

Abstract
Selective pressures imposed by pathogens have varied among human populations throughout their evolution, leading to marked inter-population differences at some genes mediating susceptibility to infectious and immune-related diseases. A common polymorphism resulting in a C529 versus T529 change in the Cadherin-Related Family Member 3 (CDHR3) receptor is associated with rhinovirus-C (RV-C) susceptibility and severe childhood asthma. Given the morbidity and mortality associated with RV-C dependent respiratory infections and asthma, we hypothesized that the protective variant has been under selection in the human population. Supporting this idea, a recent cross-species outbreak of RV-C among chimpanzees in Uganda, which carry the ancestral ‘risk’ allele at this position, resulted in a mortality rate of 8.9%. Using publicly available genomic data, we sought to determine the evolutionary history and role of selection acting on this infectious disease susceptibility locus. The protective variant is the derived allele and is found at high frequency worldwide, with the lowest relative frequency in African populations and highest in East Asian populations. There is minimal population structure among haplotypes, and we detect genomic signatures consistent with a rapid increase in frequency of the protective allele across all human populations. However, given strong evidence that the protective allele arose in anatomically modern humans prior to their migrations out of Africa and that the allele has not fixed in any population, the patterns observed here are not consistent with a classical selective sweep. We hypothesize that patterns may indicate frequency-dependent selection worldwide. Irrespective of the mode of selection, our analyses show the derived allele has been subject to selection in recent human evolution.
Introduction
There is accumulating evidence to suggest that immunity-related genes are preferential targets of natural selection (reviewed in 1–5), supporting the notion that infectious diseases have been important selective forces on human populations6. However, for most candidate loci, the mechanisms and phenotypic effects underlying the observed signatures of selection remain elusive.
Human rhinoviruses (RV) are found worldwide and are the predominant cause of the common cold. While many RV infections cause only minor illness, type C strains of the virus are associated with higher virulence. Unlike RV-A and RV-B, RV-C utilizes the Cadherin Related Family Member 3 (CHDR3) receptor to gain entry into host cells. A missense variant in CDHR3 (rs6967330, C529T) results in a 10-fold difference in virus binding and replication in vitro 7, and experimental evidence points to varied cell surface expression between the two forms of the receptor as a likely mechanism8. Adding clinical support that CDHR3 functions as an RV-C receptor, the ancestral allele at rs6967330 has been found to be associated with an increased risk of respiratory tract illness by RV-C in two birth cohorts9. The allele is also associated with a severe form of childhood asthma, in accordance with the known role of RV, particularly RV-C, in triggering asthma exacerbations10. Taken together, these studies suggest that host genetics mediate differing susceptibility to RV-C infection and asthma by affecting interactions between the virus and its receptor (e.g. whether or not the protein is expressed on the cell surface and is thus visible to the virus).
A recent outbreak of lethal respiratory illness among wild chimpanzees was attributed to human RV-C crossing species boundaries. All members of the infected chimpanzee population were invariant for the ancestral variant at the homologous position corresponding to the human rs6967330 variant. The outbreak resulted in a staggering 8.9% mortality rate 11, and suggests that RV-C infection can result in a high mortality rate in a susceptible population. As respiratory infections, particularly prior to the availability of modern medical interventions, represent significant threats to human health12–15, CDHR3, and in particular rs6967330, represent a promising candidate target of natural selection. In the present study, we investigated the evolutionary history of this locus for which there already exists strong experimental and clinical data linking genotype with phenotypes that appear to modulate disease susceptibility.
Results & Discussion
Long-term evolution of the CDHR3 locus
Examination of the locus in an alignment of 100 vertebrate genomes16,17 revealed that the CDHR3 locus is highly conserved, with homologs present in 85 species (Figure 1). Tyrosine is the ancestrally encoded amino acid at the homologous position 529 in the human protein sequence. There appears to have been a mutation leading to an A529T amino acid change introduced in a common ancestor of opossums, Tasmanian devils, and wallabies that results in the encoding of a phenylalanine at the homologous position. Numerous vertebrates (elephant, horse, rabbit, pika, naked mole-rat, rat, mouse, golden hamster, Chinese hamster, prairie vole, and lesser Egyptian jerboa) spread throughout the species tree also encode a different amino acid (histidine) at this position. Whether these mutations are fixed in each of these species requires further sequencing, and the effect of these nonsynonymous substitutions on protein expression and function remain to be explored.
