

Abstract
The maternal microbiota affects the development of the offspring potentially by microbial metabolites translocating to the foetus through the placenta. We compared placentae, foetal intestine and brain from germ-free (GF) and specific pathogen free (SPF) mouse dams by non-targeted metabolomic profiling. 100 annotated metabolites and altogether 3680 molecular features had significantly different levels in the placental and/or foetal tissues of GF and SPF mice. More than half of the annotated and differentially expressed metabolites had decreased levels in the GF tissues, suggesting their microbial origin or a metabolic response of the host to the presence of gut microbiota. These include known or suggested microbial metabolites, such as 5-aminovaleric acid betaine, (β-)alanine betaine, trimethylamine N-oxide, catechol-O-sulfate, and hippuric acid. Several metabolites had increased levels in the GF mice. These could be precursors of microbial metabolites or indicators of a metabolic response to the absence of gut microbiota. 99 molecular features were only detected in the SPF mice, suggesting the existence of unidentified microbially modified metabolites that potentially influence fetal development. Only a few molecular features showed significantly different levels in the placental tissues but not in other tissues, indicating that the potential microbial metabolites mostly pass through the placenta into the foetus.
Introduction
The intestinal microbiota has a great impact on the life and wellbeing of the host. Its cell numbers are estimated to at least equal and its gene pool exceed that of its host (Sender et al., 2016; Lloyd-Price et al., 2017). Microbes residing in the gut participate in digestion and metabolic modification of nutrients, producing substances that are absorbed by the host (Zhang and Davies, 2016). In a comparison of the serum metabolome of conventionally colonized and germ free (GF) mice, 3.5% of the > 4000 molecular features detected were unique for conventional mice and 10% of the shared molecular features had significantly different levels between the groups (Wikoff et al., 2009).
While a majority of the compounds originating in microbial metabolism detected in mammalian tissues still remain uncharacterized, some of these substances and their effects on the host are well documented. These include vitamins and short chain fatty acids (SCFAs) which the host utilizes as an essential part of its metabolism (LeBlanc et al., 2017). The SCFAs produced from complex carbohydrates by microbes residing in the alimentary tract are an important source of energy for the host (Bergman, 1990). The SCFAs have also been shown to contribute to the maintenance of the gut epithelium and regulation of the immune responses by facilitating regulatory T cell generation in the colonic mucosa (Roediger and Moore, 1981; Inan et al., 2000; Arpaia et al., 2013; Furusawa et al., 2013; Smith et al., 2013). Gut-residing microbes are also known to modify endogenous primary bile acids creating molecular species, such as deoxycholate, stimulating serotonin production by colonic enteroendocrine cells and thus affect the regulation of the intestinal function of the host (Yano et al., 2015). Other microbial metabolites or their host-produced derivatives, such as trimethylamine N-oxide (TMAO) and 5-aminovaleric acid betaine (5-AVAB), are known to modify specific host metabolic reactions of lipids (Wang et al., 2011; Koeth et al., 2013; Kärkkäinen et al., 2018).
The intestinal and absorbed levels of nutrients, particularly amino acids are also modified by microbiota (Wikoff et al., 2009; Mardinoglu et al., 2015; Yamamoto et al., 2018). Notably, microbial metabolism can diversify the fates of the amino acid tryptophan, both by production of an array of microbial indole metabolites in the gut lumen and indirectly by modifying the host metabolism of tryptophan to serotonin by enterochromaffin cells and to kynurenine by immune and epithelial cells (Clarke et al., 2012; Zelante et al., 2013; Reigstad et al., 2015; Yano et al., 2015). The microbially produced tryptophan metabolites act as aryl hydrocarbon receptor (AhR) ligands expressed on gut epithelia and many types of immune cells. They have a significant role in modifying the host mucosal immune system to promote the survival of commensal microbiota and provide protection against pathogens (Zelante et al., 2013). Modulation of the levels of tryptophan, serotonin and kynurenine by microbiota may have effects on both the enteral and central nervous systems and the systemic state of inflammation, linking disturbances in the intestinal microbiota to a multitude of disorders, such as inflammatory bowel diseases, metabolic syndrome and obesity, and neuropsychiatric disorders including autism and depression (Agus et al., 2018).
