

Abstract
Staphylococcus aureus (S. aureus) is a major cause of life-threatening systemic infection in humans. To cause blood stream infections such as sepsis and endocarditis, the bacteria must overcome the host’s endothelial barrier. The serine protease-like proteins (Spls) of S. aureus are known to contribute to pneumonia and allergic airway inflammation in animal models, but their role in endothelial damage is unknown. Here we demonstrate that SplB induces proinflammatory cytokine release in primary human vascular endothelial cells (HUVECs) in vitro. Mechanistically, we show that SplB selectively cleaves and activates human proteinase-activated receptor-2 (PAR2), and induces biased signaling via Gα13, Gαi1/oB (NF-kβ), and β-arrestin-1/-2. This activation did not trigger Gαq/11-mediated calcium release nor ERK phosphorylation. Inhibition of PAR2 in HUVECs reduced the SplB-mediated cytokine release. Intravital microscopy of cremaster muscles in mice demonstrated that administration of SplB causes microvascular leakage. Genetic deletion of PAR2 in mice or neutralization of SplB with a monoclonal antibody preserved the endothelial barrier. This study identifies PAR2 as a receptor and substrate for SplB and highlights its role in mediating endothelial damage.

Introduction
Staphylococcus aureus (S. aureus) is a ubiquitous Gram-positive bacterium that transiently or persistently colonizes the human nares and skin (1, 2). S. aureus causes many diseases ranging from soft tissue infections to systemic infections such as pneumonia, endocarditis, and sepsis. Systemic S. aureus infections are often associated with high morbidity and mortality. In addition, over the decades, S. aureus has developed resistance to several antibiotics and poses a challenge to clinicians and a burden to the global health care systems (3–7).
The endothelial barrier offers the first line of defense against invading pathogens. S. aureus secretes a wide range of virulence factors, toxins, and proteases that interact with various receptors on the endothelium (8–10), enabling invasion of host tissues by disruption of endothelial barrier. S. aureus interaction with the endothelium leads to the production of proinflammatory cytokines and triggers cell death (11, 12). Pore-forming toxins, leucocidin ED (LukED) and γ-Hemolysin AB (HlgAB) induce endothelial permeability via activating the Duffy antigen receptor for chemokines (DARC), which results in organ dysfunction and death in mice (13). Cell wall components of S. aureus, peptidoglycan G and lipoteichoic acid are also potent inducers of endothelial damage (ED) (14). It is also well known that proteases from both host and pathogen contribute to barrier disruption via interacting with a four-member family of G protein-coupled receptors (GPCRs) called proteinase-activated receptors (PARs, PAR1-4) (15, 16).
PARs are widely expressed in various cell types, including endothelial cells (17–19). PARs are activated through proteolytic cleavage of the receptor N-terminus at specific sites and unmasking of a tethered ligand (20–23). Activation of PARs leads to G-protein activation, β-arrestin recruitment and triggering of intracellular cascades via effectors including extracellular signal-regulated kinase (ERK-1/2) mitogen-activated protein kinases (MAPK) and the nuclear factor-κβ (NF-κβ). This induces the release of cytokines and chemokines, thereby recruiting leukocytes to promote the innate immune response (24, 25).
PARs are also known as central players in allergy. PAR activation in epithelial cells of patients with inflamed airways promotes the induction of allergic asthma (26–30). Activation of PAR2 on mast cells induces degranulation, while eosinophils respond with superoxide production (31). PARs can be activated by various proteases like thrombin (PAR1, 3 and 4) and mast cell-derived tryptase (PAR2) (32) that are generated following injury or infection. Pathogen-derived proteases such as allergens of house dust mite (Der p 1, −3, −9) and mold Penicillium citrinum (Pen c 13) also activate PAR2 (33, 34). The activation of PAR2 on epithelial cells and macrophages by serine proteases from German cockroach feces extract also contributes to airway hypersensitivity (34).
The species S. aureus harbors a broad spectrum of extracellular virulence factors, among them several proteases, namely aureolysin, V8 protease (SspA), the staphopains A and B (ScpA and SspB), the exfoliative toxins A and B, and the serine protease-like proteins (Spls) A-F (35, 36). Extracellular proteases of S. aureus are known to play an important role in the pathogenesis (37–42). For example, the V8 protease and staphopains of S. aureus alter the integrity of the airway epithelial barrier, thereby promoting lung damage (43). Less is known about the functions of the serine protease-like proteins (Spls). Spls promoted the dissemination of S. aureus in a rabbit model of pneumonia (39), and SplA has been reported to cleave mucin-16 (44). We recently reported that the Spls have allergenic properties in humans (SplA, SplB, SplD, SplE) and mice (SplD) (37, 40). Administration of SplD to the trachea of mice induced allergic asthma, which was associated with an increased number of PAR2-positive infiltrating cells in the lungs compared to control animals (37). These studies highlight the interaction of Spls with the host immune system, though host substrates for the Spls remain elusive.
Here we studied the regulation of the endothelial barrier by S. aureus SplB. We found that SplB induces the production of proinflammatory cytokines in primary endothelial cells in vitro. Importantly we find that SplB mediated inflammation in endothelial cells is dependent on the cell surface GPCR PAR2, which we identify as a cellular protease sensor and host substrate of SplB. In mice, administration of SplB leads to microvascular damage in a PAR2 dependent manner. Our results indicate an important role for S. aureus-SplB in triggering endothelial inflammatory responses and identify PAR2 antagonisms as a potential therapeutic target in S. aureus infection-associated pathologies.