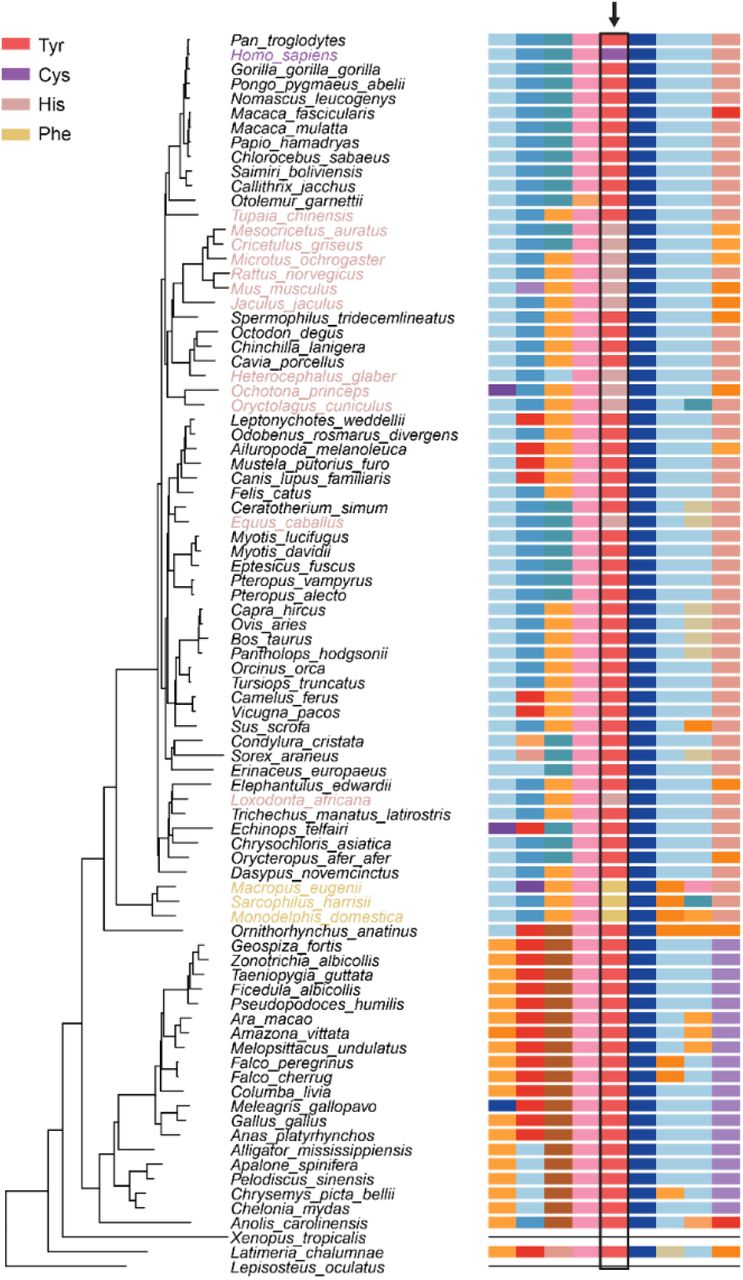
(Left) Species tree for the multi sequence alignment of 85 species in the UCSC multiz alignment. (Right) Multi sequence protein alignment surrounding position 529 of the human CDHR3 protein. Residue 529 is outlined in black and designated with an arrow. Humans carry the Tyr and Cys alleles. Data obtained from the UCSC Genome Browser16,17.
Providing further evidence that the locus has been evolutionary constrained, rs6967330 has a positive Genomic Evolutionary Rate Profiling (GERP) score of 4.39, signifying a deficit of substitutions compared to neutral expectations. Values of the statistic calculated for bases within CDHR3 range from −12.2 to 6.08 (mean = −0.15, sd = 2.59) and from −12.3 to 6.17 chromosome-wide (mean =-0.09, sd = 1.91)17,18. The apparent conservation of this locus suggests it is important for the function of CDHR3 and thus makes it an interesting candidate of natural selection.
