fluid and abdominal organs six hours following surgery (Figure 5A). Compared to THP+/+ mice, THP-/- mice have an increased number of colony forming units (CFU) of bacteria within the spleen (1.87 ± 5.01 versus 17.6 ± 18.6 CFU/mg tissue, p = 0.0136), liver (0.668 ± 0.805 versus 1.74 ± 0.697 CFU/mg tissue, p = 0.0352) and peritoneal fluid (3667 ± 4381 versus 25,125 ± 14,631 CFU/ml, p = 0.0003, Figure 5A). These results support the key role of THP in bacterial clearance, likely through enhancing macrophage activity. This defect in clearance, manifesting early during infection, could explain the premature mortality of THP-/- mice, and other states of THP deficiency, such as AKI (17).

(A) THP-/- mice have an increased bacterial burden in the spleen, liver and peritoneal fluid (right) as compared to THP+/+ mice 6 hours following CLP surgery. Representative blood agar plates shown (left).
(B) Treatment of BMDM with tTHP increases phagocytosis of pHrodo green fluorescently labeled BioParticles. Representative histogram of vehicle and tTHP treated macrophages, left, solid line indicates gates used for positive cells.
tTHP = truncated THP
To directly test whether THP could enhance macrophage phagocytic function, we treated BMDM with tTHP and measured uptake of fluorescently labeled BioParticles using flow cytometry. Compared to vehicle-treated macrophages, tTHP-treated macrophages have significantly increased phagocytosis (1 ± 0.279 versus 1.446 ± 0.36-fold change over vehicle, p = 0.0151, Figure 5B), establishing a direct effect of THP on phagocytic activity of macrophages.
THP treatment of THP-/- restores sepsis mortality to the levels of THP+/+ mice
We previously showed that tTHP can also be administered systemically in mice and reaches macrophages within the kidney (12). We then tested if treatment of THP-/- mice with tTHP could restore the protective systemic effects of THP and improve survival following sepsis. THP+/+ and THP-/- mice were injected intravenously with 0.5-0.7 mg/kg μg of tTHP (THP-/- only) or vehicle (THP+/+ and THP-/-) 1-2 hours after CLP surgery. Treatment of THP-/- mice with tTHP restored survival to the levels of vehicle-treated THP+/+ mice, while vehicle-treated THP-/- mice continued to show a significant increase in mortality (Figure 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
tTHP = truncated THP
Discussion
We have demonstrated that circulating THP increases in animal models of sepsis, as well as patients with sepsis with persistent critical illness. Circulating THP not only increased systemically, but also appears to accumulate in injured organs during sepsis, as evidenced by our results from the BAL of ARDS patients. To interpret the significance of these changes, we used a knockout mouse model and established that the absence of THP leads to worse prognosis, thereby confirming a beneficial role for this protein in sepsis. In our hands, and consistent with other studies, the use of the cecal ligation and puncture model in young mice did not cause severe kidney injury, at least early in the course of sepsis. Such experimental design helped us to separate, in the setting of sepsis, the effect of THP absence secondary to a genetic cause from THP deficiency that could be caused by AKI itself. Indeed, we previously showed that severe AKI causes acute systemic THP deficiency, a finding supported by other studies (29). The natural response of the kidney in the setting of sepsis is to increase the level of systemic THP, which provides systemic protection. Consequently, in the setting of sepsis and AKI, the loss of the protection conferred by THP could explain the increased observed mortality.
The shift in trafficking of THP towards the basolateral domain of TAL tubules within the inner stripe in the context of sepsis not only provides a plausible mechanism for increased circulating THP but is also consistent with the kidney’s response to other types of stress. In fact, basolaterally released THP also increases during the recovery phase of AKI caused by ischemia reperfusion injury (15) and is known to have protective pleiotropic effects (11) both in the milieu where it is produced and possibly on distant targets (17). Notably, the increase in basolateral trafficking occurs predominantly within the inner stripe of the medulla, as this region of the kidney contains the highest density of TAL tubules. We have proposed previously that the TALs act as a sensor to detect and respond to conditions which lead to damage of the more vulnerable S3 segments, which are located predominantly in the outer stripe (30), and have demonstrated that THP mediates a protective crosstalk between TALs and S3 segments in the setting of AKI (13).We expect that the basolateral trafficking of THP in the setting of sepsis is a protective measure by the kidney in response to systemic illness, consistent with what we have observed in both the CLP animal model and human sepsis patients. Taken together, these findings imply that a canonical response of the kidney facing stress is to promote basolateral release of THP in order to promote an adaptive recovery. When THP production is increased in sepsis, the kidney is contributing to the activation and priming of mononuclear phagocytes (in the kidney and other organs) required to fight infections.
