

Abstract
Antibiotic resistance in pathogens is extensively studied, yet little is known about how antibiotic resistance genes of typical gut bacteria influence microbiome dynamics. Here, we leverage genomes from metagenomes to investigate how genes of the premature infant gut resistome correspond to the ability of bacteria to survive under certain environmental and clinical conditions. We find that formula feeding impacts the resistome. Random forest models corroborated by statistical tests revealed that the gut resistome of formula-fed infants is enriched in class D beta-lactamase genes. Interestingly, Clostridium difficile strains harboring this gene are at higher abundance in formula-fed infants compared to C. difficile lacking this gene. Organisms with genes for major facilitator superfamily drug efflux pumps have faster replication rates under all conditions, even in the absence of antibiotic therapy. Using a machine learning approach, we identified genes that are predictive of an organism’s direction of change in relative abundance after administration of vancomycin and cephalosporin antibiotics. The most accurate results were obtained by reducing annotated genomic data into five principal components classified by boosted decision trees. Among the genes involved in predicting if an organism increased in relative abundance after treatment are those that encode for subclass B2 beta-lactamases and transcriptional regulators of vancomycin resistance. This demonstrates that machine learning applied to genome-resolved metagenomics data can identify key genes for survival after antibiotics and predict how organisms in the gut microbiome will respond to antibiotic administration.
Importance The process of reconstructing genomes from environmental sequence data (genome-resolved metagenomics) allows for unique insight into microbial systems. We apply this technique to investigate how the antibiotic resistance genes of bacteria affect their ability to flourish in the gut under various conditions. Our analysis reveals that strain-level selection in formula-fed infants drives enrichment of beta-lactamase genes in the gut resistome. Using genomes from metagenomes, we built a machine learning model to predict how organisms in the gut microbial community respond to perturbation by antibiotics. This may eventually have clinical and industrial applications.
Introduction
Antibiotic use has been steadily increasing over the past several decades and is correlated with the prevalence of antibiotic resistance in bacteria1. Widespread antibiotic resistance, in combination with the decline in development of new antibiotics, presents a major threat to human health2. The gut microbiome is a reservoir for antibiotic resistance genes3 and may be involved in the spread of resistance genes to pathogens4-6. Additionally, antibiotics are often prescribed to treat infections without considering how the drug will affect the gut microbial community, which can lead to negative consequences for the human host7. It is therefore important to study how the antibiotic resistance genes harbored by organisms in the gut microbiome impact community dynamics.
The preterm infant gut resistome is considered a research priority because premature infants are almost universally administered antibiotics during the first week of life8. Early life is a critically important time for community establishment9, and neonatal antibiotic therapies have both transient and persistent effects on the gut microbial community. Included among the many ways that antibiotics have been shown to affect the microbiome are lower bacterial diversity10, enrichment of Enterobacteriaceae10,11, reduction of Bifidobacterium spp.12, and enrichment of antibiotic resistance genes13, including those that have no known activity against the particular antibiotic administered14. Previous studies have shown that the community composition of the infant microbiome is affected by diet, with artificial formula selecting for Escherichia coli and Clostridium difficile15, and breast milk selecting for certain strains of Bifidobacterium16. The effect of birth mode on the microbiome is contested, with most studies finding that it has an effect on the gut microbiome17-19 although some show no effect20,21. Gender22 and maternal antibiotics before or during birth23-25 also influence microbiome assembly.
Here we use genome-resolved metagenomics coupled with statistical and machine learning approaches to investigate the gut resistome of 107 longitudinally sampled premature infants. We show that certain antibiotic resistance genes in particular genomes affect how clinical factors influence the gut microbiome and, in turn, how the antibiotic resistance capabilities of a gut organism influence its growth and relative abundance.