Material and methods
In vitro experiments with HUVECs
Primary human umbilical vein endothelial cells (HUVECs) were isolated by collagenase treatment as previously described (45). The investigation conforms to the principles outlined in the Declaration of Helsinki for the use of human tissues (46). Informed consent was obtained from all the mothers donating umbilical cords in accordance with the responsible Ethics Committee. Cells were maintained at 37 °C and 5% CO2 in MCDB 131 medium (Pan Biotech) supplemented with 10% fetal calf serum (FCS, Life Technologies), 1 ng/mL bovine fibroblast growth factor (bFGF), 0.1 ng/mL epidermal growth factor (EGF), 1 μg/mL hydrocortisone, 1% penicillin and streptomycin, 2 mM Glutamine, 500 μL heparin, and 2 mL endothelial cell growth supplement (ECGS). All other supplements were obtained from Gibco (Thermo Fisher Scientific). The cells were passaged 3-5 times before use. For stimulation experiments, cells were seeded into either in 12- or 24-well plates at a density of 2.5 x 105 cells/mL and serum-starved overnight in medium containing only 1% FCS. On the following day, cells were incubated in medium containing 2% FCS with or without the PAR2 negative allosteric modulator (AZ3451, Sigma Aldrich) or a NF-κβ inhibitor (Bay11-0782, Invivogen) 45 minutes before stimulation with recombinant SplB or SLIGRL-NH2 (PAR2 activating peptide, PAR2-AP, Tocris, Wiesbaden-Nordenstadt, Germany). Cell-free supernatants were collected and analyzed for cytokines by ELISA or multiplex assay (LegendPlex™) according to the manufacturer’s instructions.
Immunofluorescence
To determine the effect of SplB in vitro, we used multiorgan tissue flow (MOTiF) biochips (Microfluidic ChipShop GmbH, Jena, Germany). The composition of the model and the perfusion of the system was described elsewhere (47). Briefly, HUVECs were seeded at a density of 2.7 x 105 / cm2 on the endothelial side of the chip and grown in endothelial cell growth medium (PromoCell, Heidelberg, Germany). On the opposite chamber, 2.7 x 105 / cm2 of NCI-H441 (ATCC, Manassas, USA) were cultured in RPMI-1640 (Lonza, Cologne, Germany) supplemented with 10 % FCS. In addition, monocyte-derived macrophages were seeded on the NCI-H441 cell layer with a density of 0.9 x 105 macrophages / cm2. The alveolus-on-a-chip was stimulated with 100 μg/mL SplB for 6 h. After cutting the membranes, endothelial cell monolayer was washed with PBS (containing Ca2+ and Mg2+) and blocked with 3 % normal donkey serum (Dianova, Germany) containing 0.1 % Triton X-100 (Sigma-Aldrich, Germany). Cells were incubated over night with the primary antibodies to vascular endothelial cadherin (VE-cadherin) (BD Biosciences, Heidelberg, Germany), endothelial cadherin (E-cadherin) (BD Biosciences, Heidelberg, Germany), CD68 (BD Biosciences, Heidelberg, Germany), and intracellular adhesion molecule 1 (ICAM-1) (Invitrogen, Carlsbad, CA, USA). Following several wash steps, membranes were incubated for 30 min at RT with DAPI (Life Technologies) and the following secondary antibodies: donkey immunoglobulin (Ig)G anti-mouse IgG-Cy3, donkey IgG F(ab’) anti-rabbit IgG-AF488 and donkey anti-goat IgG-Cy5 (all obtained from Dianova, Hamburg, Germany). Images were acquired using an AXIO Observer Z1 fluorescence microscope (Carl Zeiss AG, Jena, Germany).
PAR cleavage assays with SplB
PAR-reporter cells were generated as previously described (48–50). Briefly, Chinese Hamster Ovary (CHO) cells were stably transfected with constructs expressing human PAR1, PAR2 or PAR4 tagged with an N-terminal nano luciferase (nLuc) and a C-terminal enhanced yellow fluorescent protein eYFP. Cells were routinely cultured in Ham’s F-12 Nutrient Mix with 1 mM sodium pyruvate, 100 U/mL penicillin, 100 μg/mL streptomycin, 1 mM L-glutamine, and 10% heat-inactivated fetal bovine serum (FBS, Gibco, ThermoFisher Scientific) and 600 μg/mL G418 sulfate (Invitrogen, ThermoFisher Scientific). The PAR-reporter cells were seeded in a 96-well culture plate at a density of 1 x 104 cells per well and cultured for 48 h. Cells were rinsed three times with Hanks’ Balanced Salt Solution (HBSS, Gibco ThermoFisher Scientific) containing CaCl2 and MgCl2 and incubated with 100 μL HBSS at 37 °C for 15 minutes. 50 μL of the supernatant from each well was transferred to a 96-well plate to obtain the basal luminescence. The cells were then treated with SplB (5 μg/mL) or increasing concentrations of human plasma thrombin (PAR1 and PAR4 agonist, Calbiochem-EMD Millipore) or porcine trypsin (PAR2 agonist, Type IX-S, 13000-20000 BAEE units/mg protein, Sigma Aldrich) or HBSS in a volume of 50 μL for 15 minutes at 37 °C. 50 μL of the supernatant was removed from treated wells and the luminescence values were again recorded on a plate reader (Mithras LB 940, Berthold Technologies) in the presence of the nano luciferase substrate furimazine (2 μL/mL, Promega).