The metabolic coexistence between the animal and the bacteria begins already before birth. While it is still unclear whether small numbers of live microbes exist in the healthy fetus, hundreds of microbial metabolites originating from the dam pass through the placenta (Gomez de Aguero et al., 2016; Walker et al., 2017). Very little is known of their properties and physiological effects (Ganal-Vonarburg et al., 2020). Microbe-derived AhR ligands and microbially regulated retinoids are essential for the fetal development of the immune system (van de Pavert et al., 2014; Gomez de Aguero et al., 2016; Grizotte-Lake et al., 2018). Maternal SCFAs are also readily transmitted to the fetus, programming the fetal metabolic and neural systems (Kimura et al., 2020). Other maternally derived microbial metabolites have been primarily studied in the context of toxicology (Ganal-Vonarburg et al., 2020). These observations suggest that whole bacteria are not necessarily required to inflict inflammatory immune responses by the host cells (Horn et al., 2000).
To examine the extent of the cross-placental transfer of microbial metabolites during pregnancy, we compared fetal and placental tissues from germ-free and conventional murine dams using a broad non-targeted metabolomics approach. Ultra-high performance liquid chromatography (UHPLC) coupled with quadrupole time-of-flight (QTOF) mass spectrometry allowed the detection of thousands of differentially abundant molecular features in the tissue samples.
Materials & Methods
Fetal and placental mouse tissues
Fetal and placental mouse tissues from pregnant germ-free (GF) and specific pathogen free (SPF) C57BL/6J dams were obtained from the EMMA Axenic Service at Instituto Gulbenkian de Ciência, Portugal. The GF and SPF statuses were regularly monitored by culture and 16S qPCR. The GF dams were 3-4 months old and the SPF dams 4-5 months old. All dams were fed identical RM3-A-P breeding diets (SDS Special Diet Services, Essex, UK), autoclaved at 121 °C. The SPF feed was autoclaved for 20 minutes and the GF feed for 30 minutes due to logistical reasons. The dams were euthanized 18.5 days post coitum. The fetal tissues were immediately frozen in liquid nitrogen, stored at −80 °C and shipped on dry ice to the research laboratory.
Sample processing
Frozen tissue samples were thawed in +8 °C for two hours and then weighed (approx. 100 mg) in homogenizer tubes. For the metabolite extraction, cold methanol (80 % v/v) was added in a ratio of 500 µl per 100 mg of sample. The samples were homogenized (TissueLyser II bead mill, Qiagen, Hilden, Germany) using metal beads at 6 m/s for 30 seconds. The samples were then shaken for 5 minutes in room temperature and centrifuged at 14,000 rpm at +4 °C for 10 min. After the centrifugation, the samples were kept on ice for 5 to 10 min, after which the supernatant was filtered (Acrodisc 0.2 µm PTFE membrane, Pall) into HPLC vials for analysis. The pooled quality control (QC) sample was prepared by collecting 20 µl from each sample vial and combining the material to two vials.
LC–MS analysis
The samples were analyzed by liquid chromatography–mass spectrometry, consisting of a 1290 Infinity Binary UPLC coupled with a 6540 UHD Accurate-Mass Q-TOF (Agilent Technologies), as described previously (Klåvus et al., 2020). In brief, a Zorbax Eclipse XDB-C18 column (2.1 × 100 mm, 1.8 µm; Agilent Technologies) was used for the reversed-phase (RP) separation and an Acquity UPLC BEH amide column (Waters) for the HILIC separation. After each chromatographic run, the ionization was carried out using jet stream electrospray ionization (ESI) in the positive and negative mode, yielding four data files per sample. The collision energies for the MS/MS analysis were selected as 10, 20 and 40 V, for compatibility with spectral databases.
Data analysis
Peak detection and alignment were performed in MS-DIAL ver. 4.00 (Tsugawa et al., 2015). For the peak collection, m/z values between 50 and 1500 and all retention times were considered. The amplitude of minimum peak height was set at 2000. The peaks were detected using the linear weighted moving average algorithm. For the alignment of the peaks across samples, the retention time tolerance was 0.05 min and the m/z tolerance was 0.015 Da. The heatmaps were produced with Multiple Experiment Viewer (MeV) version 4.9.0. MetaboAnalyst 4.0 was used for the pathway analysis of the annotated metabolites (Chong et al., 2018).