Results and Discussion
Antibiotic resistance of the premature infant microbiome
107 premature infants were longitudinally sampled during the first three months of life, resulting in a total of 902 samples that were sequenced and analyzed. All 107 infants received gentamicin and ampicillin during the first week of life, and 36 of those infants received additional antibiotics in the later weeks due to disease (Table 1). All samples were subject to Illumina short-read shotgun sequencing and the sequence data assembled using idba-ud (see methods for details). Resfams26 annotations of predicted amino acid sequences from the resulting scaffolds revealed that the most abundant resistance mechanisms were resistance-nodulation-division (RND) efflux pumps and ATP-binding-cassette (ABC) transporters (Fig 1). A slight, yet significant, decreasing trend in total antibiotic resistance potential is observed over time (p < 0.005). During the first week of life, empiric antibiotic therapy perturbs the microbiome though selection for antibiotic resistant organisms. This is consistent with prior results showing temporarily elevated resistance gene levels after administration of antibiotics17. Microbial community recovery begins following this period.
All 107 infants in this analysis were in the neonatal intensive care unit (NICU) of the Magee-Women’s Hospital in Pittsburgh, PA. The infants in the received post-week antibiotics column (right) were administered antibiotics while in the NICU beyond the first week of life due to late-onset sepsis, necrotizing enterocolitis, or another disease. The antibiotics administered included ampicillin, gentamycin, vancomycin, cefotaxime, cefalozin, amoxicillin, clindamycin, naficillin, ofloxacin, and piperacillin-tazobactam.

Antibiotic resistance genes of the premature infant gut microbial community. Genes were annotated through hidden Markov model searches of the Resfams database and categorized based on resistance mechanism. During the time period studied, the total resistance content of the premature infant gut microbiome has a slight negative correlation with age (Pearson’s r = −0.1, p = 0.003.)
Formula feeding influences the gut resistome through strain-level selection
Permutational multivariate analysis of variance (PERMANOVA) tests, which discern and isolate the effects of factors through partitioning of variance27, were performed to investigate the effect of feeding regimen, delivery mode, gender, maternal antibiotics, and the infant’s current antibiotic therapy on the resistome. Tests were performed on the resistomes of samples taken at weeks two, four, and six (see methods for details). At week two, formula-fed infants did not have a significantly different distribution of antibiotic resistance genes compared to infants that received breast milk. However, a difference was detected at weeks four and six (p < 0.05), accompanied by an increase in effect size as assessed by PERMANOVA F-statistic (Table S1). This signals divergence of the resistomes of formula-fed and breast-fed infants over time. Interestingly, other factors such as birth mode and antibiotic administration were not associated with a significant difference in resistance gene distribution.
Random forest models were used to classify resistomes as either belonging to a formula-fed baby or a breast-fed baby, and we used the trained model’s feature importance scores to select resistance genes for further study (Table 2). One out of the four selected resistance genes was significantly associated with a feeding group: Class D beta-lactamase was enriched in formula-fed infants (p < 0.05) (Fig 2A). The mexX gene, which encodes for an efflux pump subunit, exhibited a comparatively higher feature importance score, but there was no significant difference in its abundance between feeding groups (Table 2). This suggests that the oft-used practice of utilizing feature importance scores of random forests or other machine learning ensemble methods to identify markers of differences between groups28-30, although potentially informative, may not necessarily rank features in the order that most meaningfully reflects their contributions. This is likely due to a preference for correlated predictor variables31.

Formula feeding affects the resistome. (A) Class D beta-lactamase is enriched in formula fed infants at four weeks of age (Mann-Whitney U = 66, Bonferonni corrected p = 0.031). (B) Phylogenetic tree of Clostridium difficile genomes based on the ribosomal protein S3 gene. Names of genomes harboring a Class D beta-lactamase are colored green and labeled with an asterisk. (C) The relative abundance of C. difficile genomes with Class D beta-lactamase in formula-fed and breast-fed infants (top, n = 38) and the relative abundance of C. difficile genomes lacking Class-D betalactamase in formula-fed and breast-fed infants (bottom, n = 29).
Mann-Whitney U tests were performed on features selected by the random forest model gini importance metric after training on resistomes of formula-fed infants and breast-fed infants. Bonferonni corrections were applied to the p-values to adjust for multiple testing.