Population diversity patterns of the CDHR3 locus
Excluding Homo sapiens, sequencing data from the remaining extant species comprising all hominids (great apes) are invariant at the position corresponding to rs696733019. Genotyping of an additional 41 chimpanzees whose community experienced a severe respiratory outbreak of RV-C in 2013 revealed that all individuals were homozygous for the ancestral allele11. In examining hominin genomes, we find that Neanderthals and Denisovans carry the ancestral allele, while ancient specimens of anatomically modern humans carry both the ancestral and derived alleles. In low coverage sequencing data of H. sapiens estimated to have lived between 5000-8000 years ago (using sequencing reads from 45 aDNA samples for which we felt confident making diploid genotype calls) we estimate the allele frequencies at rs6967330 to be 34.4% A (ancestral) and 65.5% G (derived) (Table S1)20. Higher coverage aDNA extracted from a 7,000 year old skeleton found in Germany and an 8,000 year old skeleton from the Loschbour rock shelter in Luxembourg reveals heterozygotes at the locus21. Finally, a 45,000 year old early H. sapiens from western Siberia is a homozygote for the derived allele22. Collectively, these data suggest that the derived allele likely arose in the evolutionary branch leading to anatomically modern humans, although population data for Neanderthals and Denisovans remains scarce.
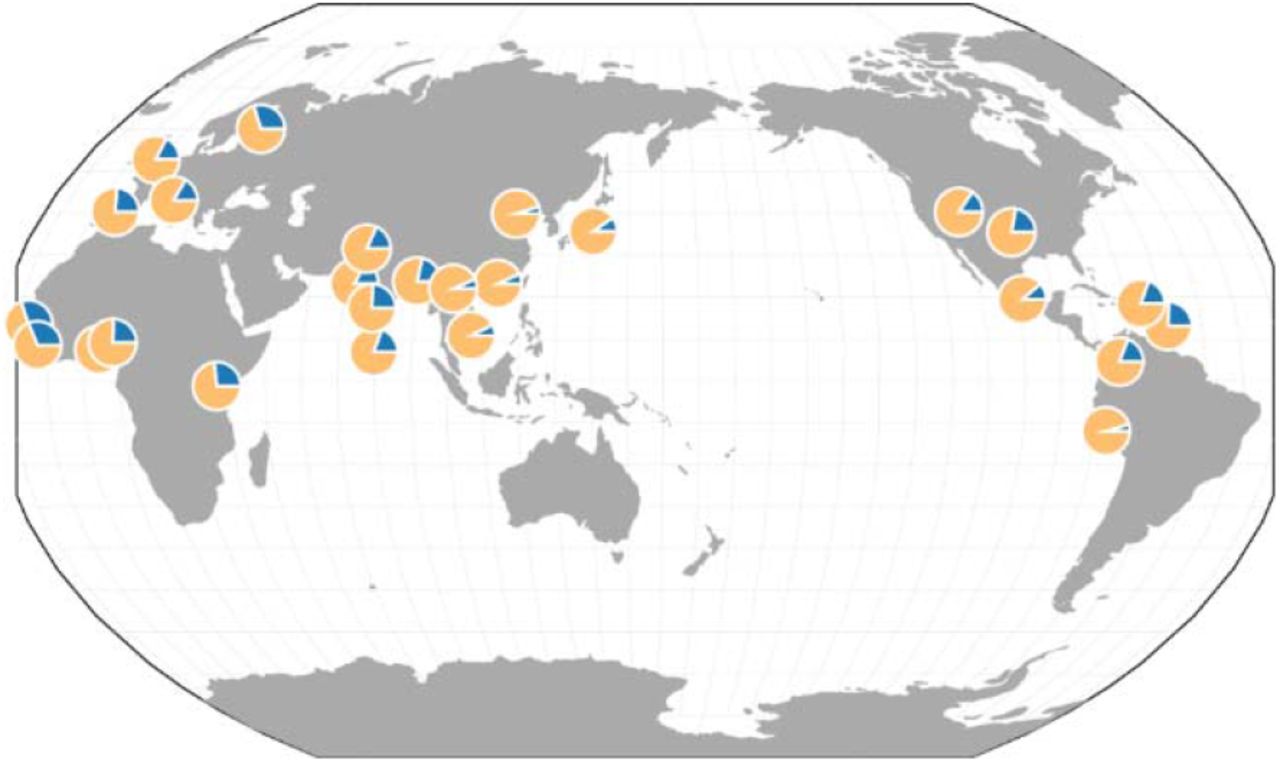
The locus represents a shared polymorphism in contemporary worldwide populations. Based on whole genome sequence data for 2504 individuals from 26 different populations23, we find that the derived (‘protective’) G allele is, on average, very common in East Asian populations (EAS, 93.0%), followed by admixed American populations (AMR, 85.7%), South Asian populations (SAS, 80.0%), and European populations (EUR, 79.2%). It is least common in African populations (AFR, 73.5%), albeit also at high frequency. At the individual population level, allele frequencies of the derived G allele range from 68.8% (“Mende in Sierra Leone”, MSL) to 95.3% (“Peruvians from Lima, Peru”, PEL) (Figure 2).