Calcium signaling
Calcium mobilization after PAR activation was measured as previously described with some modifications (51). Briefly, HEK293 cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, Thermoscientific) supplemented with 1 mM sodium pyruvate, 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine, and 10% heat-inactivated FBS. Cells were seeded at a density of 2 x 104 cells per well in a poly-D-lysine coated 96-well black cell culture plate and cultured for 48 h. The cells were rinsed twice with phosphate-buffered saline solution (PBS, Invitrogen) and incubated with Fluo-4 NW calcium indicator (50 μL per well, Life Technologies) for 30 minutes at 37°C in the dark and for an additional 15 minutes at RT. Changes in intracellular fluorescence was measured as an indicator of elevated cytoplasmic Ca2+ using a microplate reader FlexStation3 (Molecular Devices). The baseline fluorescence values were recorded for 20 s before the addition of agonists and fluorescence spectra were recorded for an additional 180 s. In some experiments, cells were pre-treated with Gαq/11 inhibitor (YM-254890, 1 μM) (52) for 10 minutes at RT before treating with agonists. Calcium ionophore (A23187, 6 μM, Sigma-Aldrich) was used as a control in all experiments to establish proper dye loading and to determine the maximum response possible in each plate of cells.
G protein and β-arrestin recruitment assays
Recruitment of Gα12, 13, i1 or oB, and β-arrestin-1 or −2 was measured using bioluminescence resonance energy transfer (BRET) assay (53, 54). Briefly, to measure Gα12, 13, i1 or oB recruitment, HEK293 cells (2 x 104 cells per well) were transiently transfected using X-tremeGENE™ 9 (Sigma) with plasmids encoding eYFP tagged PAR2 (PAR2-eYFP) (50) and renilla luciferase (rluc8) tagged Gα subunits (eYFP:rluc8, 10:1 ratio) in an opaque white 96-well plate. pcDNA5/FRT/TO-Gα12/13/i1/oB-RLuc8 plasmids were a gift from Dr. Bryan Roth [Addgene plasmid: 140985 (Gα12-rluc8); 140986 (Gα13-rluc8); 140973 (Gαi1-rluc8); 140977 (GαoB-rluc8)] (55). 48 h after transfection, the culture medium was replaced with HBSS (60 μL) and coelenterazine-h substrate (10 μL, 50 μM, Nanolight Technology) was added to the plate. eYFP (530 nm) and rluc8 (470 nm) emissions were measured for 240 s on a FlexStation3 plate reader immediately after the addition of 30 μL trypsin (100 nM), SplB (5 μg/mL) or HBSS injected using the integrated automatic pipettor.
To measure β-arrestin-1 or −2 recruitment, HEK293 cells were transiently transfected using calcium phosphate with plasmids encoding PAR2-eYFP and rluc tagged β-arrestin-1 or −2 (eYFP:rluc, 10:1 ratio) for 24 h. β-arrestin-1 and −2 plasmids were a kind gift from Dr. Michel Bouvier. Cells were transferred to a white 96-well plate at a density of 2.5 x 104 cell/mL and cultured for an additional 24 h before incubation with HBSS alone or different concentrations of trypsin (10 nM) or SplB (5 μg/mL) for 20 minutes at 37 °C. The rluc substrate coelenterazine-h (5 μM) was added 10 minutes before taking the measurement. Emission of eYFP and luminescence of rluc was measured on a Mithras LB 940 plate reader (Berthold Technologies).
ERK/MAPK Western blot analysis
PAR2-knockout (KO) HEK293 cells were generated using CRISPR/Cas9 as previously described (53). Western blot analysis of ERK MAPKinase was done as previously described (53). Briefly, HEK293 or PAR2 KO HEK293 cells were seeded at a density of 4 x 105 cells/mL in a 12-well plate and serum-starved for 2 h to reduce basal levels of ERK phosphorylation. Cells were then stimulated with either SplB (5 μg/mL) or trypsin (10 nM) for 10 minutes. Control wells received only buffer (HBSS). Cells were harvested in an NP-40 based lysis buffer supplemented with protease and phosphatase inhibitor cocktail. Proteins in the cell lysates were separated on 4-12% Bis-Tris precast gels (Invitrogen) in MES buffer, blotted with primary antibody to phosphorylated ERK (pERK; p44/42 Thr202/Tyr204) or total ERK (tERK; p44/42) followed by incubation with HRP-conjugated secondary antibodies (1:1000). Chemiluminescence was measured using ECL detection reagent (GE Healthcare) on an iBright CL1000 (Invitrogen). All antibodies were obtained from Cell signaling Technology, Massachusetts, USA.
SplB cleavage site on PAR2
To determine the cleavage site(s) of SplB on PAR2, a peptide representing the hPAR2 N-terminal domain spanning the tethered ligand region, GTNRSSKGRSLIGKVDGTSHV-NH2, was synthesized using Fmoc solid-phase peptide synthesis on a peptide synthesizer (Syrowave, Biotage). The purity of the peptide was 98% as assessed by analytical reversed-phase HPLC-MS. Peptide and enzyme solutions were dissolved and diluted in 25 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES, Fisher Scientific). The peptide (100 μM in a total volume of 100 μL) was incubated with either trypsin (10 nM) or SplB (50 μg/mL) for 60 minutes at 37 °C. The reaction was terminated by adding ice-cold 0.1% trifluoroacetic acid (TFA) in H2O (100 μL) as described previously (56) and subjected to MALDI-MS analysis (AB Sciex 5800 TOF/TOF MS, UWO MALDI MS Facility, Western University). Cleavage sites (//) were identified from any additional m/z peaks (peptide fragments) observed in the mass spectrum of the enzyme-treated peptide.
