

Abstract
Parasitoid life style represents one of the most diversified life history strategies on earth. There are however very few studies on the variables associated with intraspecific diversity of parasitoid insects, especially regarding the relationship with spatial, biotic and abiotic ecological factors. Cotesia sesamiae is a Sub-Saharan stenophagous parasitic wasp that parasitizes several African stemborer species with variable developmental success. The different host-specialized populations are infected with different strains of Wolbachia, an endosymbiotic bacterium widespread in arthropods that is known for impacting life history traits notably reproduction, and consequently species distribution. In this study, first we analyzed the genetic structure of C. sesamiae across Sub-Saharan Africa, using 8 microsatellite markers, and 3 clustering software. We identified five major population clusters across Sub-Saharan Africa, which probably originated in East African Rift region and expanded throughout Africa in relation to host genus and abiotic factors such as climatic classifications. Using laboratory lines, we estimated the incompatibility between the different strains of Wolbachia infecting C. sesamiae. We observed an incompatibility between Wolbachia strains was asymmetric; expressed in one direction only. Based on these results, we assessed the relationships between direction of gene flow and Wolbachia infections in the genetic clusters. We found that Wolbachia-induced reproductive incompatibility was less influential than host specialization in the genetic structure. Both Wolbachia and host were more influential than geography and current climatic conditions. These results are discussed in the context of African biogeography, and co-evolution between Wolbachia, virus parasitoid and host, in the perspective of improving biological control efficiency through a better knowledge of the biodiversity of biological control agents.
Introduction
Understanding the extraordinary biodiversity of insects requires both analyzing large scale beta diversity patterns (Heino et al. 2015) and unraveling mechanisms of genetic differentiation among populations including geographic, abiotic or biotic interactions (Roderick 1996). Parasitoid wasps are one of the most diverse groups of insects (Grimaldi 2005). Coevolutionary interactions are likely major diversifying forces in host–parasitoid systems due to the strength of reciprocal selection pressures (Van Valen 1973; Henry et al. 2008). As strong insect antagonists, they are the most used agents for biological control programs, which provide one of the best alternatives to chemical control of insect pests (Harvey 2011). There are theoretical expectations that host parasitoid coevolution generates diversity because several traits related to host specificity, such as specific virulence and host recognition, are mechanistically linked to reproductive isolation, especially when the parasitoid mates on the host just after emergence (Dupas et al. 2008; Hoskin & Higgie 2010). Other biotic interactions, particularly those involving microorganisms affecting reproduction such as Wolbachia sp., are expected to drive diversification of parasitoids (Bordenstein et al. 2001; Branca et al. 2009). To distinguish between the different ecological factors responsible for population structure, a combination of, on the one hand, laboratory data on reproductive incompatibility and, on the other hand, field data on the geographic structure of ecological drivers and population differentiation are needed.
Cotesia sesamiae
Cameron (Hymenoptera: Braconidae) is a parasitoid wasp widespread in Sub-Saharan Africa that has been used in biological control for controlling Busseola fusca (Fuller) (Lepidoptera: Noctuidae), a major stemborer pest of maize and sorghum crops (Kfir 1995; Kfir et al. 2002). Cotesia sesamiae is a stenophagous parasitoid that successfully parasitizes diverse host species (Ngi-Song et al. 1995; Branca et al. 2011). However, a variation in parasitism success on different hosts has been shown among populations of parasitoids (Mochiah et al. 2002a; Gitau et al. 2010). In contrast to the C. sesamiae population from Mombasa - coastal Kenya (avirulent towards B. fusca), the C. sesamiae population from Kitale – inland Kenya (virulent towards B. fusca) is able to develop in B. fusca, but both develop in Sesamia calamistis Hampson (Lepidoptera: Noctuidae), the main host of C. sesamiae in coastal Kenya (Ngi-Song et al. 1995). These differences in host acceptance and development have been linked to the observed polymorphism of a candidate gene, CrV1, located on the bracovirus locus (Dupas et al. 2008; Gitau et al. 2007; Branca et al. 2011). Bracoviruses are symbiotic polydnaviruses integrated to the genome of braconid wasps, contributing to their adaptive radiations (Whitfield 2002; Dupuy et al. 2006). The viruses constitute the major components of the calyx fluid of the wasp and are expressed in parasitoid host cells, regulating its physiology, development and immunology (Beckage 1998). In particular, the CrV1 gene, has been shown to contribute to immune suppression by active de-structuration of the cytoskeleton of host immune cells (Asgari et al. 1997). A comparative genomics study of the virus between Cotesia species and C. sesamiae populations, virulent and avirulent against B. fusca, showed patterns suggesting important role for positive selection, gene duplication and recombination among viral genes in the adaptive diversification process (Jancek et al. 2013). Whilst host resistance puts likely a strong selective pressure on local adaptation of the wasp, other ecological and geographic factors must be considered and analyzed for the development of scenario of C. sesamiae response to environmental changes. Climatic differences or geographical barriers might weaken the capacity of some C. sesamiae populations to colonize areas where the most prevalent host is suitable for parasitoid larval development, even if parasitic wasps have been shown to disperse quite efficiently, sometimes beyond the capacity of their associated host (Antolin & Strong 1987; Ode et al. 1998; Van Nouhuys & Hanski 2002; Assefa et al. 2008, Santos & Quicke 2011). Other factors such as Wolbachia might act as a barrier to gene flow through reproductive incompatibility (Werren 1997; Jaenike et al. 2006), which can be especially problematic in the context of biological invasions by preventing crosses between ecological or geographic populations along the range expanded area of the invasive pest host. Wolbachia is a widespread bacterium infecting the majority of insect species that can induce reproductive incompatibilities (Werren 1997; Hilgenboecker et al. 2008). Several Wolbachia strains have been identified in C. sesamiae expressing cytoplasmic incompatibilities (CI) between populations of parasitoids (Mochiah et al. 2002b). The different populations of C. sesamiae, virulent and avirulent against B. fusca, are infected with different strains of Wolbachia (Branca et al. 2011). Reproductive isolation can prevent adapted parasitoid populations to expand across their host range, a phenomenon that could be particularly relevant in biological control programs. In this study, our objective is to analyze the relative importance of neutral geographic factors and major selective forces, biotic (i.e. host species and Wolbachia strain), abiotic (i.e. climate) shaping the genetic structure of the parasitoid Cotesia sesamiae across Sub-Saharan Africa. First, the genetic structure was assessed using 8 microsatellites markers with several genetic clustering approaches, each using different pertinent hypotheses in an effort to reach the broadest picture of the structure. Second, we tested the cross incompatibility between differentially Wolbachia-infected C. sesamiae populations to infer their potential influence on limiting gene flow. Third, we estimated the amount and direction of gene flow in between genetic clusters of selected C. sesamiae populations to see if Wolbachia infection can affect the parasitoid metapopulation dynamics. Finally, we interpreted geographic patterns of C. sesamiae genetic structure in the context of African climate, Wolbachia infection and host occurrence.
Material and methods
Insects field collection
Infected stemborer larvae were collected in 142 localities of 9 sub-Saharan African countries. GPS positions were recorded at each locality. Stemborer larvae were identified using a larval picture library (corresponding to adult moth identifications), and according to the host plant, as most stemborers are host plant specific (Le Ru et al. 2006). Larvae collected from the field were reared on an artificial diet (Onyango & Ochieng’-Odero 1994) until pupation or emergence of parasitoid larvae. After the emergence of cocoons, adult parasitoids were kept in absolute ethanol. Morphological identification of parasitoids was based on genitalia shape following the method of Kimani-Njogu et al. (1997). Total genomic DNA of one female per progeny was extracted using the DNeasy Tissue Kit (QIAGEN). If only male were present then analyses were performed on one male. Because wasps are haplodiploids, the haploid genotype of males was converted to homozygous diploids for analyses to avoid discarding data. This should not bias the results because of a very low level of heterozygosity due to very high inbreeding. Total genotyped individuals were 590 females and 47 males discarding individuals with too many missing genotypes (more than 2 over 8 loci).
Insects rearing
For crossing experiments, females of both virulent and avirulent C. sesamiae strains against B. fusca were obtained from laboratory-reared colonies. The virulent, thereafter named Kitale (Kit) C. sesamiae strain was obtained from B. fusca larvae collected from maize fields in Kitale, Western Kenya, in 2006, while the avirulent C. sesamiae strain thereafter named Mombasa (Mbsa), was obtained from S. calamistis larvae collected from maize fields in coastal Kenya in 2007. The two lines have different Wolbachia infection status: Kitale line is infected with Wolbachia WCsesB1 strain while Mombasa line is infected with two strains of Wolbachia WCsesA and WCsesB2 (Table 1). Twice a year, both colonies were rejuvenated by field collected parasitoids. The wasps of both strains were continuously reared on larvae of S. calamistis as previously described Overholt et al. (1994). Parasitoid cocoons were kept in Perspex cages (30 x 30 x 30 cm) until emergence.
Status of the Kitale and Mombasa strains (Mochiah et al. 2002a)
Adults were fed a 20% honey–water solution imbibed in a cotton wool pad and kept under artificial light for 24 h to mate. In all experiments, only 1-day-old females, putatively mated and unexperienced to oviposition were used. The experiments were carried out at 25 ± 2 °C, 50– 80% RH, and a 12:12 h (L:D) photoperiod.
The stemborer species, B. fusca and S. calamistis, were continuously reared on artificial diet as previously Onyango & Ochieng’-Odero (1994). For each species, three times a year, several stemborer larvae were added to rejuvenate the colonies. Fourth larval instars were introduced into jars (10 x 20 cm), each containing pieces of maize stem, and left for 48 h to feed and produce frass to facilitate host acceptance by the parasitoid wasps for parasitism experiments. Thereafter, the larvae were used in the experiments.
Genetic markers sequencing and genotyping
Eleven previously developed microsatellites markers (Jensen et al. 2002; Abercrombie et al. 2009) were amplified and fragment size determined. Amplifications were performed in 10 μL with approximately 5 ng of genomic DNA, 1 × HotStarTaq PCR buffer, 2 μL Q-Solution 5× (QIAGEN), 1.6 mM of dGTC, dTTP and dCTP, 50 μM dATP, 5 pM of each primer, 0.25 U Taq polymerase (HotStarTaq, QIAGEN) and 0.01 U of [33P]-dATP. The ‘touchdown’ PCR (Mellersh & Sampson 1993) was used as follows: initial activation step at 95°C for 15 min, 18 cycles at 94°C for 30 s, 60 to 51°C for 30 s (-0.5°C/cycle), 72°C for 30 s, 29 cycles at 94°C for 30 s, 54°C for 30 s, 72°C for 30 s and a final elongation step at 72°C for 10 min. Results were visualized using an ABI 310 and a AB3130 sequencer with fluorescent size standard (GeneScan 600 Liz, Applied Biosystem). Amplifications were made following conditions previously described using fluorescent labeling (Pet, Vic, Ned or 6Fam) of the forward primer.