Genome-resolved analysis revealed that Class D beta-lactamase genes are most frequently carried by Clostridium difficile. Of the 67 C. difficile genomes in the dereplicated dataset, 38 of these organisms harbor a Class D beta-lactamase gene. Phylogenetic analysis reveals that these 38 organisms are very closely related (Fig 2B). To ascertain if this C. difficile strain is involved in the enrichment of Class D beta-lactamase in the formula-fed infant gut resistome, the relative abundance of C. difficile with and without a Class D beta-lactamase gene in the gut microbiome of breast-fed and formula-fed infants was assessed. In infants that only receive formula, C. difficile with Class D beta-lactamase is consistently more abundant than C. difficile lacking this gene; while in infants that receive breast milk, both types of C. difficile are low in relative abundance (Fig 2C). Prior studies have reported an increased abundance of C. difficile in the gut microbiomes of formula-fed infants15, but here we reveal that formula feeding selects for a particular C. difficile strain.
Class D beta-lactamase hydrolyzes beta-lactam antibiotics32, and there is no known connection between host diet and its antibiotic resistance function. Since it is thus unlikely that Class D beta-lactamase offers a selective advantage to organisms in the gut of formula-fed infants, other possibilities were explored. Pairwise correlations of the Resfams and KEGG metabolism modules in C. difficile genomes revealed that one KEGG module, the cytidine 5′-monophospho-3-deoxy-D-manno-2-octulosonic acid (CMP-KDO) biosynthesis module, was perfectly correlated with the presence of the Class D beta-lactamase gene. CMP-KDO catalyzes a key reaction in lipopolysaccharide biosynthesis33. Further inspection of the KEGG annotations revealed that only one gene from this module was present in C. difficile: arabinose-5-phosphate isomerase. This gene typically occurs in Gram-negative bacteria, where it plays a role in synthesis of lipopolysaccharide for the outer membrane34, yet a recent study identified arabinose-5-phosphate isomerase in a Gram-positive organism, Clostridium tetani35. Although the function of this gene in Gram-positive bacteria is unknown, it is hypothesized to be a regulator and may modulate carbohydrate transport and metabolism35. If so, C. difficile (Gram-positive) strains with arabinose-5-phosphate isomerase may have a competitive advantage because they are able to rapidly respond to availability of the carbohydrates that are abundant in formula.
Major facilitator superfamily pumps are associated with increased replication
A previous analysis revealed that antibiotic administration is associated with elevated replication rates of gut organisms, which was hypothesized to be due to high resource availability after elimination of antibiotic susceptible strains36. We show here that a sample’s mean replication index in the days following antibiotic treatment is positively correlated with total resistance gene content (p < 0.05) (Fig 3A). The replication index, or iRep value36, is influenced by the fraction of cells in the population that are replicating. Our results suggest that the elevated iRep values after antibiotic treatment are likely not solely explained by resource availability, but rather appear to be influenced by the organisms’ resistance gene sets.