Intravital microscopy of cremaster muscles
The surgical preparation of the cremaster muscle was performed as described elsewhere (57). Briefly, C57BL/6 WT or PAR2-KO mice were anesthetized with ketamine/xylazine (100 mg/kg ketamine and 10 mg/kg xylazine; i.p), and the left femoral artery was cannulated in a retrograde manner using a polyethylene-10 catheter (inner diameter 0.28 mm, Portex) for the administration of substances. The right cremaster muscle was exposed through a ventral incision of the scrotum. The muscle was opened ventrally in a relatively avascular zone and spread over the transparent pedestal of a custom-made microscopy stage. Epididymis and testicle were detached from the cremaster muscle and replaced in the abdominal cavity. The muscle was superfused with warm-buffered saline throughout the procedure. Leakage of intravenously applied FITC dextran (Sigma-Aldrich) to the perivascular tissue was analyzed in animals after intrascrotal stimulation with 10 μg of SpIB. In selected experiments, animals also received monoclonal antibodies directed against SpIB. All animal experiments were carried out in accordance with German animal protection laws and approved by local animal protection authority (Regierung von Oberbayern, Munich, Germany).
Statistical analyses
The cytokine data in Figure 1 are expressed as mean ± SD of three independent experiments. The t-test was utilized to determine statistical significance vs control, *p<0.05. The luminescence measurements in the PAR cleavage assay are presented as a percentage of the maximum response to thrombin (3 U/mL) or trypsin (100 nM). The cleavage data are presented as the mean ± SEM of at least three independent experiments performed in duplicates. Two-way ANOVA was utilized to determine statistical significance relative to the HBSS control, ***p<0.001. BRET ratio (eYFP/rluc) was calculated for each condition, and the net BRET ratios were obtained after normalizing to the buffer condition. The BRET data are represented as mean ± SEM of at least three independent experiments performed in duplicate. The Western blot analysis data are expressed as ratio pERK to tERK mean ± SEM of three independent experiments. Two-way ANOVA was utilized to determine statistical significance vs relative response obtained with HBSS in the same cell line, *p<0.05. The immunofluorescence data are expressed as mean MFI ratio of VE-Cadherin and ICAM-1 mean ± SD of three independent experiments. The t-test was utilized to determine statistical significance vs control, *p<0.05. The vascular leakage data in mice are expressed MFI of FITC mean ± SD of 4-5 animals per group. The t-test was utilized to determine statistical significance, *p<0.05 SplB vs. Par2−/−-SplB.

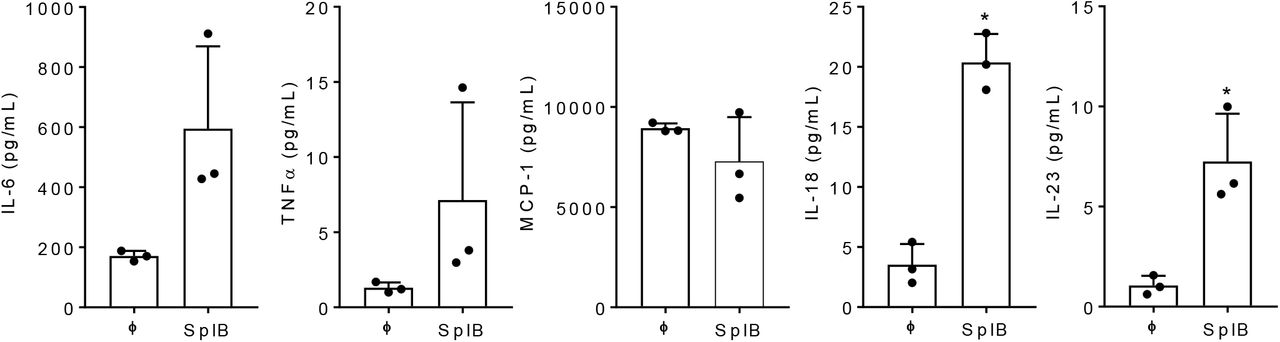
(A) Primary human endothelial cells were left untreated or treated with SplB (10 μg/mL) for 24 h and the supernatants were analyzed for indicated cytokines by cytokine bead array. Data are mean ± SD of three independent experiments performed in duplicates.
Results
SplB induces proinflammatory cytokines in primary human endothelial cells
To assess whether SplB has the ability to induce endothelial inflammation in vitro, we stimulated primary human umbilical vein endothelial cells (HUVECs) with SplB for 24 h. As shown in Figure 1, SplB significantly enhanced the interleukin (IL)-6 release and tended to increase the release of TNFα, IL-18, and IL-23.