Peaks identifying fragment sizes were assessed using GeneMapper 4.0 Software. Locus B1.42 presented peaks difficult to analyze with multiple bumps preventing any accurate measure of fragment size and was thereby discarded. Loci B1.155 and B5.126 were also not considered in the analyses because they presented a high percentage of missing genotypes (respectively 14.6% and 27.0%) probably reflecting the occurrence of null alleles. Eight loci were then genotyped per individual.
Wolbachia
infection status was checked using the protocol developed in Branca et al. (2011).
Cross-mating experiment
To obtain Wolbachia-free parasitoids colonies (named cured lines), the gravid females of each aforementioned parasitoid line were reared on larvae of S. calamistis previously fed on artificial diet Onyango & Ochieng’-Odero (1994), enriched with 2000 mg/L rifampicine (Dedeine et al. 2001). This process was repeated for three generations of female wasps to create cured colonies of Mombasa (Mbsa) and Kitale (Kit) C. sesamiae.
Cross experiment tests were conducted between both Mbsa and Kit C. sesamiae lines to assess the mating incompatibilities due to the presence of different Wolbachia types. Individual parasitoids were allowed to emerge singly by separating single cocoon from each cocoons mass. Individual male and female parasitoids from each colony (i.e. Kit C. sesamiae cured and uncured as well as Mbsa cured and uncured) were used for cross-mating experiments. Sixteen possible cross-mating combinations were investigated (Table 4). Each cross-mating combination was repeated at least 20 times.
After mating, females were presented 4th instar larvae of S. calamistis for oviposition using the method of Overholt et al. (1994). Thereafter, the larvae were reared and observed daily for mortality or parasitoid emergence. The developmental time of the progeny (egg to adult), the brood size, the sex ratio and the mortality outside and inside the host were recorded.
The presence of Wolbachia infections, in all C. sesamiae populations used in the cross-mating experiments, was tested using PCR techniques on ftsZ and wsp genes as described by Ngi-Song & Mochiah (2001). DNA was extracted from about 50 individuals (a mixture of males and females) from each population previously stored in 99% ethanol.
To test the effect of mating direction on each reproductive trait, a non-parametric Kruskal-Wallis test was applied with crosses as factor. ANOVA was not used because none of the data were normally distributed and had homoscedastic variance. Following Kruskal-Wallis test, a pairwise Wilcoxon’s rank sum test was conducted with false discovery rate (FDR) correction for multiple testing. Data were split into four groups for statistical analyses: crosses between Kit wasps, crosses between Mbsa wasps and crosses between populations in both directions. For all crosses, CI is expected between infected males and uninfected or differentially infected females. CI should lead to a reduction in female production either by female mortality (FM phenotype, diminution of the size of the progeny and the number of females) or male development (MD phenotype, only diminution of the proportion of females) (Vavre et al. 2000).
Statistical analyses for Wolbachia crosses experiments were performed in R 3.2 (R Core Team 2013).
Genetic structure inference
To infer population structure from genetic data we used three different Bayesian methods for population partitioning: INSTRUCT, based on Hardy-Weinberg equilibrium with inbreeding (Gao et al. 2007), TESS3, taking into account spatial autocorrelation based on tessellation (Caye et al. 2016) and DAPC, a statistical partitioning method based on PCA (Jombart & Ahmed 2011). First, Instruct software was used with the Adaptive Independence Sample algorithm using inbreeding coefficient at population level as a prior model (mode 4, option v) (Gao et al. 2007), since C. sesamiae is known to have a highly inbred reproductive system (Ullyett 1935; Arakaki & Ganaha 1986). The number of clusters corresponding to the strongest genetic structure was determined using the method of Evanno et al. (2005). Each inference had a total number of iterations of 200,000 with a burn-in period of 100,000 iterations. Other parameters were kept as default value except the significance level of the posterior distribution of parameters, which was set to 0.95. The posterior probability of assignation of each individual was re-calculated over 10 MCMC runs using the CLUMPP software (Jakobsson & Rosenberg 2007) with greedy algorithm and 10,000 random permutations. Second, TESS3 was run using admixture with the BYM model (Durand et al. 2009). To identify the strongest structure, the model was run with K ranging from 2 to 9 using 100,000 sweeps with a 10,000 burning period. Degree of trend was assessed by running the algorithm with a varying value from 0 to 3 by 0.5 steps. The degree of trend showing the best DIC was kept. Genetic structure was then assessed for K=5, the best K, and T=1.5, the best degree of trend, with MCMC chain run for 1,000,000 sweeps with a 100,000 burn-in period. Third, we used a PCA-type approach with DAPC in R package adegenet (Jombart & Ahmed 2011) which is hypothesis-free since it just clusters individuals to maximize the explained genetic variance within the data.