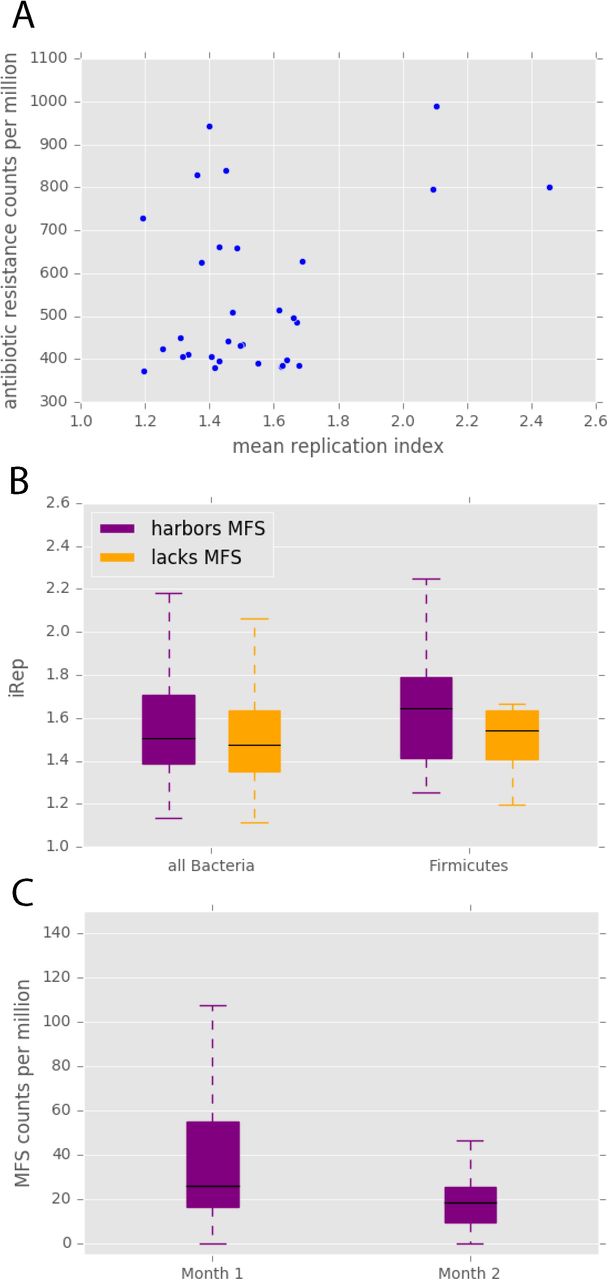
Antibiotic resistance and replication. (A) In samples taken within five days after antibiotic treatment, the antibiotic resistance potential of that sample is correlated with its mean replication index (Pearson’s r = 0.39, p = 0.03). (B) In infants that did not receive antibiotics after the first week of life, bacteria harboring at least one major facilitator superfamily (MFS) transporter gene have significantly higher iRep values (Mann-Whitney U= 827176.0, p = 1.55 × 10−5), and this pattern is apparent within the Firmicutes phylum (Mann-Whitney U = 136756.0, p = 0.0002). (C) MFS transporters are enriched in the first month of life compared to the second month of life (Wilcoxon signed-rank W = 165, p = 0.02). All p-values are Bonferonni corrected.
To characterize the effect of antibiotic resistance genes on iRep values in isolation from the confounding effects of antibiotics, we studied infants that did not receive any antibiotics after the first week of life. In these infants, organisms carrying genes for major facilitator superfamily (MFS) transporters have significantly higher iRep values than those that do not have MFS genes (p < 5 x 10−5) (Fig 3B). As there are known differences in median iRep values among phyla36, the comparison was repeated within each phylum that contained members with and without MFS genes. The trend of higher iRep values for organisms with MFS was most apparent in Firmicutes (p < 5 x 10−4) (Fig 3B). Therefore, the presence of these antibiotic resistance genes appears to inherently increase replication, even when no antibiotics are being administered. This could be due to protection from antibiotics being produced at a low level by other gut organisms37 or a result of MFS pumps’ naturally beneficial physiological functions38. We also acknowledge that this finding may simply reflect high incidence of organisms with MFS genes during periods of fast replication without a causal link.
We discovered that MFS gene levels in the resistome are significantly lower in the second month of life compared to the first (p < 0.05) (Fig 3C). MFS is the only resistance mechanism category for which this is true. The comparative enrichment of MFS genes in early in life may be a response to the empiric antibiotics (ampicillin and gentamicin) administered to all premature infants during the first week. The decline in MFS genes supports the established phenomenon of an eventual decrease of antibiotic resistance gene levels in the microbiome after the cessation of antibiotic therapy17.