Data clean-up (for each mode separately) and statistics (for all signals remaining after clean-up) were performed in R version 3.5.1. Low-quality features were flagged and discarded from statistical analyses. Molecular features were only kept if they met all the following quality metrics criteria: low number of missing values, present in more than 70% of the QC samples, present in at least 60% of samples in at least one study group, RSD* (the non-parametric version of relative standard deviation) below 20%, D-ratio* (non-parametric measure of the spread of the QC samples compared to the biological samples) below 10%. In addition, if either RSD* or D-ratio* was above the threshold, the features were still kept if their classic RSD, RSD* and basic D-ratio were all below 10%. Drift correction was applied to the data.
The cleaned data matrices of the four modes were combined before imputation. Features were then imputed using random forest imputation, with an OOB error of 0.009. QC samples were removed prior to imputation to prevent them from biasing the procedure.
Differential features between the treatment (GF) and control (SPF) were determined using a simple linear model (Student’s t-test) fit separately for each feature. The results were adjusted for multiple comparisons using Benjamini–Hochberg false discovery rate (FDR). FDR-adjusted p-values (Q-values) below 0.05 were considered significant.
For the MS Peaks to Pathways analysis in MetaboAnalyst, the data was first normalized by medians, cube root transformed, automatically scaled, and parametric statistical significances calculated with equal variances and P-value (FDR) cutoff 0.05. In Peaks to Pathways, the molecular weight tolerance was set to 10 ppm, primary ions enforced, and adducts set based on the experimental data.
Compound identification
The chromatographic and mass spectrometric characteristics (retention time, exact mass, and MS/MS spectra) of the significantly differential molecular features were compared with entries in an in-house standard library and publicly available databases, such as METLIN and HMDB, as well as with published literature. The annotation of each metabolite and the level of identification was given based on the recommendations published by the Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI): level 1 refers to confirmed identifications based on reference standards analyzed with the same instrument with identical conditions; level 2 means putative annotations with matching m/z and MS/MS fragmentation spectra with publicly available databases; level 3 signifies a putative characterization of compound class based on the observed physicochemical characteristics of the molecular feature; and level 4 covers all the remaining (unknown) signals (Sumner et al., 2007).
Results
Differences in all observed molecular features
The non-targeted metabolomics data consisted of a total of 12166 molecular features after data cleanup. The metabolomic profiles were clearly different in all studied tissues (Fig. 1 and Supplementary Figure 1). The GF and SPF mice clustered separately in t-distributed stochastic neighbor embedding (t-SNE) analysis, especially when each tissue was analysed individually (Fig. 1). The clearest separation between GF and SPF tissues was observed in the placenta. The gender of the fetus did not have an effect on the separation (not shown).

Metabolic profiles of germ-free (GF, red) and specific pathogen free (SPF, blue) placentae and fetal tissues, analysed by t-Distributed stochastic neighbor embedding (TSNE). The results are shown for the whole dataset including all the signals after data cleanup (n = 12 166) and separately for each tissue.
The clustering by tissue is also evident in the heatmap of all observed molecular features (Fig. 2). At this level, the difference in signal abundance related to germ-free status can be observed from a few relatively small clusters of molecular features in the placental and intestinal tissues.

Heat map of the normalized abundances of all the signals from the four studied modes (RP+, RP–, HILIC+, HILIC–) after data cleanup (n = 12 166) in each studied sample. A hierarchical clustering was applied to arrange the metabolites based on their similarity of the abundance between the samples. SPF = Specific Pathogen Free, GF = Germ Free.
The concentrations of 3680 molecular features differed between germ-free (GF) and specific pathogen free (SPF) mice in at least one of the tissues investigated (adjusted P < 0.05 and Cohen’s d > 0.8).
There were 2200 features which were more abundant in SPF mice in at least one tissue (Fig. 3a). These were most numerous in the fetal intestine. 168 features were more abundant in SPF mice in all three tissues investigated.