The influence of various factors on the genetic variance was described using multiple correspondence analysis (MCA, package FactoMineR), and assessed using the adonis function in vegan R package (Oksanen et al. 2013). This corresponds to an extension of AMOVA (Excoffier et al. 1992) for crossed factors and in a non-hierarchical pattern (McArdle & Anderson 2001). The factors considered were: host genus, the Wolbachia infection status, spatial cluster of samples and the Köppen-Geiger climate type (Kottek et al. 2006). As the sampling was not done randomly across Sub-Saharan Africa, we tested the effect of spatial structuration by defining spatial cluster grouping localities close to each other. The spatial cluster of samples was obtained with hierarchical clustering from latitude and longitude data (Mclust function) (Fraley & Raftery 2002; Fraley et al. 2012). Genetic distance between individual were generated using Smouse Peakall’s formula (Smouse & Peakall 1999) in GenoDive (Meirmans & Van Tienderen 2004).
A Bayesian analysis of population sizes and reciprocal migration rates between the consensus genetic clusters obtained from partitioning methods was performed using software Migrate (Beerli & Palczewski 2010). Migrate-n software version 3.6.6 was run using the microsatellite model set to Brownian motion and the gene flow model to asymmetric. Since asymmetric gene flows can only be estimated pairwise, we run independently the software for each pairwise cluster comparison. Prior distributions of θ and M were chosen to get posterior distributions that are not truncated. Five chains of different heat from 1 to 10 were run for 500,000 generations with a burn-in period of 10,000.
Results
Genetic structure
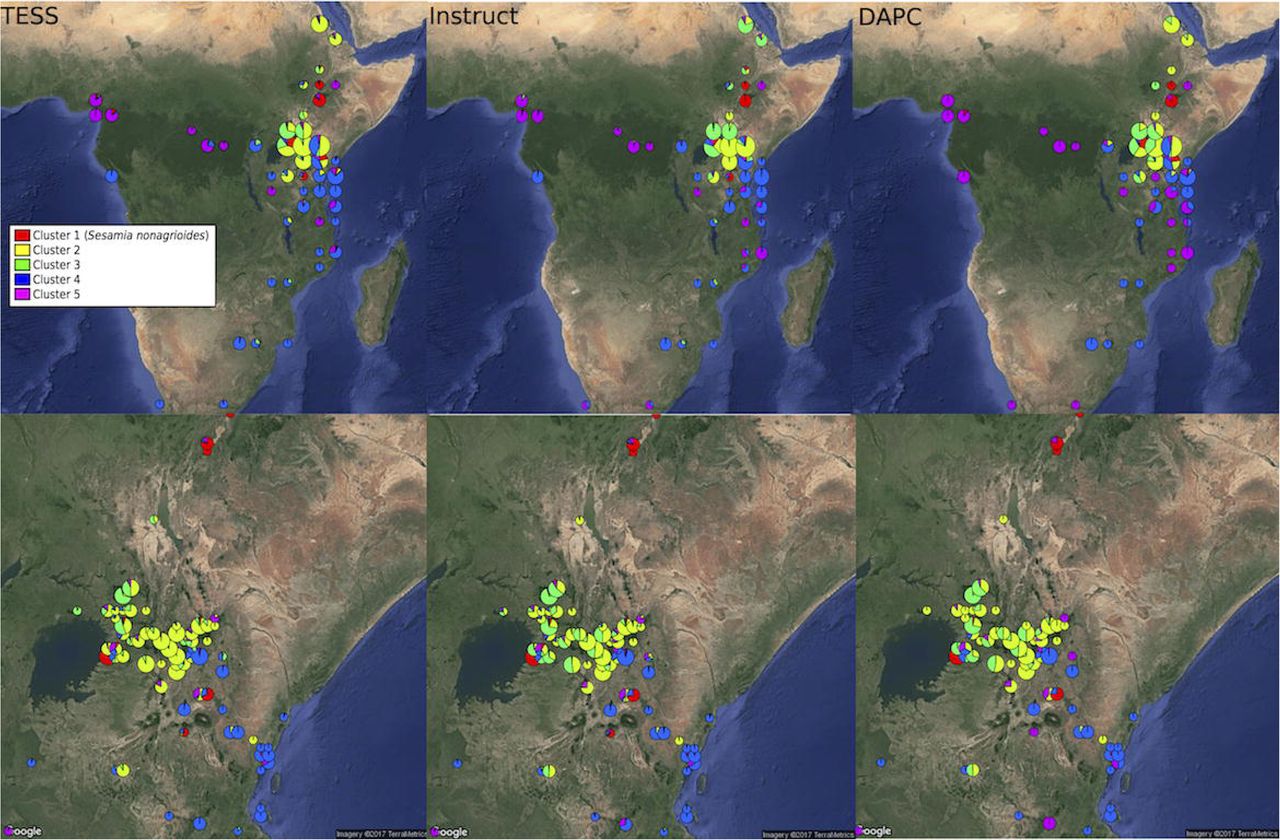
The three clustering methods, Instruct, DAPC and TESS3 used in this study gave similar results regarding the population structure of C. sesamiae populations. For each method, the best fit was observed for five clusters (maximum delta-K for Instruct, Figure S1, diffNgroups criterion for DAPC and Deviance Information Criterion for TESS3). Regarding the structuration in relation to the host species, cluster 1 of all three methods was found exclusively on Sesamia nonagrioides (Figure 1, in red), clusters 2 and 3 were found mainly on Busseola ssp. (Figure 1, in yellow and green, respectively), cluster 4 on Sesamia and Chilo spp. (Figure 1, in blue) and finally cluster 5 was recovered from five host genera (Figure 1, in purple). Geographically, the three methods provided similar picture of genetic structure with some difference in admixture proportion. Cluster 1 population was scattered in between central Ethiopia, western Kenya and Northern Tanzania, and even Cameroon (Figure 2 in red). This corresponded to the population found on S. nonagrioides. One discordance appeared with the DAPC method, which failed to assign one individual from Arusha (Tanzania) into Cluster 1. Cluster 2 extended from Eritrea to Western Kenya in Instruct, but was restricted to Western Kenya in TESS3 and DAPC (Figure 2, yellow). Conversely, cluster 3 was only present in Western Kenya and central Tanzania in the three methods but extended to Eritrea in TESS3 and DAPC (Figure 2, green). Cluster 4 extended from South Africa to East Kenya and Rwanda in the three methods (Figure 2, blue). In Instruct and TESS3 analyses, a very high posterior probability of cluster 4 was also observed further west in the coast of Congo-Brazzaville. Cluster 5 extended from Tanzania to Cameroon in all three methods but was found much more spread in DAPC analysis, until South-Africa, and to a lesser extent in Instruct (Figure 2, purple). Overall, there seem to be a clear delimitation between cluster 2 and 3, which extend form Tanzania to Eritrea, and cluster 4 and 5, which were found from Cameroon to South Africa. Delimitation within these two groups of two clusters seemed to be shallower and influenced by the method used.