SplB cleaves PAR2 but not PAR1 and PAR4
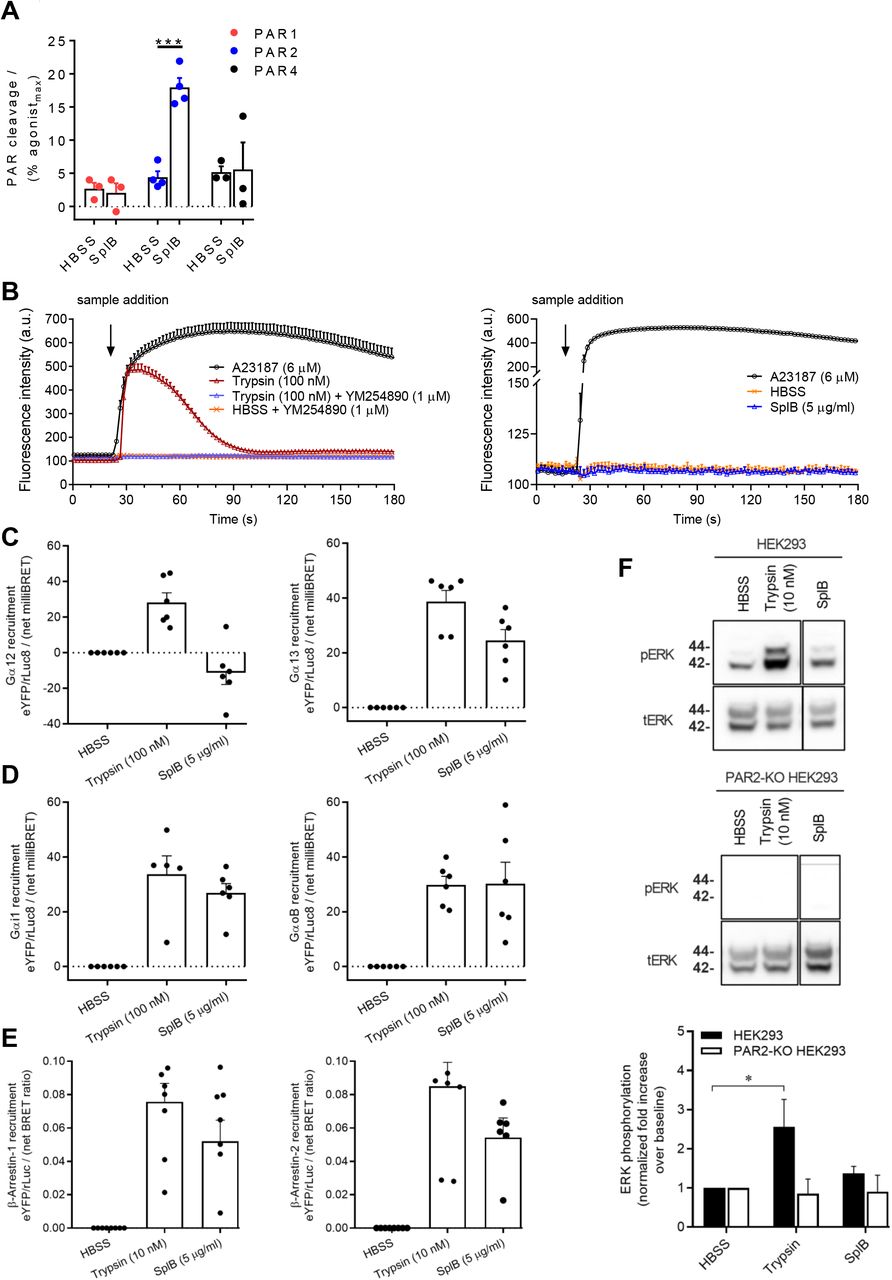
Since PARs are major targets of many bacterial proteases and mediate the production of cytokines in various host cells, including endothelial cells (58, 24), we hypothesized that SplB cleaves and activates PARs. To study this, CHO cells expressing nLuc-PAR1-eYFP, nLuc-PAR2-eYFP, or nLuc-PAR4-eYFP were treated with SplB and the cleavage of the N-terminal nLuc tag was monitored (Figure 2A). Thrombin (PAR1 and PAR4 activating enzyme) and trypsin (PAR2 agonist) were used as positive controls and cleaved the N-terminal tags over the expected range of concentrations (Supplementary Figure 1A-C). Cells treated with HBSS only served as negative controls. As shown in Figure 2A, SplB selectively cleaved PAR2 with no cleavage evident for the PAR1 and PAR4. SplB (5 μg/ml) displayed significant activity at PAR2 producing a nLuc cleavage signal that was ~20% of the maximal cleavage of PAR2 elicited by trypsin (100 nM).

(A) nLuc-hPAR1/2/4-eYFP CHO cells were left untreated (HBSS) or treated with SplB (5 μg/mL) and the PAR cleavage was measured. Data are mean ± SEM of at least three independent experiments performed in duplicates. (B) PAR2 triggered calcium mobilization was measured in HEK293 cells treated with either trypsin (100 nM) in the presence or absence of YM254890 (1 μM), or SplB (5 μg/mL) or A23187 (6 μM). Data are mean ± SEM of three independent experiments. Recruitment of (C) Gα12 and Gα13, (D) Gαi1 and GαoB and (E) β-arrestin-1 and −2, to PAR2 in response to SplB (5 μg/mL) or trypsin (10 or 100 nM). BRET ratios (eYFP/rluc) were obtained after normalizing to the HBSS buffer condition. Data are mean ± SEM of at least three independent experiments performed in duplicates. (E) WT or PAR2-KO HEK293 cells were treated with SplB (5 μg/mL) or trypsin (10 nM). The cell lysates were subjected to SDS-PAGE and blotted for total ERK (tERK) or phosphorylated ERK (pERK). Data are mean ± SEM of three independent experiments performed in duplicates. A and F: Two-way ANOVA was utilized to determine statistical significance, ***p<0.001 (A) and *p<0.05 (F) vs buffer control. A23187: Calcium ionophore; YM254890: Gαq/11 inhibitor
SplB activates Gα13, Gαi1/oB and β-arrestin-1/2 recruitment to PAR2 but does not activate Gαq/11 or MAPK
Following the observation that SplB can cleave PAR2, we sought to determine whether this could activate PAR2 signaling. It is known that PAR2 activation by trypsin triggers robust calcium release, which is dependent on the Gαq/11 signaling pathway (59). HEK293 cells that endogenously express PAR2 were treated with increasing concentrations of trypsin or SplB as indicated (Figure 2B). As expected, trypsin triggered calcium mobilization from the cells in a dose-dependent manner (Supplementary Figure 2), which was dependent on Gαq/11 as inhibition of the latter completely abolished calcium mobilization (52) (Figure 2B). To our surprise, SplB (5 μg/mL) did not induce calcium mobilization from the cells (Figure 2B) despite the significant cleavage of the PAR2 N-terminus by this enzyme (Figure 2A). These results demonstrate that SplB does not activate PAR2 signaling via the Gαq/11 pathway.