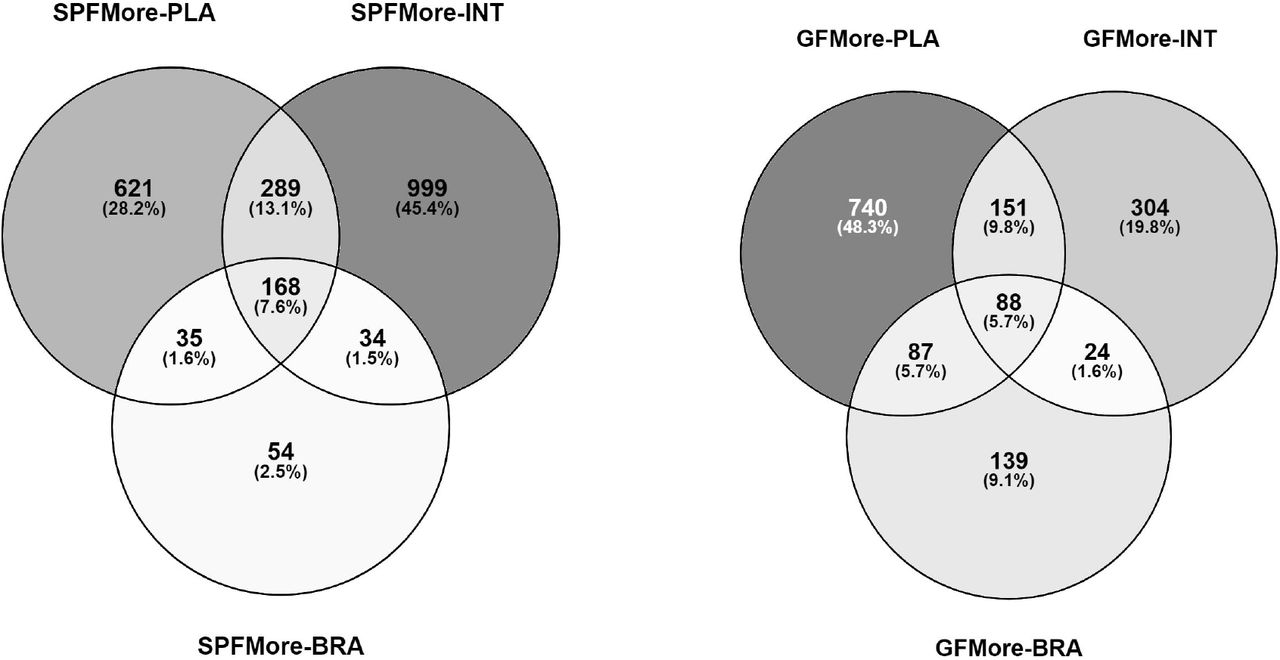
Venn diagrams showing the distribution of molecular features which were more abundant (FDR-adjusted P < 0.05; Cohen’s d < −0.8 or > 0.8) A) in specific pathogen free mice in at least one tissue, B) in germ-free mice in at least one tissue. SPF = Specific Pathogen Free, GF = Germ Free, PLA = PLAcenta, INT = INTestine, BRA = BRAin.
Similarly, 1533 features were more abundant in GF mice in at least one tissue (Fig. 3b). These were most commonly observed in placenta and also more commonly in the fetal brain than the features enriched in SPF mice. 88 features were more abundant in GF mice in all tissues.
A total of 99 features were only observed in SPF mice (Fig. 4). These were most commonly detected in all three SPF tissues (n = 37) or at least in both placenta and fetal intestine. None were detected only in both fetal tissues or only in the fetal brain.
In contrast, only 6 features were exclusively observed in GF mice (not shown).

Venn diagram showing the distribution of molecular features (n = 99) which were observed only in specific pathogen free mice (signal-noise ratio > 5 in SPF mice and < 5 in GF mice; FDR-adjusted P < 0.05, Cohen’s d < −0.8 for GF vs. SPF). SPF = Specific Pathogen Free, GF = Germ Free, PLA = PLAcenta, INT = INTestine, BRA = BRAin.
Annotated metabolites
One hundred of the differentially abundant molecular features could be putatively annotated and 89 of these identified (Table S1). Heatmaps of significantly differential annotated metabolites are shown in Figures 5 and 6.