Posterior probability of assignment of each individual of Cotesia sesamiae wasps to each of the 5 Instruct clusters and post-processed with CLUMPP (Cluster 1 in red, cluster 2 in yellow, cluster 3 in green, cluster 4 in blue, cluster 5 in purple). Individuals are grouped by the host genus where they were found. Individuals found on an unidentified host are not represented.

Distribution of genetic clusters of Cotesia sesamiae wasps for DAPC with K=5 (A), TESS3 software (B) and the Instruct software CLUMPP consensus with K=5 (C). For each clustering method, only individual with posterior probability of assignment above 0.5 are represented for each analysis. Distribution in Sub Saharan Africa is represented at the top and a zoom in Kenya at the bottom.
Wolbachia strains distributions
A rather good concordance was observed between genetic structure at microsatellites level and Wolbachia strain distributions (Figure 3). Cluster 1 was associated exclusively with the Wolbachia wCsesA strain, cluster 2 and 3 with wCsesB1, cluster 4 and 5, with the bi-infection wCsesA/wCsesB2.

Distribution of Wolbachia infection in Cotesia sesamiae wasps across Sub-Saharan Africa. Red: wCsesA,: blue: wCsesA/wCsesB2, yellow: Absent, purple: wCsesB2, green: wCsesB1.
Relative influence of biotic and abiotic factors
The individuals belonging to the cluster found exclusively on Sesamia nonagrioides were interpreted as a distinct species by Kaiser et al. (2015, 2017), based on eco-phylogeny analyses and cross-mating experiments, and corresponding to a host and plant-host driven ecological speciation event. As it has now been described as the species Cotesia typhae (Kaiser et al. 2017), it was not considered in these analyses to prevent an overestimation of host effect. Multiple correspondence analysis (Figure 4) suggested the presence of structure in relation to all the factors considered (spatial cluster, Köppen-Geiger climate classification, Wolbachia infection status and host genus). The full models tested with adonis function (Table 2) confirmed that all neutral (geography) and selective forces, abiotic (climate type and geography) and biotic (host genus, Wolbachia infection), contribute significantly to the genetic variance of the microsatellite genotypes. Because the adonis method tests factors sequentially, it is important to consider each factor as either first term or marginal (last) term to see the extent of the effect. When added first in the sequence of factors in the adonis function, the biotic factors had higher R2 than the abiotic factors (0.43 and 0.38 for Wolbachia and host genus, respectively and 0.28 and 0.21 for Köppen-Geiger Climate type, and localization, respectively) (Table 3). In addition, all the factors had significant marginal effects (Table 3). Pairwise interactions between factors were weak (R2<0.04), but significant for all the possible interactions (Table 2). None of the tripartite interactions was significant.
Analysis of molecular variance using microsatellite distance matrices and a full model containing all terms and interactions.
Sum of squares and partial R2 of Host genus, Wolbachia infection status, Köppen-Geiger climate and localization taken either as marginal effect or as the first term when adding them sequentially.
Brood size, sex ratio, developmental time and mortality outside and inside the host of populations of different crosses on Sesamia calamistis (N = number of replicates).
Note. Cs Kit, Cotesia sesamiae from the inland Kitale area of Kenya; Cs Mbsa, Cotesia sesamiae from the coastal Mombasa area of Kenya; cured, Wolbachia-free parasitoids colonies (i.e. cured lines); in crosses within each population and between populations, values with different letter are significant (q-value <0.05; pairwise Wilcoxon’s rank sum test, q-value = FDR corrected p-value).

Estimates of gene flow between four geographic genetic clusters of Cotesia sesamiae wasps identified by Instruct. Each circle represents the infection status of individual found assigned to each cluster and the colour are corresponding to the one on figure 3. The fifth genetic cluster found only infecting Sesamia nonagrioides was excluded of the analysis.