Sepsis is caused by factors related to the particular invading pathogen(s) and to the status of the immune system of the host. By serving an immunomodulatory role in enhancing phagocytosis of macrophages, circulating THP controls the pathogen factors (i.e. their burden), thereby ameliorating the prognosis of recuperation and survival. However, we expect that THP’s beneficial effects in sepsis extend beyond engulfment of pathogens. Improving phagocytic function will likely also increase clean-up of damaged/dying cells and neutrophil-derived extracellular traps NETs (31), further improving survival outcomes. This vigilant cleanup of cell debris by active phagocytes is essential to recovery, as apoptotic cells contribute to multiple organ dysfunction or failure. Other previously described functions of THP might also improve survival in sepsis, including regulation of inflammatory cytokines, granulopoiesis and reactive oxygen species (12, 14, 17, 32). In our previous work we also demonstrated that interstitial THP inhibits pro-inflammatory signaling, such as neutrophil – chemokine release and granulopoiesis (14). Both of those inhibitory activities could contribute to the enhanced survival observed in septic mice. We have recently also demonstrated that THP reduces renal and systemic ROS burden via inhibition of TRPM2 channel (17) and oxidative stress is a key driver of sepsis pathology (33). THP is reported to regulate circulating and renal cytokines by acting as a cytokine trap (32), which could also affect the outcome systemic inflammatory response in sepsis. It is therefore possible that some of THP’s protective effect in sepsis extends beyond its immunomodulatory role. Although we showed that THP induces a transcriptional program for macrophage activation and directly enhances phagocytosis, further studies are needed to elucidate a potential receptor for THP on these immune cells, and this is currently an active area of investigation in our lab.
The function of THP in the lung within the setting of ARDS is unclear. Given the protective nature of systemic THP in an animal model of sepsis, it is expected that THP’s presence in the lungs would be beneficial to activate macrophages, but this cannot be inferred from the current data. Other factors are likely important modifiers of the role of THP in the lungs, such as the timing of THP measurements in the course of disease and the state of immune activation and/or exhaustion of alveolar macrophages. The small sample size, variability in the timing of BAL sampling and the lack of longterm follow-up data from this cohort limit any additional insights, but these findings are foundational for a future prospective trial where BAL samples can be assayed for THP and alveolar macrophage signaling and activity.
The successful rescue of THP-/- mice with tTHP paves the way for development of THP as a therapeutic agent in the setting of sepsis. There are several steps that are needed to move to the next level in therapeutic development. Our studies need to be independently reproduced. Additional studies are also needed to establish the pharmacokinetics and pharmacodynamics of tTHP so that the dosing strategy could be optimized in various settings and models of sepsis and AKI. Furthermore, it is still unclear if a beneficial effect will be present in pharmacological dosing when endogenous THP is still being produced. Furthermore, production of recombinant forms of THP with proven efficacy are needed to extent these studies into a path for clinical applications. Despite these outstanding challenges, findings from these studies, and previous demonstration of benefits of systemic THP in AKI recovery, provide a compelling and promising venue that deserves further study.
Our study has limitations. The sepsis cohort has a small number of patients and the findings need to be reproduced in a larger study. The measurements of THP on admission to the ICU do not inform us about baseline THP because these patients are likely to be ill before admission to the ICU. We also do not have data addressing genetic variability in the UMOD promoter, an important driver of THP production by the kidney (34). In this context, the direction of change is more relevant than absolute levels. We cannot exclude that constitutive THP deficiency in THP-/- mice could predispose them to worse outcomes in sepsis, especially since THP is needed to proper functioning of TAL cells and its deficiency could lead to salt wasting over time (35), which could lead to fluid loss and circulatory failure in sepsis (36). However, the fact that systemic THP rescue improves outcomes argues against a local effect of THP within the TAL cells and support a systemic protective effect, which is consistent with our other findings.
We did not have serum samples from the patients with ARDS to compare the level of THP in the blood and BAL fluid from the same patients. However, the comparison of THP: albumin allowed the comparison of the level of relative enrichment in BAL fluid compared to plasma. The mechanisms of how THP is concentrated in the lungs are not clear and require further investigation. Our results could be the basis of future studies of THP levels in the plasma and BAL fluid as a biomarker for sepsis and lung injury, and we anticipate that our preliminary findings will spur large studies to evaluate such novel applications.
In conclusion, we demonstrated that circulating THP is a key protective molecule released by the kidney during sepsis, and its deficiency worsens the outcomes. We propose that THP potentiates a protective crosstalk between the kidney and other organs during sepsis. This work warrants further studies to investigate the use of THP as a biomarker in critical illness, and its potential therapeutic use in sepsis with AKI or other states of THP deficiency.