Heat map of the normalized signal abundances of the significantly differential annotated metabolites (n = 89) in each studied sample. A hierarchical clustering was applied to arrange the metabolites based on their similarity of the abundance between the samples.

Heat map of the normalized signal abundances (as group averages) of the differential annotated metabolites (n = 89). A hierarchical clustering was applied to arrange the metabolites based on their similarity of the abundance between groups. SPF = Specific Pathogen Free, GF = Germ Free.
58 of these metabolites were more abundant in SPF mice in at least one tissue, most of these in all three tissues or in intestine and/or placenta (FDR-adjusted P < 0.05, Cohen d > 0.8; Fig. 5, Table S1). 23 metabolites were more abundant in all three SPF tissues. These included betaines (5-AVAB, alanine / β-alanine betaine and TMAO), solanidine, catechol-O-sulphate, hippuric and pipecolic acid, amino acids and their derivatives (such as kynurenine and aminoisobutyric acid) and small peptides. Five of the annotated compounds were observed exclusively in SPF mice: benzamide, 4-hydroxybenzenesulfonic acid, two unidentified alkaloids and a triterpenoid.
40 annotated metabolites were more abundant in GF mice in at least one tissue, primarily in placenta and/or brain (Fig 7). These included several acylcarnitines, phosphatidylcholine, amino acids (such as ergothioneine) and several small peptides.
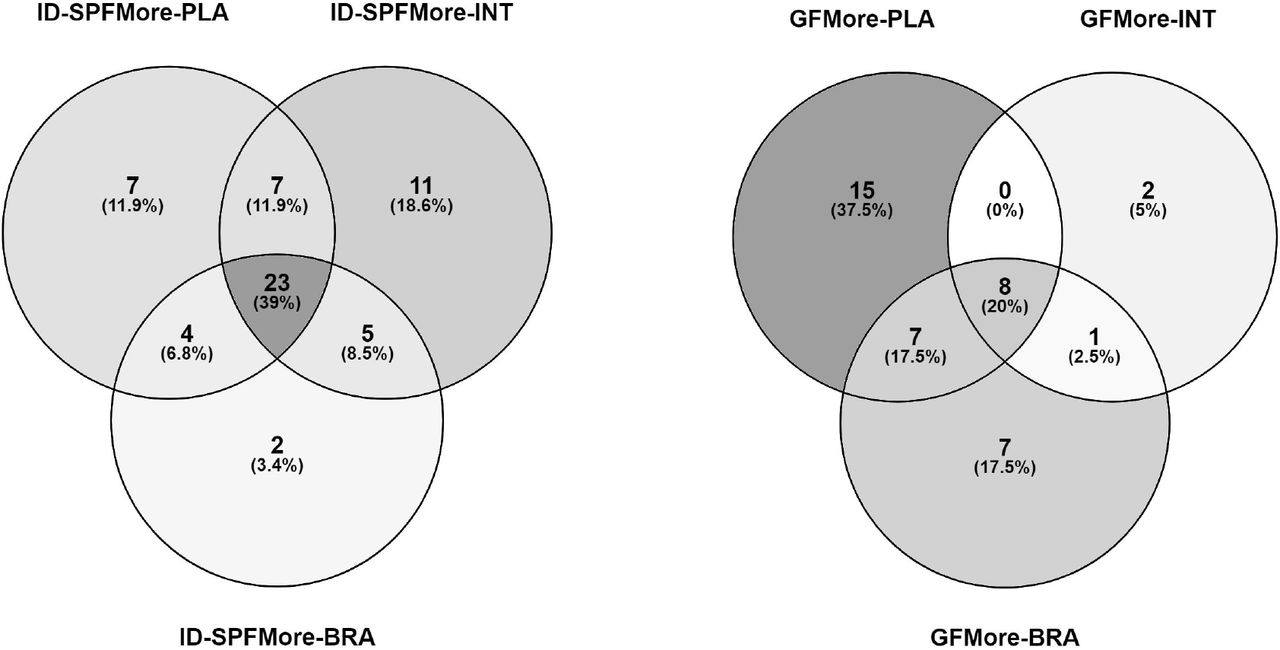
Venn diagrams showing the distribution of annotated metabolites which were more abundant (FDR-adjusted P < 0.05; Cohen’s d > 0.8) A) in specific pathogen free mice in at least one tissue, B) in germ-free mice in at least one tissue. SPF = Specific Pathogen Free, GF = Germ Free, PLA = PLAcenta, INT = INTestine, BRA = BRAin.