Wolbachia crosses experiment
For crosses within each population, the brood size dropped in crosses involving infected males and cured females (i.e. Cs Kit x Cs Kit-cured and Cs Mbsa x Cs Mbsa-cured) from 34-36 to 23 for Kitale population and from 32-42 to 21 for Mombasa population (Table 4). Although for these both potentially incompatible crosses the sex ratio (or %females) decreased significantly for Kitale population and no significant change was detected for Mbsa population, the overall number of females was reduced in these both crosses from 45-62% to 44% and from 57-64% to 55% for Kitale and Mbsa population, respectively. No significant changes in the developmental time and in the mortalities outside and inside the host through dissection were detected between these incompatible crosses and the other crosses.
To the contrary, in crosses potentially showing bidirectional CI, i.e. crosses involving individuals from different populations and infected with different Wolbachia strains (i.e. Cs Kit x Cs Mbsa and Cs Mbsa x Cs Kit), we only found a significant decrease of the percentage of female from 47-67% to 11-0% in the cross involving Mbsa males and Kit females (Table 4). In this cross, almost no females were recovered despite a normal overall progeny size, suggesting a complete incompatibility with pure male development (MD) phenotype (Vavre et al. 2000). By contrast, in the cross for Kit male (wCsesB1) with Mbsa female (wCsesA/wCsesB2), CI expressed only when Mbsa females were cured and only partially since females were recovered. No significant changes in the developmental time and in the mortalities outside and inside the host through dissection were detected between these incompatible crosses (i.e. Cs Kit x Cs Mbsa-cured, Cs Kit x Cs Mbsa, Cs Mbsa x Cs Kit-cured and Cs Mbsa x Cs Kit) and the other crosses.
Migration patterns
For Bayesian analyses of pairwise migration rates, acceptance rate ranged between 0.20 and 0.56 with an effective MCMC sample size from ∼500 to ∼2700. Clusters defined by Instruct were used except Cluster 1 for the main reasons as exposed above. Mostly symmetric gene flow was found between Cluster 2 and 3, which are mainly infected with the same wCsesB1 Wolbachia strain (Figure 5); they were found mainly on Busseola, at least in one contact zone in Central Kenya (Figure 2). Otherwise, asymmetric gene flow between clusters were found. All the gene flow with cluster 5 were orientated toward Cluster 5. Gene flow between Cluster 4 (found mainly on Sesamia and Chilo) and Cluster 2 was the lowest despite the presence of a contact zone in Kenya (Figure 2). Kit population from the laboratory colony was assigned to Cluster 2 and Mbsa population from the laboratory rearing to cluster 5 as inferred in Instruct clustering (Table 1). Therefore, migration was more orientated from wCsesB1-infected population toward wCsesA/wCsesB2 bi-infected populations, mainly because of an asymmetric gene flow in that particular direction between Cluster 3 and Cluster 4.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Multiple correspondence analysis between microsatellite markers distance between individuals and ecological variables
Discussion
Geographic, ecological and biotic factors determining the genetic structure of Cotesia sesamiae
The five major clusters inferred by the three different genetic clustering methods, TESS, Instruct and DAPC exhibited very similar geographic partition. However, TESS3 and Instruct admixture models were more concordant. DAPC results differed by the many geographic areas assigned to just one cluster. The DAPC algorithm optimizes a model without admixture that assigns individuals and not portion of their genomes to clusters. It seeks linear combination of genetic variables that maximizes between clusters component of the genetic distance between individuals. Models without admixture are not robust to the inclusion of admixed individual in the sample, reciprocally, models with admixture are less able to detect barriers when admixture is limited (François et al. 2010). In the absence of intrinsic biological reproductive barriers between the populations, we would expect the admixture model is the best suited because the five clusters are all represented in Kenya and Tanzania with a geographic continuum in both countries. However, the presence of reproductive isolation mechanisms, may limit admixture in this continuum of populations. Indeed, the results of Instruct non-spatial admixture model (Figure 2) shows that populations maintained their integrity; admixture being limited to the hybridization zones despite the ability of C. sesamiae to expand throughout Africa. We will discuss below the factors that may limit admixture in this continuous environment in the light of our results on experimental crosses, Wolbachia strains distribution, host ecological specialization, climate, and on the biology of C. sesamiae.