PAR2 like many other GPCRs can couple to multiple signalling effectors including Gα12/13, Gαi1/oB and β-arrestin-1/2. We assessed recruitment of each of these effectors to PAR2 using a BRET assay. HEK293 cells transiently expressing PAR2-eYFP and rLuc8 tagged G proteins (Gα12, Gα13, Gαi1 or GαoB) were treated with either trypsin (100 nM) or SplB (5 μg/mL). As observed in previous studies (54), trypsin triggered significant recruitment of all four G-proteins Gα12, Gα13, Gαi1, and GαoB to PAR2, whereas SplB treatment resulted in the recruitment of Gα13, Gαi1 and GαoB as indicated by an elevated BRET signal compared to the baseline in HBSS treated controls (Figure 2C, 2D).
Next, we assessed β-arrestin-1 and/or −2 recruitment to PAR2 using a BRET assay. For this, HEK293 cells expressing PAR2-eYFP and rLuc-β-arrestin-1 or −2 fusion proteins were treated with either SplB (5 μg/mL) or trypsin (10 nM). As expected, trypsin treatment resulted in the recruitment of β-arrestin-1 and −2 (Figure 2E). SplB also triggered a significant increase in recruitment of both β-arrestins (Figure 2E) compared to the HBSS control.
In recent work, we demonstrated that PAR2-mediated ERK phosphorylation in HEK293 cells occurs downstream of Gαq/11 signaling (59). Nevertheless, we assessed ERK phosphorylation following the cleavage of PAR2 by SplB. HEK293 cells that endogenously express PAR2 or PAR2-KO HEK293 cells were treated with either SplB or trypsin and p42/44 (ERK) MAPK phosphorylation was assessed by Western blot. SplB did not trigger a significant increase in ERK phosphorylation compared to untreated cells (Figure 2F). In contrast, trypsin activation of PAR2 resulted in a significant increase in ERK phosphorylation compared to untreated controls (Figure 2F). As expected, neither trypsin nor SplB was able to induce ERK phosphorylation in cells lacking PAR2 (Figure 2F). Overall, these data are in line with our observation that SplB stimulation of PAR2 does not induce Gαq/11 mediated signaling.
SplB induced IL-6 release is dependent on PAR2-NF-κβ signaling cascade
We next sought to determine whether SplB triggered cytokine production in endothelial cells was mediated by PAR2. We treated primary HUVECs with SplB or a PAR2-AP with or without a PAR2 negative allosteric modulator AZ3451 and assessed IL-6 levels in the culture supernatants. Treatment of cells with SplB induced production of IL-6, while the response to the PAR2-AP was more modest but elevated in comparison to the high basal levels of IL-6 seen in untreated cells (Figure 3A). Inhibition of PAR2 in SplB treated cells lowered the cytokine release to the background level (Figure 3A). It has been reported that some PAR2 activating proteases induce cytokine production via the NF-κβ pathway (60). In addition, activation of Gαi1/oB by PAR2 can activate NF-κβ, leading to IL-6 release. Blockade of NF-κβ signaling completely abolished the spontaneous and SplB triggered IL-6 release in HUVECs (Figure 3B), indicating that SplB induces IL-6 cytokine release in endothelial cells in an NF-κβ-dependent fashion. Since PAR2 activates Gαi1/oB in response to SplB, it is likely that IL-6 release here is mediated by Gαi1/oB.

Primary human endothelial cells (HUVECs) were stimulated with SplB or PAR2-AP in the presence or absence of inhibitors to PAR2 (F) or Bay11-0782 (G) for 45 min. After 24 h, cell-free supernatants were analyzed for IL-6 by ELISA. Data are mean ± SD of two independent experiments performed in duplicates or triplicates. G: One-way ANOVA was utilized to determine statistical significance, **p<0.0001 vs buffer, ###p<0.01, SplB vs SplB-Bay11-0782. AP: Activating peptide; Bay11-0782: NF-κβ signaling inhibitor; iPar2: PAR2-inhibitor
Taken together, these results show that SplB selectively cleaves PAR2 and induces the downstream signaling through Gα13, Gαi1/oB, and β-arrestin-1/2 pathway. In addition, SplB action on endothelial cells occurs at least in part through PAR2, and culminates in NF-κβ activation and cytokine production.
SplB does not cleave PAR2 at the trypsin cleavage site
Since SplB and trypsin activated PAR2 show a partially overlapping signaling profile (Figure 2), we sought to examine whether SplB cleaves PAR2 at the trypsin cleavage site. To determine this, we synthesized a peptide representing the hPAR2 N-terminal domain spanning the trypsin cleavage site, GTNRSSKGRSLIGKVDGTSHV-NH2 (Supplementary Figure 3A), and incubated with trypsin or SplB for 60 minutes. The digestion mixture was then subjected to MALDI-MS analysis to identify the possible peptide fragments. Consistent with previous findings (61, 60), trypsin cleaved the synthetic peptide at the expected R36//S37 (major site) and K34//G35 sites within the 60 minutes reaction time (Supplementary Figure 3B). However, SplB did not cleave this peptide as no peptide fragmentation was detectable (Supplementary Figure 3C). This result indicates that the SplB cleavage site on hPAR2 lies outside of this region and needs to be investigated further in future peptide mapping and mutational studies.