Pie charts represent the relative allele frequencies of the ancestral A allele (blue) and the derived G allele (yellow) in each of the 26 populations of the 1000 Genomes Project23. This image was generated through the Geography of Genetic Variants browser52.
We next investigated linkage disequilibrium (LD) patterns around CDHR3 with Haploview24. A tight LD block was identified on chromosome 7 from 105,657,078-105,659,873, with only moderate LD decay extending up to 105,680,022. Considering only biallelic SNPs with a minor allele frequency (MAF) > 0.01, haplotypes within these blocks were identified with the Pegas package in R25,26. Relationships among haplotypes occurring at >1% were inferred using network analysis (Figure 3). Eight haplotypes at frequency ≥ 1% were identified from the region of high LD, with the majority of individuals in all populations carrying the same haplotype with the derived G allele (n = 3848); two less frequent haplotypes carrying the derived allele are found in various geographic regions. The ancestral allele is found in the remaining five haplotypes, which vary in their distributions across regions. Twenty-eight haplotypes were found using the same criteria of a MAF > 0.01 and haplotype frequency of > 1% in the larger genomic block containing moderate LD. Phylogenetic reconstruction of the resulting haplotypes in this region resulted in a clear separation of those carrying the ancestral and derived alleles. Unexpectedly, Chimp, Neanderthal and Denisovan haplotypes are closer to H. sapiens A-haplotypes suggesting that natural selection shaped the haplotype pattern (Figure S1).

(Top) Haplotype network from chromosome 7 between 105,657,078 and 105,659,873. Haplotype network was constructed from phased genome sequences of 2504 individuals with variation at 16 sites in the 2,795bp region. Only haplotypes occurring at > 1% are shown. Colors reflect super population designation of individuals. (Bottom) Unrooted tree from chromosome 7 between 105,657,078 and 105,680,022. Haplotypes of the same 2504 individuals were derived for a larger haplotype block. Again, only haplotypes occurring at > 1% are shown and colors reflect super population designation of individuals. The tree is based on 83 SNPs (68 informative) from the 22,945bp region.
World-wide Selection at the locus
Given the morbidity and mortality associated with viral respiratory infections13,15 and severe childhood asthma12,14 particularly prior to the availability of modern medical interventions, we hypothesized that the frequency of the protective G allele (resulting in a Cys529) at rs6967330 might have increased more rapidly than under neutrality. We performed genome-wide scans for selection in the 26 populations of the 1000 Genomes Project23. To do so, we used the integrated haplotype score (iHS)27 and the number of segregating sites by length (nSL)28, which are designed to capture rapid increases in frequency of selected variants and have been applied in the context of detecting recent positive selection 27,29,30. We calculated these neutrality statistics in each population independently and normalized by frequency-bin for all loci with a DAF ≥ 0.2 (Figure S2). Fourteen populations presented extreme values of iHS and nSL (≥ 95th percentile of the distribution in the population) at rs6967330, with 2 and 5 populations falling in the 99th percentile for the two statistics, respectively. CDHR3 lies in a region of high recombination (1.8cM/Mb) and rs6967330 resides in a male specific recombination hotspot17,31. This may explain the slightly different patterns observed in nSL and iHS statistics, as an excess of extreme values of iHS have been observed at regions of low recombination28.
To investigate whether selection has acted on all populations simultaneously, we implemented a multi-population (MP) combined statistic approach for iHS and nSL. For each SNP in each population, we combined the percent ranks of iHS and nSL using Fisher’s method, and defined the MP-iHS and MP-nSL as the resulting χ2 value. We obtained a MP-iHS score of 156 and a MP-nSL score of 157 for rs6967330. Both scores are in the 99th percentile, regardless of whether we include only segregating sites in 2 or more populations or limit it to those SNPs segregating in all 26 populations examined. This suggests that the derived allele at rs6967330 is advantageous and that non-neutral processes have acted on this locus across global populations.