There are at least three strains of Wolbachia infecting C. sesamiae populations across Sub-Saharan Africa (Branca et al. 2011). We did not find bidirectional incompatibility between populations as a result of different infection. Only individuals infected with wCsesA and wCsesB2 strains showed incompatibility with cured or wCsesB1 infected Kit individual. Infected individuals wCsesA/wCsesB2 were already found highly incompatible in a previous study (Mochiah et al. 2002b), but incompatibility was not assessed for wCsesB1-infected individuals. The results for Wolbachia crosses involving wCsesB1 infected males and cured females does not present normal CI phenotype because there was no increase in male proportion in the progeny; however, we observed a reduction in progeny size (males and females) leading to a reduced number of females. This result is coherent with Wolbachia invasion theory since Wolbachia fitness is linked to the fitness, which female progeny size is a proxy, of Wolbachia-infected females relative to non-infected counterparts (Werren, 1997). However, this means that there is an unknown mechanism leading to high mortality of male eggs in incompatible crosses. Possibly, part of the male progeny includes in fact diploid males that could be affected in incompatible crosses, as diploid males are common in Cotesia wasps (Zhou et al. 2006; De Boer et al. 2007). A direct effect on development, not related to fertilization, could be also considered. In a similar way, surprisingly, no strong incompatibility was observed between Mbsa wCsesA/wCsesB2 cured female and Mbsa infected males as no biased sex ratio was found in the progeny. However, as in the case of Kit, a reduction in progeny size was observed which means that probably CI expresses differently between individual of the same genetic background (Kit or Mbsa), than when incompatible crosses occur between different genetic backgrounds. In the inter-population crosses studied here, a MD phenotype is very likely, as male-biased sex ratio was not associated to significant progeny size reduction. The consequence of this unidirectional incompatibility will be asymmetric gene flow between differentially infected populations (Jaenike et al. 2006; Telschow et al. 2006). Indeed, CI is an efficient mechanism for Wolbachia to spread within populations by giving infected females a higher fitness. We should therefore expect the spread of individuals infected with wCsesA/wCsesB2 across C. sesamiae geographical range, reflected by higher migration rate from wCsesA/wCsesB2-infected populations toward other populations. However, using microsatellite markers, we observed conversely a lower migration rate from wCsesA/wCsesB2 -infected genetic clusters toward the other clusters (Figure 5), except for the migration between cluster 4 and 2. This unexpected result may be explained by local adaptation. Regions where C. sesamiae are infected with wCsesA/wCsesB2 are indeed dominated by avirulent parasitoids and susceptible hosts, whereas regions where C. sesamiae are infected with wCsesB1 are dominated by virulent parasitoid attacking resistant host. Females migrating from bi-infected to wCsesB1 regions are maladapted and killed by encapsulation, but females migrating from wCsesB1 to regions with wCsesA/wCsesB2-infected individuals are able to develop on the host. Yet, males infected with wCsesB1 can reproduce with bi-infected females wCsesA/wCsesB2, which would allow some gene flow from wCsesB1 to wCsesA/wCsesB2. In conclusion, Wolbachia incompatibility favors the expansion of avirulent parasitoid wasp that are not capable to survive in some areas, and, in the opposite, the spread of virulent parasitoid is limited by area where parasitoid population are dominated by individuals infected with highly incompatible Wolbachia. This situation should lead to potentially stable contact zone between populations and current genetic structure.
To disentangle the effects of geography, Wolbachia infection, parasitoid host, and other ecological factors, a statistical model was optimized using adonis R function. The biotic and abiotic factors including geography analyzed in our statistical model explained more than 75% of the genetic variance. When looking at the factors most correlated to the genetic structure, our results are consistent with the hypothesis that ecology plays a significant role in reinforcing C. sesamiae population structure across evolutionary time. Indeed, adonis analysis showed that the strongest determinant of genetic variance was Wolbachia infection followed by the host species and the least contributing factors were localization and climate. An illustration of the dominant effect of the host is the particular status of the population represented by cluster 1, consistently collected on Sesamia nonagrioides. This population shows also higher Fst when compared to the other populations in every clustering method confirming it constitutes a new species as it has recently been proposed (Kaiser et al. 2015, 2017). Another population corresponding to cluster 5 expands from Cameroon to East-Africa and Uganda, trough Democratic Republic of Congo; this region corresponds to the great Equatorial forest of Africa, which is characterized by hot and wet climatic conditions. The cluster 4, located from Eastern Kenya to Mozambique along the Coast, is situated in a much drier area than cluster 5. This area is also important regarding host, since B. fusca, characterized as a resistant host, is rare in those regions (Le Ru et al. 2006; Moolman et al. 2014). The cluster 2 and 3 are located both in North-Eastern Sub-Saharan Africa but their positions differed according to clustering algorithm (i.e. West Kenya, Ethiopia and Eritrea). In terms of climatic conditions, these regions are very similar but the observed clusters might reflect two sympatric populations with recurrent gene flow as they are infected with the same Wolbachia strains (Figure 3).
These C. sesamiae populations show some geographic similarities with the genetic structure observed in the known resistant host B. fusca (Dupas et al. 2014), with five clusters observed across Africa, and a strong structure observed in East African Rift Valleys regions, contrasting with reduced structure observed in South and Central African regions. The cluster 3 of C. sesamiae located between Eastern and Western Rift Valley has an overlapping distribution with “H” cluster of B. fusca. The cluster 2 on the East of Eastern Rift overlaps with “KE” cluster of B. fusca. The cluster 4 of C. sesamiae ranges in East Africa at lower altitudes where B. fusca is rare or absent (Dupas et al. 2014) and to the south. The clusters 4 and 5 exhibit large distributions that overlap with the “S” cluster of B. fusca from South to East and Central Africa (Figure 2). A fifth population is also present in both species. Cluster 1 of C. sesamiae corresponds to parasitic wasp infecting S. nonagrioides that has been described as a new species, C. typhae. Cluster “W” of B. fusca is only present in West Africa and isolated from the other B. fusca populations (Figure 2). These results suggest that B. fusca and C. sesamiae share a common phylogeographic history that explain the current genetic structure of both species. For instance, the highest diversity for both species has been found in the East African Rift Valley. The East African Rift valley also explained the differentiation observed between two C. sesamiae lineages based on 6 mitochondrial and nuclear markers (Kaiser et al.,2015). One lineage corresponds geographically and ecologically to clusters 2 and 3, and the second one to cluster 4. The East African Rift Valley has already been observed as a center of diversification for several species (Odee et al., 2012; Habel et al, 2015; Freilich et al, 2016; Mairal et al., 2017). This observed biological diversity has been related to both topological heterogeneity and variable climatic conditions that occurred since the formation of the East African Rift Valley ca. 20 Mya, with the alternation of arid and wet periods (Sepulchre et al., 2006). Therefore, we could explain this observed pattern either by first the colonization of the East African Rift Valley followed by diversification or that the origin of both species lays in the East African East Valley which has been followed by further extension with admixture across Africa, except in West Africa, where C. sesamiae is absent and where B. fusca is totally isolated with zero migration observed to date (Sezonlin et al. 2006; Dupas et al. 2014).