Pathway analysis
In order to predict metabolic pathways affected by the lack of microbiota in germ-free mice, we performed the MS Peaks to Pathways analysis in the MetaboAnalyst pipeline (Chong et al., 2018). This module utilizes the mummichog algorithm and gene set enrichment analysis (GSEA), which fit the mass spectrometry peak data into known metabolic pathways without pre-existing compound annotations (Chong et al., 2018).
In terms of KEGG and BIOCYC pathways, the metabolism of several essential amino acids was significantly affected in both fetal tissues (Supplementary Table 1, Kanehisa et al., 2011; Karp et al., 2019). In the fetal brain, aminoacyl-tRNA and bile acid biosynthesis were also affected. In the fetal intestine, phosphonate & phosphinate metabolism, caffeine metabolism, glycolysis, degradation of putrescine and nicotine, and methionine salvage were affected. In placenta, the affected pathways included folate biosynthesis, lactose degradation, methionine salvage and nicotine degradation.
The MS Peaks to Pathways analysis suggested an annotation for four of the 32 unknown metabolites which were only detectable in SPF mice. These were pyridoxamine, anthranilate, octanoate and 5α-pregnane-3α,20α-diol.
Discussion
This is the first study probing the metabolomic effects of a complete maternal microbiota in mammalian fetuses. We compared fetuses of germ-free (GF) and conventional (SPF, specific pathogen free) murine dams utilizing non-targeted metabolomics. We used LC-MS, which provides high sensitivity, selectivity, dynamic range and accuracy for detection of small molecular metabolites (< 1500 Da), excluding short chain fatty acids (SCFAs), which have been extensively studied already previously (Roediger and Moore, 1981; Inan et al., 2000; Smith et al., 2013; Reigstad et al., 2015; LeBlanc et al., 2017). SCFAs are known to cross the placenta to fetal tissues and have an impact on the fetal development (Shekhawat et al., 2003; Priyadarshini et al., 2014). In our study, we focused on the less studied metabolites to find new putative targets for further investigations.
The lack of maternal microbiota affected the metabolite profile of the fetal tissues and placenta. A total of 2200 detected molecular features were more abundant in SPF mice in at least one tissue, while more than 1500 showed higher levels in the GF mice. Approximately one hundred molecular features could be detected only in the SPF tissues. The numbers of compounds depleted in GF mice were largest in the fetal intestine and/or placenta. These observations indicate that the maternal microbiota strongly affects the host metabolism in placenta and also in the fetus, not only by directly producing metabolites, but also pervasively impacting host physiology.
LC-MS signals are annotated based on mass spectral databases. In this study, one hundred of the differentially abundant metabolites could be annotated based on current databases. Several betaines, amino acids and their derivatives, small peptides, certain alkaloids, catechol-O-sulphate, hippuric acid and pipecolic acid were more abundant in SPF mice. Several acylcarnitines, some amino acids, several small peptides and phosphatidylcholine were more abundant in GF mice.
Betaines
The betaines TMAO, 5-AVAB and (β-)alanine betaine had considerably lower levels in all the studied tissues of the GF mice (Figure 5). These are zwitterions containing a positively charged trimethylammonium group and a negatively charged oxygen. They have been confirmed or suggested as gut microbial metabolites in recent studies.
In humans, TMAO is the end product of dietary phosphatidylcholine, choline, and carnitine, which are metabolized first by gut microbes into trimethylamine (TMA) and then in the liver into its N-oxide form. In our study, some of the potential precursors of TMAO, including phosphatidylcholine and several acylcarnitines, had higher levels in all of the GF mouse tissues, which indicates that they were not metabolized in the gut of the dam due to the lack of gut microbiota. This is supported by a recent study, where fetuses from microbially depleted mice and GF mice had more than a twofold reduction of TMAO relative to SPF controls (Vuong et al., 2020). Reintroducing bacteria to the dams resulted in the rise of TMAO levels in the fetuses.