A complex evolutionary scenario
We originally hypothesized that selection pressures imposed by RV-C infection may have resulted in a classic selective sweep at the locus; however, our results here are not consistent with such a model. Preliminary dating estimates of RV-C point to a recent origin in the last few thousand years8. We infer that the protective (derived) allele was already present in ancestral populations at intermediate frequencies during this time frame (65.5% in aDNA dated 5,000-8,000 years ago). Haplotype-based statistics such as iHS and nSL have low power to detect selection occurring on standing variation with intermediate frequency32. This suggests that patterns of variation observed at rs6967330 are unlikely to have resulted from selection imposed by RV-C itself; our findings suggest instead that an alternative selective agent(s) may have been/be acting on the CDHR3 locus prior to emergence of RV-C. These findings well parallel those observed at the chemokine receptor gene-5 (CCR-5), where an unknown historical selective pressure maintained a deletion in this gene that attenuates infectivity and disease progression of HIV (reviewed in 33), because that mutation clearly predates the emergence of AIDS.
Frequency-dependent Selection
Several lines of evidence point to the derived allele at rs6967330 arising before the out-of-Africa dispersal, including a lack of haplotype structure (Figure 3) and the presence of the allele at homozygous state in aDNA from anatomically modern humans dating back to ~45,000 year old20–22. While we find signatures of selection at the locus, the protective variant has not fixed in any of the contemporary populations examined (Figure 2), contrary to expectations for a strongly positively selected variant that arose prior to human migrations out of Africa34. We therefore wondered if balancing selection could explain observed patterns of variation at the locus, as balancing selection can maintain functional diversity over long periods of time through frequency-dependent selection, heterozygote advantage, pleiotropy, and fluctuating selection35. Until recently, evidence for balancing selection in the human genome was limited to a few classical cases such as the heterozygous advantage conferred by the HbS sickle cell mutation against malaria36,37, genes of the major histocompatibility complex/human leukocyte antigen complex38, and the ABO blood group39. Balancing selection, however, has recently been recognized as more prevalent than previously thought, particularly in shaping human immune system phenotypes35,40,41. To test whether the rs6967330 locus has been under long-term balancing selection (i.e. selection occurring over at least hundreds of thousands of generations 38,42,43), we examined the distribution of β, a recently developed summary statistic designed to detect clusters of alleles at similar frequencies, in 1000 Genomes Project data44. β was similar across all populations around the rs6967330 locus, and values were not indicative of long-term balancing selection (Figure S3).
Balancing selection operating over shorter timescales clearly also plays a role in shaping human diversity (e.g. HbS sickle cell mutation36,37). Such a short-term balancing selection scenario could explain the signals we detected with haplotype-based statistics, as genomic signatures of short-term balancing selection are predicted to be indistinguishable from incomplete sweeps of positive selection 27,41. In both of these scenarios, the selected allele rapidly increases in frequency, with the allele eventually fixing under positive selection or oscillating around a steady state in the case of balancing selection. As a reminder, haplotype-based selection methods suggest that rs6967330 rose in frequency faster than under neutrality, both at the global and the individual population level. Furthermore, population differentiation as measured with FST at this locus is low relative to other SNPs with the same frequency (Figure S4), a finding that is also consistent with short-term balancing selection across multiple populations35,38,41. Based on our observation that the derived variant at rs6967330 was present at high frequencies in ancient human specimens and in the homozygous state in the case of a 45,000-year-old fossil, we posit that this allele may have started to increase in frequency in ancestral populations before reaching its current equilibrium frequency independently in worldwide populations. The derived allele ranges in frequency from 68.8-95.3% in modern human populations, which is compatible with an equilibrium frequency being high. It is possible that as the allele frequency of the derived mutation reaches high frequencies in a population, circulation of the viruses exploiting CDHR3 (past and present) are affected, with attendant impacts on the selective pressures they impose on human populations. Such a frequency-dependent selection scenario seems more plausible than that of a heterozygote advantage given that in Danish children with severe asthma, having even one copy of the risk variant was associated with increased risk of exacerbation and hospitalization10. If there were another virus that exploited this receptor in past human populations, we might assume that heterozygotes would similarly not be conferred protection.
In conclusion, our analyses combined a priori knowledge of a genetic variant underlying variable susceptibility to RV-C infections7,9,11 with population genetic analyses of whole genome sequence data to investigate the evolutionary history of the locus in CDHR3. The conservation of the protein, combined with its complex evolutionary history, exemplifies the biological importance of CDHR3, which may or may not be ultimately relevant to its function as the cellular surface receptor for RV-C. We detected a worldwide signature of selection at this locus, but also found that patterns of variation do not conform to the classic selective sweep model. Instead, we posit the possibility of frequency-dependent balancing selection operating at this locus which warrants more investigation into genotypic and biochemical effects of the variant (e.g. whether there is any phenotypic benefit to being heterozygous, trade-offs of different genotypes, etc.). Irrespective of the mode of selection, our analyses show that the derived allele has been advantageous and selected for in recent human evolution.