A model that predicts an organism’s response to vancomycin and cephalosporins
We modeled the relationship between gene content of a gut organism and its direction of change in relative abundance (increase vs. decrease) after a premature infant is administered a combination of glycopeptide (vancomycin) and beta-lactam (cephalosporin, either cefotaxime or cefazolin) antibiotics. Principal component analysis was performed on Resfams26 and KEGG39 annotations to generate a low-dimensional representation of each organism’s metabolic potential and resistance potential. The first five principal components (PCs) cumulatively explained 48% of the variation in the dataset. Using these PCs as input, the AdaBoost-SAMME algorithm40 was applied, with decision tree classifiers as base estimators. The model, trained on 70% of the data, performed extremely well on the validation set, with a precision of 1.0 and recall of 1.0, indicating that every genome was correctly classified. Because the validation set was utilized for testing during the preliminary stages of model development, the model was also evaluated with a final test set, on which it achieved 0.9 precision and 0.7 recall.
Of the features that acted as the strongest contributors to each of the PCs, five genes with a tendency to occur in microbes that increase in relative abundance after antibiotic treatment were identified (Fig 4). One of these is subclass B2 beta-lactamase. Subclass B2 beta-lactamase of Aeromonas spp. has been shown to hydrolyze carbapenems and displays much lower levels of resistance to cephalosporins41. Due to its substrate specificity for carbapenems, it was surprising that this beta-lactamase was among the top predictors of an organism’s ability to persist after treatment with cephalosporins. However, the substrate specificity of an antibiotic resistance gene can depend on the context of that gene42, and a single base substitution in a beta-lactamase gene can alter substrate specificity43. Our results imply that in some gut organisms, beta-lactamases falling into the B2 subclass may confer resistance to cephalosporins.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The tendency for genes to occur in the class of genomes that increased in relative abundance after antibiotics. Genes and modules strongly contributing to the principal components used in the machine learning model were identified, and class tendency was calculated using the ratio of the gene’s prevalence in the increase group to its prevalence in the decrease group. Genes associated with the increase class of genomes are colored red.
Furthermore, our model shows that a gene linked to vancomycin resistance, vanR, is among the genes predictive of an organism’s propensity to increase in relative abundance after antibiotic treatment (Fig 4). VanR is the transcriptional activator of an operon encoding genes involved in peptidoglycan modification (VanH, VanA, and VanX)44. This gene cluster confers resistance because peptidoglycan is the target of vancomycin, and the antibiotic cannot bind to the modified version. VanR is essential for the initiation of the vancomycin resistance operon promoter45, which may explain why this gene was so crucial in our model.
In addition to genes specifically encoding for resistance to beta-lactams or glycopeptides, efflux pumps and transporters were also strong contributors to the PCs used as input to the model. Mex genes (of the resistance nodulation cell division family of drug efflux pumps) and ATP-binding cassette (ABC) transporter genes were associated with microbes that increase in relative abundance after antibiotics (Fig 4). Multidrug efflux pumps are essential for the intrinsic drug resistance of many bacteria, and overexpression of the genes for these pumps leads to elevated resistance levels46. Among possible directions for future research is inclusion of transcriptomics data to develop a model with even more robust predictive capability.
Previous studies have utilized data from 16S rRNA gene amplicon sequencing or read-based metagenomics of the human microbiome to predict life events of the human host using machine learning or other modeling techniques47,48. However, read-based metagenomics lacks resolution at the genomic level, and due to strain-level differences in antibiotic resistance48, taxonomy data from marker gene studies cannot be used to predict how particular organisms in a community will respond to antibiotics. Here, for the first time, we utilize the high-dimensional data obtained by reconstructing genomes from metagenomes to make predictions about the future states of individual gut microbes. This has tremendous potential for application in the fields of medicine and microbial ecology. For example, such a model can be used before administering drugs to a patient to verify that a particular combination of antibiotics will not lead to overgrowth of an undesirable microbe. In other environmental systems, various types of perturbations can be modeled to make predictions about how they will influence key members of microbial communities. The trained models can then be used to inform decisions in areas like agriculture and contaminant removal, where individual organisms may be of great importance51,52. Our study serves as a proof of concept for this application of machine learning used in conjunction with genome-resolved metagenomics to derive biological insight.