Materials and Methods
Study approval
Plasma samples from sepsis patients were obtained after written informed consent was obtained, in accordance with the Declaration of Helsinki. Bronchoalveolar lavages samples from ARDS and control patients were obtained within the framework of the Indiana BioBank after written informed consent was obtained, in accordance with the Declaration of Helsinki. All mice were bred, housed, and handled in the Association for Assessment and Accreditation of Laboratory Animal Care–accredited animal facility of Indiana University School of Medicine. All animal procedures were performed in accordance with the protocol approved by the Institutional Animal Care and Use Committee at Indiana University and the Veterans Affairs.
Sepsis Cohort
Inclusion criteria for the cohort included proven or suspected infection in at least one site, organ dysfunction in one or more organ system as measured by the Sequential Organ Failure Assessment Score and two or more of the following: a core temperature above 38 or below 36°C, a heart rate greater than 90 beats per minute, a respiratory rate greater than or equal to twenty breaths per minute or PaCO2 less than or equal to 32 mmHg or use of mechanical ventilation for an acute process, and a white blood cell count greater than or equal to 12,000/ml or less than or equal to 4,000/ml or immature neutrophils greater than 10%. Patients were excluded if they were below eighteen years of age, if they were judged to be in a terminal state or if sepsis developed after a proven or suspected head injury that was clinically active at the time of admission, if more than 24 hours had passed since inclusion criteria were met, if they had stage 5 chronic kidney disease or end stage renal disease and if they were included in other experimental studies. Of 416 patients who were screened for eligibility, 41 met criteria for inclusion. Of these, 26 patients consented to participate in the trial, 6 failed to consent and 9 refused participation. After informed consent, all the patients enrolled into the study based on the criteria had approximately 100 μl of serum isolated at admission to ICU and 48 hours later. Additionally, the following data was extracted from electronic medical records at those same time to calculate the Sequential Organ Failure Assessment Score: baseline demographics, comorbid diseases, presence of CKD and state, complete blood count, basic metabolic panel, liver function test, heart rate, systolic and diastolic blood pressures, body temperature, microbiology data and site of infection, Glasgow Coma scale rating, arterial blood gas values, supplementary oxygen and ventilator settings, urine outputs, urinalysis and need for renal replacement therapy. AR
DS Cohort
Bronchoalveolar lavages (BAL) samples from ARDS patients with a clinical diagnosis of sepsis and control patients were obtained from a clinical BAL lab at Indiana University which has IRB approval to perform research on residual specimens (PI – Homer Twigg, M.D.). The lab receives clinical specimens from multiple physicians and hospitals for cellular differential analysis. Thus, the BAL technique was not standardized among subjects. ARDS samples for this study were selected based on the presence of respiratory failure, diffuse infiltrates on chest imaging, and a marked neutrophilic alveolitis on BAL analysis, and the diagnosis was adjudicated by chart review for these patients. Control subjects consisted of BAL from normal subjects participating in other studies in the PI’s lab.
Mice
Animal experiments and protocols were approved by the Indianapolis VA Animal Care and Use Committee. Age matched 8-12 week-old Tamm-Horsfall protein (THP) knockout male mice (129/SvEv THP-/-) and wild type background strain (129/SvEv THP+/+) were used (22). Cecal ligation and puncture was performed according to accepted protocols (18). In brief, mice were anesthetized with isoflurane and placed on a homeothermic table to maintain core body temperature at 37°C. The cecum was exposed via a midline incision and ligated with sutures. The ligated cecum was perforated with a 21-gauge needle. A small amount of fecal material was expressed into the abdominal cavity prior to closure of the incision. All animals received a subcutaneous analgesic (buprenorphine, 0.03 mg/mL, administered at 0.1 mg/kg) at the end of surgery and at the beginning/end of each subsequent day prior to sacrifice. Animals treated with either 0.5-0.7 mg/kg tTHP (purification described below) or vehicle (5% dextrose) were injected intravenously within 1-2 hours after surgery. For bacterial burden analysis, organs were removed aseptically and homogenates prepared in cold sterile PBS using the PreCellLys tissue homogenizer. Serially diluted homogenates were plated on blood agar and incubated for twenty-four hours at 37° C. Blood agar plates were imaged and individual colonies enumerated. Peritoneal fluid was harvested by injecting 3 ml of sterile PBS into the peritoneal cavity and removing prior to harvest. Peritoneal fluid was serially diluted and plated as described for tissue homogenates. Animals with evidence of cecal rupture were removed from the analysis.