Methods
Datasets
1000 Genomes Project
Individual level phased sequencing data from the 1000 Genomes Project Phase 3 dataset were downloaded from ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/23.
Ancient H. sapiens
BAM files for each of the 230 individuals included in the study by Mathieson et. Al.20 were downloaded and converted to MPILEUP format using samtools45. Low coverage sequencing data at asthma susceptibility loci were manually inspected for each individuals for which at least one read had mapped to the site. Manual diploid genotyping calls were made for aDNA samples for which we felt confident making diploid genotype calls (Table S1).
Neandertal and Denisovan
Geontypes for the Vindija, Altai, and Denisovan genomes46 generated using snpAD, an ancient DNA damage-aware genotyper, were downloaded from http://cdna.eva.mpg.de/neandertal/Vindija/VCF/.
Great Apes
Genotypes of primates sequences were obtained from https://eichlerlab.gs.washington.edu/greatape/data/ and converted to the corresponding human regions with the LiftOver software17.
Haplotype Networks
Indels and multi-allelic sites were filtered out with bcftools45. Variants having a Hardy-Weinberg equilibrium exact test p-value below 1×10−5 as calculated using the – hwe midp function in PLINK1.947 in any of the 26 populations were removed from all populations. The core haplotype surrounding rs6967330 was identified using biallelic markers within 100kb of rs6967330 in Haploview24 from all 26 populations in the 1000 Genomes Phase 3 release. A large haplotype block was defined on Chromosome 7 from 105,657,078 to 105,680,022, and a smaller haplotype of Chromosome 7 from 105,657,078 to 105,659,873. Haplotypes within the defined haplotype blocks were extracted from biallelic markers with a minor allele frequency (MAF) > 0.01 with the Pegas package25,26. Haplotypes occurring at >1% (at least 26 individuals) in the total 1000 Genomes Project dataset were constructed into networks. Genotypes from two high quality Neanderthal genomes and a Denisovan genome were similarly extracted and used in network analyses.
Haplotype-Based Selection Scans
We computed iHS and nSL using the method implemented in Selink, a software to detect selection using whole-genome datasets (https://github.com/h-e-g/selink). As iHS and nSL are sensitive to the inferred ancestral/derived state of an allele, we computed these statistics only when the derived state was determined unambiguously32. Results were normalization by derived allele frequency (DAF) bins (from 0 to 1, increments of 0.025)27,32. We also minimized the false-positive discovery by excluding SNPs with a DAF below 0.2, as the power to detect positive selection has been shown to be limited at such low frequencies27,32. For each statistic, we considered the percent rank at rs6967330 relative to the genome-wide distribution in each population.
In order to test selection shared among populations, we combined iHS and nSL into a single multi-population (MP) combined selection score, MP-iHS and MP-nSL. The rationale behind these composite approaches48–50 is that neutrality statistics, though expected to be correlated among populations under neutrality, are more strongly correlated for positively selected variants than for neutral variants. Indeed, under global positive selection, iHS and nSL tend to become negative in all populations while false positives will only be negative in a few populations. Consequently, candidates genuinely selected in several populations should harbor extreme values for MP-iHS and MP-nSL. For each SNP and each population, we determined the empirical p-value of iHS and nSL, i.e. the rank of each statistic in the genome-wide distribution divided by the number of SNPs. We then used the combine_pvalues function implementation of Fisher’s method51 of the SciPy python package to compute MP-iHS and MP-nSL for every SNP (passing the criteria above to compute iHS and nSL) found in at least two populations.
β Test for Long-Term Balancing Selection
β scores for each population in the 1000 Genomes Project were obtained from https://github.com/ksiewert/BetaScan.
Population Differentiation
Global FST was calculated with Selink (https://github.com/h-e-g/selink).
Author Contributions
MBO, CSP, and ACP conceived the project. MBO, GL, and JCT performed the analyses. MBO drafted the paper and made the figures. All authors provided critical feedback, reviewed, and edited the manuscript.
Competing interests
The authors declare no competing interests.
Acknowledgements
We wish to thank Andrew Kitchen for his advice on phylogenetic analyses and Lluis Quintana-Murci for feedback on the manuscript.





{kind=link}
{kind=link}
{kind=link}