Materials and Methods
Sample collection, sequencing, assembly, and gene prediction
Fecal samples were collected from 107 infants of the NICU at the Magee Women’s hospital during the first three months of life. Briefly, DNA was extracted using the PowerSoil DNA isolation kit (MoBio Laboratories, Carlsbad, CA, USA) and sequenced using the Illumina HiSeq platform. Details on sample recovery, extraction, library preparation, and sequencing have been previously reported 53-55. Using default parameters for all the programs, the reads were trimmed with Sickle (https://github.com/najoshi/sickle), cleared of human contamination following mapping to the human genome with Bowtie256, and assembled with idba_ud57. Additionally, idba_ud was used to generate co-assemblies for each infant by simultaneously assembling all the samples belonging to the infant. Prodigal58 run in the metagenomic mode was used for gene prediction.
Genome recovery and relative abundance calculation
For each infant, reads from all samples from that infant were mapped to all individual assemblies from that infant as well as the infant’s co-assembly using SNAP59.
Coverage of scaffolds was calculated and used to run concoct60 with default parameters on all individual assemblies and co-assemblies. To remove redundant bins, all bins recovered from each infant were dereplicated using dRep61 v0.4.0 with the command: dRep dereplicate_wf–S_algorithm gANI-comp 50-con 25-str 25-l 50000-pa .9-nc .1.
Using Bowtie256, the reads from each sample were mapped to the set of genomes that were recovered from that particular infant. The read mapping output files were used to calculate the average coverage of each genome in each sample, and the coverage values were converted to relative abundance by utilizing the read length, total number of reads in the sample, and genome length.
iRep calculation
For each sample, a set of representative genomes was first chosen from the complete collection of dereplicated genomes. First, all genomes were clustered at 98% ANI using dRep61. A pangenome was then generated for each of these clusters using PanSeq62, creating a list of fragments representing the entire sequence-space of each cluster. All pangenomes of all clusters were merged, and reads from all samples were mapped to the resulting pangenome set using SNAP59. By analyzing the coverage of all fragments in the pangenome set, the breadth of each genome in each sample was calculated (number of genome fragments > 1x coverage / total genome fragments). Genomes with less than 85% breadth were removed from analysis. For all remaining genomes, the genome from each cluster with the highest breadth was added to that sample’s representative genome list.
Next, reads from each sample were mapped to its representative genome list using bowtie256 default parameters. iRep36 was run on the resulting mapping files using default parameters and without GC correction. Only values that passed iRep’s default filtering and were < 3 were considered for analysis.
Annotation
The amino acid sequences of genes predicted by the metaProdigal gene finding algorithm58 were searched against Resfams26, an antibiotic resistance gene specific profile hidden Markov model (HMM) database using the hmmscan function of HMMER v 3.1b263. The –cut_ga option was used to set the reporting and inclusion limits as the profile-specific gathering threshold, which have been manually optimized on a profile-by-profile basis to ensure Resfams prediction accuracy26. The Resfams annotation output and the coverage of each scaffold that had a hit to a Resfams profile were used to generate sample resistance gene summaries. Each sample resistance gene summary, which represents the antibiotic resistance potential of a particular infant gut microbiome at a particular point in time, contains the counts per million (CPM) for each of the 170 antibiotic resistance gene families in the Resfams database. Additionally, genome resistance gene profiles that indicated the count of each resistance gene were developed for each genome.
To gather general metabolism data, all binned sequences were searched against Kyoto Encyclopedia of Genes and Genomes (KEGG)39 HMMs and the results were parsed for genome profiling. This resulted in a KEGG metabolism profile for each organism that displayed the fraction of each KEGG module encoded by that genome.