Real time PCR
RNA extraction from mouse kidneys was done by Trizol extraction paired with Precellys homogenization (Bertin Instruments) according to the manufacturer’s protocol, and reverse transcribed as previously described (15). Real-time PCR was performed in an Applied Biosystems (AB) ViiA 7 system using TaqMan Gene Expression Assays also from AB. The following primers were used: THP: Mm00447649_m1, GAPDH: Mm99999915_g1. Cycling parameters were: 50°C for 2 min, then 95°C for 10 min followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. Expression values were normalized to GAPDH endogenous control and reported as fold change compared to control using the delta-delta CT method, according to the manufacturer’s instructions.
Western Blot
Protein extraction from mouse kidneys was done using RIPA buffer paired with Precellys homogenization (Bertin Instruments) according to the manufacturer’s protocol. Samples were separated on a 4-12% Bis-Tris Bolt Gel (Invitrogen Life Technologies) before transferring to a 0.45 μm PVDF membrane. Blots were probed with a polyclonal sheep anti-mouse THP (LifeSpan Bio, LS-C126349) and imaged using Super Signal Femto West Maximum Sensitivity Substrate (ThermoFisher Scientific). Blots were stripped using Restore Western Blot Stripping Buffer (ThermoFisher Scientific) and reprobed with mouse anti-beta actin (Millipore Sigma, MAB1501)
Tamm-Horsfall protein purification
Tamm-Horsfall protein was purified from normal human urine according to the method described by Tamm and Horsfall (10). THP was subsequently treated with 8M urea and resolved using size exclusion chromatography to recover a non-aggregated form in the range of 60-120 kiloDaltons (kDa), as we described recently (12). The purified tTHP was kept in a 5% Dextrose water solution. In all in vitro experiments, the concentration of endotoxin was < 0.02 EU/ml, measured using Limulus Amebocyte Lysate (LAL) assay.
Immuno-fluorescence confocal microscopy and 3D tissue cytometry
Immuno-fluorescence labeling with sheep anti-mouse THP (LifeSpan Bio, LS-C126349) DAPI (Sigma Aldrich, Cat #D9542) and FITC-phalloidin (Molecular Probes, Cat. #D1306 and #F432) was performed on 50 μm vibratome sections of kidneys fixed with 4% paraformaldehyde as described previously (15). Briefly, immunostaining was done at room temperature, overnight, with primary antibody in PBS + 2% BSA + 0.1 % Triton X-100 and secondary antibody + FITC-phalloidin + DAPI in the same buffer. After mounting on glass slides, stained sections were viewed under Olympus Fluoview laser-scanning confocal microscope. Images were collected under X20 and X60 magnification. Quantitation of the cellular distribution of THP fluorescence in thick ascending limb tubules (identified by presence of THP staining and absence of Oregon 488-green phalloidin staining) was performed on high power fields (3-4 fields per section, 1-2 sections per mouse, 4 mice per experimental group), using ImageJ. Apical and basolateral membrane associated THP average intensity was measured along the appropriate border in a 3 pixel (1.17 μm) spanning region of interest using a brush tool in ImageJ. The ratio of basolateral membrane associated THP: apical membrane associated THP was calculated for each tubule.
Preparation of Bone Marrow Derived Macrophages
Bone marrow derived macrophages were generated as described previously (37). Briefly, THP+/+ mice were euthanized by CO2 treatment followed by cervical dislocation. Using aseptic technique, the skin was peeled from the top of each hind leg and down over the foot. The foot was removed, and the hind legs were cut at the hip joint with scissors, leaving the femur intact. Femurs were placed in plastic dishes containing sterile PBS/1% FBS. Excess muscle was removed, and the leg bones were severed proximal to each joint. A 25-gauge needle was used to flush each bone with cold sterile wash medium (PBS/1% FBS). Cells were centrifuged for 10 min at 500 x g (room temperature). Supernatant was discarded and the cell pellet resuspended in RPMI 1640 with L-glutamine, 10% FBS and 1X pen/strep. Bone marrow cells were differentiated into macrophages by plating them at a density of of 2-4 x 105 cells/ml in RPMI 1640 with L-glutamine, 10% FBS and 1X pen/strep supplemented with 50 ng/ml M-CSF. Partial media changes were performed every three days after seeding. Macrophages were used for experiments within 7-10 days after seeding.