Wolbachia and biological control
It is widely acknowledged that a better understanding of tritrophic interactions between plants, phytophagous insects and associated antagonists can help to develop better pest management strategies by identifying bottom-up and top-down effects in the food chain (Agrawal, 2000). Wolbachia can be considered as a fourth trophic level in such system but, the impact of Wolbachia on parasitoid host plant interactions has not received much attention. It was shown that a Wolbachia strain invasion temporarily reduces the impact of the parasitoid on its host (Branca & Dupas 2006). But this impact can be sustained in the case of stable contact between incompatible strains in “hybrid” zones. Conversely, Wolbachia can reinforce adaptive divergence between locally adapted populations to the benefit of the parasitoid (Branca et al. 2009). Cotesia sesamiae is a good model to test the effect of Wolbachia on host parasitoid assemblages as the four consensus genetic clusters differed for their Wolbachia and Lepidoptera host associations. In hybrid area, maladaptive gene flow may be observed and limited by Wolbachia strain bidirectional incompatibility. This is the case between coastal (Mbsa) and inland (Kit) populations of the parasitoid (Dupas et al. 2008). The maladaptation may be the strongest in the AS Köppen Geiger Climate Zone (corresponding to dry mid altitude agroclimatic zone) in wet seasons when B. fusca represents half of the host community (Ong’amo et al. 2006), whereas avirulent C. sesamiae toward B. fusca dominates parasitoid populations (Dupas et al. 2008). Strong counter counter-selection of avirulent alleles is expected in B. fusca abundance peaks. Busseola fusca is dominant in some seasons in mid altitude areas where virulent alleles dominate (Dupas et al. 2008). Although avirulent parasitoids are able to select host at contact, which may reduce maladaptation in the field, using contact cues to select host is risky because the host can bite and kill the parasitoid before oviposition can be made; 25% of C. sesamiae entering the stem tunnel are killed by S. calamistis larvae upon contact (Potting et al. 1999). The presence of partially incompatible Wolbachia strains in the virulent and avirulent parasitoid populations may favor their cohesiveness in balancing host communities across seasons. Hence, reducing gene flow between locally adapted populations toward their host, in absence of premating isolation, might reduce maladaptation in hybrid zone and our study confirms Wolbachia can reinforce this process. For instance, very few heterozygous females between virulent and avirulent alleles on the bracovirus CrV1 locus has been found in a previous study, since they are likely maladapted (Branca et al., 2011). Therefore, we would expect a lack of recombination and strong diversification on genes, particularly at the bracovirus locus, related to host specificity in C. sesamiae, pattern that has yet to be investigated at the genome level.
Thompson (2005), in his seminal book on coevolutionary mosaics stressed that gene flow had an ambivalent influence on coevolutionary interactions. Gene flow is essentially maladaptive, bringing locally maladapted genes to populations in interaction (Nuismer 2006), but in the presence of negative frequency dependent dynamics of coevolutionary interactions, rare new variants originating from other populations may be adapted. Our results show some congruence between C. sesamiae and B. fusca genetic structure (Dupas et al. 2014). Congruence with host structure is therefore observed at different ecological levels, not only at the level of host genus as shown by adonis analyses but also at the level of host populations. This may reduce maladaptation of C. sesamiae toward B. fusca and favor local coevolutionary interactions.
Conclusion
Our study presents a unique comprehensive case for assessing the determinant of genetic structure in a parasitoid species, including multiple interactive biotic and abiotic forces. The parasitoid, like its main host B. fusca, likely diversified across the East African Rift Valleys where all the genetic clusters are found. Despite their wide distribution across Sub-Saharan Africa, some populations have maintained their integrity as shown by the non-spatial admixture model. Two important results pinpoint toward the strong influence of host on parasitoids population dynamics and population genetics at a large geographical scale: (1) although the species genetic clusters appear to have diversified across East African Rift Valleys refuges, host species that are distributed across Africa became then the strongest factor determining genetic structure, rather than climatic selection and geographic isolation (2) migration rate inferred from Bayesian analysis of microsatellite data suggests a limitation of gene flow due more to host adaptation than to Wolbachia infections. This result has a fundamental importance in the context of biological control program. As opposed to chemical control agents, biological control agents are expected to be able to cope with host evolution (Holt & Hochberg 1997) but other interactions may limit this evolutionary sustainability. In our case, parasitoid wasps are able to cope with host evolution despite many additional biotic and abiotic ecological forces including reproduction manipulators that would be expected to reduce local adaptation to host. The insect host dominates the piling up of all these factors and could explain why parasitoids can be very successful biological control agents even when introduced in climatically and geographically distant environments from their native settings (Stiling & Cornelissen 2005). More generally, this work supports the hypothesis of the higher impact of ecological versus neutral forces and of host versus other ecological forces on the diversification of parasitoid - host interactions.
References