Statistical and computational analysis
To evaluate the effect of feeding regimen, delivery mode, gender, maternal antibiotics, and the infant’s current antibiotic therapy, three cross-sectional PERMANOVA64 tests for weeks two, four, and six were performed using the adonis2 function of the vegan package in R65. For each infant, the first sample of each week was identified and the resistance gene summary of that sample was included in the PERMANOVA. If antibiotics were being administered on the day of sampling, the infant was labeled as currently receiving antibiotics. Infants that were exclusively fed breast milk and infants that were given breast milk at any point were both labeled as receiving breast milk. The Bray-Curtis dissimilarity metric was used and 9,999 permutations were performed to assess the marginal effects of the terms. The factor revealed to have a significant difference in antibiotic resistance gene content (p < 0.05) was selected for continued analysis. To identify antibiotic resistance genes associated with either formula feeding or breast milk during the weeks indicated by the PERMANOVA results, the infant’s diet was used to classify sample resistance gene summaries using random forest models66. Mann-Whitney U tests were performed on Resfams that had feature importance scores above 0.07 in the random forest models, as calculated by the Gini importance metric. Genomes containing resistance genes significantly associated with a particular feeding type, along with genomes of the same species lacking these genes, were further investigated. The ribosomal protein S3 (RPS3) genes for each genome were identified using rp16.py (https://github.com/christophertbrown/bioscripts/blob/master/bm/rp16.py). The RPS3 nucleotide sequences were aligned with MUSCLE67 using default parameters, and a maximum-likelihood phylogenetic tree was built with RAxML68. Pairwise Pearson correlations of Resfams with KEGG modules within these genomes were calculated.
The Pearson correlation of mean replication index (iRep) for a sample and the sample’s total resistance gene content was determined for samples collected within five days following antibiotic treatment. The replication rates of organisms harboring antibiotic resistance genes were compared to those lacking resistance genes of the same category. The first sample from a particular infant from the first month of life was matched with the same infant’s first sample from the second month of life, and Wilcoxon signed rank tests were performed to evaluate the change in abundance of each resistance mechanism. All p-values were Bonferonni corrected for multiple testing.
Infants for which there was a sample taken both before and after post-week antibiotic treatment were identified and the before and after samples were selected. Genomes from these samples were identified and labeled as either increasing or decreasing in relative abundance from the pre-antibiotic sample to the post-antibiotic sample. Using scikit-learn66, development of a machine learning model to predict the direction of change in relative abundance for each genome based on its Resfams and KEGG metabolism was attempted; yet an adequate model could not be developed, presumably due to variation in the ways that organisms respond to different antibiotic combinations. Therefore, the dataset was narrowed to include the six infants that received either cefotaxime or cefazolin (both cephalosporin antibiotics) in conjunction with vancomycin. 70% of the genomes obtained from these infant samples were used for training, 15% was used as a validation set for model improvement, and 15% was held out as a final test set. Several attempts to improve model performance through algorithm choice, feature engineering, and parameter tuning were applied, and the model that exhibited the best results with regard to precision and recall was selected. This model was then used to make predictions on the final test set. Each feature constructed for the model was a principal component of the Resfams and KEGG metabolic data, and the genes/modules contributing most strongly to each of these principal components were identified. The tendency for each of the genes and modules to occur in the increase class was calculated by adding-1 to the gene’s mean value in the increase class divided by its mean value in the decrease class.
Data availability
The dataset used is comprised of 597 previously reported samples53-55, as well as 305 new samples. These samples are available at NCBI under accession number SRP114966 (https://www.ncbi.nlm.nih.gov/sra/?term=SRP114966). The code for the analysis, along with all the data and metadata used in the analysis, is hosted at https://github.com/SumayahR/antibiotic-resistance.
Author Contributions
MJM and JFB conceived of the project. MRO processed the sequence data, including binning and de-replication of genomes. SFR performed the computational analysis and interpreted the results. SFR and JFB wrote the manuscript, and all authors contributed to manuscript revisions.
Acknowledgements
Funding was provided through the National Institutes of Health (NIH) under grant RAI092531A and the Alfred P. Sloan Foundation under grant APSF-2012-10-05. This work used the Vincent J. Coates Genomics Sequencing Laboratory, supported by NIH S10 OD018174 Instrumentation Grant. The study was approved by the University of Pittsburgh Institutional Review Board (IRB) (Protocol PRO12100487).
We acknowledge Robyn Baker for recruiting infants, and Brian Firek for performing DNA extractions. We would also like to thank David Burstein for the KEGG HMM annotation pipeline, and Christopher Brown for scripts to identify ribosomal proteins and to calculate genome coverage.
References