TMAO inhibits reverse cholesterol transport by affecting bile acid synthesis on multiple levels and increases deposition of cholesterol to arterial walls (Koeth et al., 2013). Elevated levels are associated with increased risk of cardiovascular diseases, such as atherosclerosis and thrombosis (Tang et al., 2013; Zhu et al., 2016). The only notable dietary source of this compound is seafood (Wishart et al., 2018).
5-AVAB was also recently associated with cardiovascular health: in a mouse study, it decreased the beta-oxidation of cardiomyocytes and thus may protect the heart tissue in ischemic conditions (Kärkkäinen et al., 2018). In another study conducted on the cord plasma of pre-eclamptic infants, the levels of 5-AVAB were increased in the pre-eclamptic cases compared to non-PE infants (Jääskeläinen et al., 2018). The role of (β-)alanine betaine in mammalian physiology is poorly known.
Catechol-O-sulfate, hippuric acid, methylimidazoleacetic acid and solanidine
Catechol-O-sulfate and hippuric acid are potential microbial metabolites of dietary polyphenols, such as flavonoids and phenolic acids, via degradation by gut microbiota and subsequent sulfation or glycine conjugation in the liver (Feliciano et al., 2016; de Mello et al., 2017). Hippuric acid is also produced by liver from breakdown products of dietary phenols and aromatic amino acids for excretion into urine; gut microbiota is involved in this process (Lee et al., 2012). Hippuric acid was missing from the brain tissue of the GF fetuses, and significantly reduced in placenta and fetal intestine. Decreased levels of hippuric acid have also been reported in serum of GF mice and in the urine of pseudo germ-free rats (Wikoff et al., 2009; Lee et al., 2012).
Methylimidazoleacetic acid is the main metabolite of histamine. In this study, it was missing from the brain tissues of the GF fetuses and had also considerably lower levels in other tissues compared to SPF mice. Another main metabolic pathway of histamine leads to the formation of imidazoleacetic acid, which had decreased levels in the placental and fetal intestinal tissues of the GF mice. Methylimidazoleacetic acid may be associated with miscarriage, potentially by the dysregulation of cytokine networks possibly caused by imbalance in gut bacteria (Liu et al., 2020).
Solanidine is a steroidal glycoalkaloid, which is obtained via dehydroxylation from other glycoalkaloids, such as α-chaconine and α-solanine, which are abundant in potato, a component of the RM3-A-P breeding diet. Solanidine has been detected as the main metabolite in rats after oral ingestion of α-chaconine, and the current findings support the hypothesis that solanidine is a gut microbial metabolite of dietary glycoalkaloids in mice (Norred et al., 1976).
Amino acids and related metabolites
Concentrations of several amino acids and their derivatives were significantly different. Earlier studies have shown that the GF status is associated with reduced levels of most amino acids in the intestine and plasma (Wikoff et al., 2009; Yamamoto et al., 2018). The 4 to 6-fold lower levels in GF mice fetal tissues of pipecolic acid, a degradation product of L-lysine produced by intestinal bacteria, may thus reflect reduced availability of the amino acid L-lysine in the maternal gut and/or lack of the contribution of gut microbiota to the pipecolic acid pathway of lysine catabolism in GF mice (He, 2006).
We observed reduced levels of tryptophan in the intestine of the GF fetuses. Its neuromodulatory metabolite serotonin was reduced in both placenta and the brain. Levels of hydroxyindoleacetic acid (HIAA), a breakdown product of serotonin, and kynurenine, an endogenous metabolite of tryptophan (Neavin et al., 2018), were also decreased in the GF fetal brain and intestine. HIAA may also be generated in bacterial metabolism (Wishart et al., 2018). Kynurenine has been implicated as AhR receptor ligand promoting development of regulatory T cells (Zelante, 2013). Adult GF mice have been shown to exhibit lower levels of serotonin in plasma and brain, but higher plasma levels of tryptophan, than control SPF mice (Wikoff et al., 2009; Clarke et al., 2012).