SplB induces endothelial barrier disruption in vitro and in vivo
Many proteases, bacterial toxins, and cytokines are known to disrupt the endothelial barrier. Previous studies have demonstrated that proteases that activate the PAR2 can also induce endothelial barrier disruption (62). Having demonstrated that SplB activates PAR2 and induces proinflammatory cytokines in endothelial cells, we asked whether this bacterial protease is able to damage the endothelial barrier. To address this, SplB-exposed HUVEC monolayers were subjected to immunofluorescence analysis for the expression of VE-cadherin and ICAM-1, two essential components that maintain the integrity of the endothelial barrier. In the SplB treated cells the amount of available VE-cadherin was significantly diminished while ICAM-1 expression was upregulated, resulting in a strong reduction of the mean fluorescence intensity (MFI) ratio between VE-cadherin and ICAM-1 (Figure 4A). This suggests that S. aureus SplB weakens the endothelial barrier integrity. Next, we employed intravital microscopy to observe the integrity of the endothelial barrier in vivo in a mouse model of microvascular leakage (57). Consistent with our observations in vitro, administration of SplB in mice leads to massive leakage of FITC dextran across the vasculature to the perivascular space in the murine cremaster muscle. Importantly, deficiency of PAR2 or treatment of animals with a SplB-neutralizing monoclonal antibody before SplB administration almost completely abolished the SplB effect (Figure 3B). These results demonstrate that S. aureus-SplB induces endothelial barrier dysfunction via PAR2 activation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Primary human endothelial cell-monolayers were stimulated with PBS or SplB. The cells were fixed with 4% PFA before being subjected to immunofluorescence for the indicated markers. Red: VE-cadherin, Yellow: ICAM-1. The mean fluorescence intensity (MFI) ratio of VE-Cadherin and ICAM-1 was calculated. Data are mean ± SD of three independent experiments. (B) Leakage of intravenously applied FITC-dextran to the perivascular tissue was analyzed in WT and PAR2 KO animals after intrascrotal stimulation with SpIB with or without pre-treatment with SplB-monoclonal antibody (mAb). Data are mean ± SD of 4-5 animals per group. A-B: A t-test was utilized to determine statistical significance, *p<0.05 vs buffer (A) and *p<0.05 vs SplB + mAb (B).
Discussion
Our study demonstrates that one of the extracellular S. aureus-serine protease-like proteins (Spls), SplB, induces endothelial dysfunction in vitro and in vivo. Spls are a group of six proteases, namely SplA to SplF encoded by the spl operon (63), whose functions are yet to be fully explored. The Spls show high sequence homology among themselves and structural homology to S. aureus V8 protease (63). Except for SplA, which cleaves mucin-16 on the human epithelial cells (44), no biologically relevant substrates for Spls have been identified so far. Here we report that SplB cleaves human PAR2 but not PAR1 and PAR4, which identifies the first biologically relevant and therapeutically tractable substrate and receptor for SplB.
PARs (PAR1-4) are a family of 7-transmembrane GPCRs, activated after proteolytic cleavage of the N-terminus at a specific site by extracellular proteases. This event creates a new N-terminal domain, which acts as a tethered ligand that activates the receptor (64, 60). The canonical activator, trypsin, cleaves hPAR2 between R36//S37 and K34//G35, revealing a tethered ligand with the sequence “SLIGKV” (61, 60). Similar to trypsin, cockroach E1-E3 and Pen C proteases cleave PAR2 at the canonical cleavage site, R36//S37, whereas neutrophil-derived enzymes cleave PAR2 downstream of this tethered ligand region. For example, proteinase-3 cleaves PAR2 at V48//T49, V55//E56, T57//V58, V61//D62, D62//E63, cathepsin-G at F59//S60, F64//S65 and S65//A66, and elastase at V42//D43, V48//T49, V53//T54, V58//F59, A66//S67 and S67//V68 (60). Cleavage of PAR2 by these neutrophil enzymes are known to trigger signalling responses that are distinct from those elicited by the canonical activator trypsin (64). To determine the SplB cleavage sites on hPAR2, we treated a peptide spanning the N-terminal domain of the trypsin cleavage region with SplB. We found that SplB did not cleave the peptide, indicating that SplB recognition sites are distinct from the trypsin target site as seen for the neutrophil derived enzymes. Previous studies characterizing SplB cleavage specificifity had identified preferred cleavage motifs of SplB as WELQ//Q, WELQ//N and WELQ//G (65, 66). While the entire SplB cleavage motif was not identified in the PAR2 N-terminus, a Q27//G cleavage site is present upstream of the trypsin cleavage site and needs to be investigated further in future studies as the putative SplB cleavage site on PAR2.
PAR2 can interact intracellularly with multiple heterotrimeric G-proteins to trigger various signaling pathways (21, 20). PAR2 activation by trypsin triggers robust cytoplasmic calcium release, which is dependent on Gαq/11-signaling pathway (59). We observed that SplB cleavage of PAR2 did not result in Gαq11-mediated calcium release and ERK phosphorylation (pERK) downstream of PAR2, while trypsin, a known PAR2 activating enzyme, induced robust calcium mobilization (Supplementary Figure 2) and activated pERK. PAR2 can however couple to multiple signalling effectors including Gα12/13, Gαi/oB and β-arrestins. Using a BRET assay, we demonstrated that cleavage of PAR2 by SplB triggers recruitment of Gαi, GαoB, Gα13, β-arrestin-1 and −2. It has been shown that depending on the effector being engaged, PAR2 activation leads to different intracellular signaling cascades influencing transcriptional regulation of specific genes including the induction of proinflammatory cytokines (60, 48). It is also know that PARs can elicit distinct responses referred to as “biased agonism,” where ligands or proteases of various origins activate the same receptors but induce different signaling responses (67, 53, 24). Here we identify SplB as a novel biased activator of PAR2.