Phagocytosis assay
BMDM were pre-treated with 1 μg/ml tTHP (purification described above) or vehicle (5% dextrose) for two hours. They were then incubated with pHrodo Green E. coli BioParticles (Invitrogen, Cat #P35366l, diluted 1:500 from the manufacturer’s protocol) for 15 minutes. Cells were washed and harvested by treatment with Tryple for 10 minutes at 37° C and then centrifuged at 300 x g for 5 minutes at room temperature. Cells were resuspended in PBS/1% FBS and mixed with an equal volume of 1 μg/ml propidium iodide. Samples were kept on ice before being run on the Guava easyCyte flow cytometer. Control samples (unlabeled cells/no beads/no PI, cells/beads/no PI, cells/no beads/PI, beads alone) were used to determine live/dead gates and to determine the percentage of live cells which had taken up the fluorescent BioParticles.
Single cell RNA-sequencing of BMDM
BMDM were treated with 1 μg/ml tTHP (purification described above) or vehicle (5% dextrose) for eighteen hours. Cells were washed and harvested by treatment with Tryple for 10 minutes at 37° C and then centrifuged at 300 x g for 5 minutes at room temperature. Cells were counted and resuspended in ice cold PBS/2% FBS/1 mM CaCl2 prior to dead cell removal using the Easy Sep Dead Cell (Annexin V) Removal Kit, according to the manufacturer’s direction. To remove calcium salts, the cells were washed twice with PBS/2% FBS. Samples were filtered over a 40 μm filter and run on the Chromium 10X platform. Sequencing data was analyzed using the Seurat program in R Studio (38).
Measurement of human Tamm Horsfall Protein (THP)
Concentrations of human THP in human plasma, human bronchoalveloar lavage fluid and mouse serum of animals dosed with human THP were measured using an ELISA kit from Sigma Aldrich (Cat. #RAB0751) according to the manufacturer’s protocol.
Measurement of mouse Tamm Horsfall Protein (THP)
Concentrations of mouse THP in mouse serum were measured using an ELISA kit from LifeSpan Bio (Cat. # LS-F6973-1) according to the manufacturer’s protocol.
Measurement of human serum albumin (HSA)
Concentrations of human serum albumin in human plasma (from a random subset of the sepsis cohort) and bronchoalveloar lavage fluid (from both patients and controls of the ARDS cohort) were measured using an ELISA kit from Sigma Aldrich (Cat. #RAB0603) according to the manufacturer’s protocol.
Measurement of Serum Creatinine
Concentrations of mouse serum creatinine were measured by isotope dilution liquid chromatography tandem mass spectrometry by the University of Alabama O’Brien Center Bioanalytical Core facility.
Statistical Analysis
Values of each experimental group are reported as mean ± standard deviation unless otherwise indicated. All statistical tests were performed using GraphPad Prism software unless otherwise indicated. A two tailed t-test was used to examine the difference in means for continuous data. An F test was used to compare variances; in cases where the variances were significantly different (p < 0.05), a Welch’s correction was applied to the t-test. A paired t-test was used for samples from the same patient collected at different times. The Fisher’s exact test was used to determine differences between categorical variables. Simple linear regressions were used to determine relationships between two continuous variables. Statistical significance was determined at p< 0.05. Statistical outliers were defined using the Tukey’s fences methods, unless otherwise indicated. Kaplan-Meier survival curves were analyzed using Gehan-Breslow-Wilcoxon test to determine statistical significance.
Author Contributions
KL, RM, ES and TEA designed, analyzed and interpreted experiments and wrote the manuscript. TH and PD analyzed data and edited the manuscript. SD and RNM analyzed data. Transcriptomic data acquisition and analysis was performed by KL and SW. Protein biochemistry experiments were performed by KL and RM. Experiments in the tissue culture models were performed by KL. Animal studies were conducted by KL, SK, TEA, RM and TH. Collection of human cohort specimens and data was performed by CH, VG and HT.Acknowledgements: We acknowledge the Indiana Biobank for assistance obtaining the bronchoalveolar lavage samples (https://indianabiobank.org) and the University of Alabama O’Brien Core for serum creatinine measurements.
Funding
This work was supported by The National Institute of Diabetes Digestive and Kidney Disease (NIDDK: 1R01DK111651 and P30DK079312 to TME), a Veterans Affairs Merit Award to TEA, and an American Society of Nephrology Ben J. Lipps award to KL.
Footnotes
Funding: This work was supported by The National Institute of Diabetes Digestive and Kidney Disease (NIDDK: 1R01DK111651 and P30DK079312 to TME), a Veterans Affairs Merit Award to TEA, and an American Society of Nephrology Ben J. Lipps award to KL.
Conflict of Interest, TEA and RM have applied for a patent related to this work titled “Modified Tamm-Horsfall Protein and related compositions and methods of use”, publication number US 2018/0305420 A1.
Figure 6 revised.
References