Other amino acid-related metabolites, including phenylacetylglycine, 1-methylhistidine, 3-methylhistidine, 3-hydroxybutanoic acid, Glu-Gln and Glu-Tyr, also had lower levels in the GF tissues, indicating that the presence of microbiota may increase their production. Phenylacetylglycine has been identified as a gut microbial metabolite of phenylalanine with associations to health and disease in humans (Poesen et al., 2016; Nemet et al., 2020). Certain amino acids and small peptides, such as L-threonine and two tripeptides with unknown structure, had higher levels in the GF tissues compared to SPF, suggesting that they were accumulated in the tissues due to lack of microbial metabolism.
Energy metabolism
The GF status is known to affect the energy metabolism of mice (Bäckhed et al., 2007). We found increased levels of carnitine and various acylcarnitines in placenta and fetal tissues of GF mice. Carnitine and acyl carnitines are involved in beta oxidation of fatty acids. They also have a multifaceted role in the developing brain (Ferreira and McKenna, 2017).
Pathway analyses
Pathway analysis suggested that amino acid metabolism was broadly affected by the lack of maternal microbiota, especially regarding essential amino acids. tRNA charging was also significantly affected. In the fetal intestine, the maternal microbiological status had impacts on glycolysis, degradation of putrescine and phosphinate/phosphonate metabolism. Interestingly, bile acid metabolism was affected in the fetal brain. In the placenta, folate and bile acid biosynthesis, methionine salvage and lactose degradation were affected.
The main metabolic pathways affected by the germ-free status of the dam were related to amino acids arginine, histidine, glutamine and glutamic acid (FDR-corrected p < 0.05). Other pathways involved were proline, beta-alanine, aminoacryl-tRNA, nitrogen metabolism, and pantothenic acid / CoA biosynthesis.
Unannotated molecular features undetected in GF mice
In total 3680 molecular features were differentially expressed in GF tissues and 99 were not detected in GF mice. Five of these could be at least partially annotated: benzamide, 4-hydroxybenzenesulfonic acid, two alkaloids and a triterpenoid. In addition, the MS Peaks to Pathways pipeline suggested a tentative annotation for four compounds: pyridoxamine, anthranilate, octanoate and 5α-pregnane-3α,20α-diol. Most of the compounds missing from GF mice were detected in all three tissues of the SPF mice, or at least in placenta and fetal intestine, suggesting that they are absorbed through placenta.
Benzamide is not a mammalian metabolite, but occurs in the bacterial degradation of tryptophan and indoles; anthranilate is a key metabolite in these reactions (Kanehisa et al., 2011; Consalvi et al., 2019; Naz et al., 2019).
Limitations of the study
The LC-MS method used to detect the metabolites in this study does not allow the detection of short-chain fatty acids (formic to valeric acid), many volatile organic compounds (VOCs) and molecules above 1500 Da (including lipopolysaccharides, large peptides, proteins, and nucleic acids). On the other hand, methyl, hydroxyl and amino derivatives of SCFAs are within the analytical range and have been studied less.
The current databases available for metabolite identification contain the most well-known mammalian endogenous metabolites. Reference data on exogenous (e.g. plant-derived) and microbially produced metabolites is limited. Therefore, a significant proportion of molecular features in the dataset could not be annotated. A tentative characterization of the chemical class was acquired for some of the unknowns with the prediction of molecular formula and comparison to in silico generated MS/MS fragmentation patterns.
Conclusion
The germ-free status of the dam affects the metabolism also in the fetus. The three studied tissues all have a clearly distinct metabolite profile and the germ-free status changes the profiles in all tissues, more in placental tissues compared to the fetal brain and intestinal tissues.
In the fetal tissue, 89 annotated metabolites were affected by the germ-free status of the dam. Several known and potentially microbially originated metabolites were among the differential compounds, further highlighting the impact of gut microbiota on the host metabolism. Additionally hundreds of unannotated molecular features were significantly more abundant in SPF mice or missing from GF mice, indicating that a majority of microbially processed metabolites in the fetus and placenta are still unknown and need to be researched further.
Conflict of interest
Olli Kärkkäinen and Ville Koistinen are co-founders of Afekta Technologies Ltd., company providing metabolomics services.
Acknowledgements
We thank Kirsi Lahti and Santeri Suokas for expert technical assistance.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}