Treatment of primary endothelial cells with SplB and PAR2-AP lead to proinflammatory cytokine release in a PAR2 dependent manner. The observed cytokine response was completely abolished in the presence of NF-κβ inhibitor, indicating that SplB triggers the PAR2-NF-κβ axis. Extracellular proteases of various origins activate PAR2 on endothelial cells and induce the production of proinflammatory cytokines and chemokines, the latter involved in the disruption of the endothelial barrier (68, 69) by downregulating the expression of VE-cadherin, an essential component of the barrier (9). SplB induced a similar response in human primary endothelial cells that was characterized by the release of proinflammatory cytokines and a strong reduction of VE-cadherin. Other mechanisms are also possible for PAR2 regulation of the endothelial cytoskeleton. PAR2 activation of the small Rho GTPase Cdc42 is implicated in remodelling of the actin cytoskeleton and exocytosis (70). S. aureus is similarly reported to engage Cdc42 in order to invade human endothelial cells (71). Whether S. aureus regulation of Cdc42 involves PAR2 remains to be examined.
Our results unveiled that SplB induces endothelial barrier disruption in vivo in mice. Therapeutic blockade of SplB with a monoclonal antibody preserved the barrier integrity. Overcoming the endothelial barrier is crucial for S. aureus dissemination and a wide range of virulence factors and proteases secreted by S. aureus are reported to induce endothelial barrier dysfunction (9, 43, 14, 8). Here we identified a key virulence factor of S. aureus that contributes to barrier disruption. It is plausible that SplB-induced barrier disruption in vivo is mediated by the disturbance of VE-cadherin homeostasis.
Endothelial dysfunction is also a hallmark of allergic asthma (72), and PAR2 signaling has been implicated in disturbing the endothelial barrier integrity and the development of pathology (63, 9, 27, 32, 33, 29). Many allergen-derived proteases mediate endothelial dysfunction via activating PAR2 on mucosal endothelial and epithelial cells (14). Allergens of house dust mite activate PAR2 on multiple cells in the lung (73). Mice deficient for PAR2 or subjected to therapeutic PAR2-blockade displayed a strongly reduced influx of eosinophils in bronchoalveolar lavage fluid (BALF) and lung tissue compared to WT counterparts (33, 74). Deficiency of PAR2 protected mice from cockroach allergen-induced airway inflammation and airway hyperresponsiveness (75). We previously demonstrated that administration of SplD in mice induces allergic asthma, which was associated with increased numbers of PAR2-positive infiltrating cells in the lungs, but SplD did not cleave PAR2 (40). It is plausible that SplD-induced type 2 lung inflammation triggered mast cells to release endogenous proteases, which in turn recruited and activated inflammatory cells (macrophages and T cells) in a PAR2 dependent manner. Overall, these data indicated that PAR2 may be an excellent target for development of drugs to limit S. aureus mediated inflammatory responses. Indeed, the PAR2 negative allosteric modulator AZ3451 in our hands decreased inflammation triggered by SplB. Additional work with antagonists, particularly those selectively targeting PAR2 signaling pathways such as Gαi and Gα12/13 may uncover more effective therapeutics. Antagonists that only block PAR2 coupling to Gαq/11 are unlikely to be effective in the context of SplB mediated inflammation.
Taken together, our results clearly show that S. aureus SplB activates endothelial cells via PAR2 and contributes to inflammation and the disruption of the endothelial barrier. This identifies a novel mechanism by which S. aureus invades the host.
Author contributions
Conceptualization and project design: A.C, R.R, B.M.B, and M.N.D; methodology and performance of experiments: A.C, P.E.T, B.S, L.A.M, and M.N.D; data evaluation: A.C, B.S, S.D.E, C.A.R, R.R, and M.N.D; interpretation of data: A.C, C.A.R, R.R, B.M.B, and M.N.D; writing - original draft preparation: A.C, R.R, B.M.B, and M.N.D; review, comments, and editing: all authors critically reviewed the manuscript. Par2−/− mice were provided by B.H.R. Recombinant SplB protein was expressed and purified by M.N. SplB monoclonal antibody was produced by D.M.M and J.I.
Declaration of interests
The authors declare no competing interests.
Supplementary Figure 1. Thrombin cleaves PAR1 and 4, and trypsin cleaves PAR2. (A and C) nLuc-hPAR1/4-eYFP CHO cells were treated with thrombin and (B) nLuc-hPAR2-eYFP CHO cells with treated with trypsin; and the PAR cleavage was measured. Data are mean ± SEM of at least three independent experiments performed in duplicates.
Supplementary Figure 2. Trypsin induces calcium mobilization. HEK293 cells were treated increasing concentrations of trypsin as indicated. HBSS buffer served as a control. Calcium signaling traces were obtained Fluo-4 NW calcium indicator. Data are mean ± SEM of three independent experiments.
Supplementary Figure 3. SplB does not cleave PAR2 at the trypsin cleavage site. Human PAR2 N-terminal domain peptide spanning the tethered ligand region, GTNRSSKGRSLIGKVDGTSHV-NH2, was incubated with either trypsin (10 nM) or SplB (50 μg/mL) for 60 minutes at 37 °C. The digestion mixture was analyzed for peptide fragmentation on MALDI-MS. Cleavage sites (//) were identified from any additional m/z peaks (peptide fragments) observed in the mass spectrum of the enzyme-treated peptide.
Acknowledgements
We thank Vanessa Vau and Sarah Böttcher for their assistance with the immunofluorescence experiments, and Britta Fielitz for providing primary human endothelial cells. This study was supported by Deutsche Forschungsgemeinschaft (DFG), Sonderforschungsbereich (SFB) 914, project B03 to C.A.R.
Footnotes
Figure files updated Supplemental files updated Authors' list affiliations updated Manuscript has been revised
References




