

ABSTRACT
The progressive maturation of cells down differentiation lineages is controlled by collaborative interactions between networks of extracellular signals and intracellular transcription factors. In the vertebrate spinal cord, FGF, Wnt and Retinoic Acid signaling pathways regulate the progressive caudal-to-rostral maturation of neural progenitors by regulating a poorly understood gene regulatory network of transcription factors. We have mapped out this gene regulatory network in the chicken pre-neural tube, identifying CDX4 as a dual-function core component that simultaneously regulates gradual loss of cell potency and acquisition of differentiation states: in a caudal-to-rostral direction, CDX4 represses the early neural differentiation marker Nkx1.2 and promotes the late neural differentiation marker Pax6. Significantly, CDX4 prevents premature PAX6-dependent neural differentiation by blocking Ngn2 activation. This regulation of CDX4 over Pax6 is restricted to the rostral pre-neural tube by Retinoic Acid signaling. Together, our results show that in the spinal cord, CDX4 is part of the gene regulatory network controlling the sequential and progressive transition of states from high to low potency during neural progenitor maturation. Given CDX well-known involvement in Hox gene regulation, we propose that CDX factors coordinate the maturation and axial specification of neural progenitor cells during spinal cord development.
INTRODUCTION
Differentiating cells transition from one temporary state to another, losing potency and acquiring specialized functions in the process. Each step along the differentiation pathway is defined by a unique assortment of active transcription factors (Davidson, 2006; Royo et al., 2011). This transcriptome can change over time, mostly cued by dynamic extra-cellular signaling factors (Peter and Davidson, 2013; Sandmann et al., 2007). It is the cross-regulation between transcription and signaling components that promotes the progressive acquisition of specialized functions while preventing dedifferentiation: transcription factors specify the cell’s identity and ability to respond to signaling factors (competence), and signaling factors control the sequential activity of transcription factors to promote directional acquisition of specialized traits (Davidson and Levine, 2008; Levine and Davidson, 2005; Sandmann et al., 2007). These interactions between transcription factors and signaling pathways form complex networks that have been challenging to dissect, hindering our understanding of the mechanisms regulating cellular state transitions.
The vertebrate spinal cord serves as an important accessible model to study the maturation of neural progenitors during their transition from one cellular state to the next. Maturation of spinal cord progenitors at the caudal end of the embryo follows a caudal-to-rostral organization, with undifferentiated cells localizing to the caudal regions and more mature cells localizing to the more rostral positions (Butler and Bronner, 2015; Diez del Corral et al., 2003; Diez del Corral and Storey, 2004; Wilson et al., 2009). During the early segmentation stages in chick embryos up to the point of tailbud formation (0 to 16 somites equivalent to Hamburger and Hamilton (HH) stages 6-12; Hamburger and Hamilton, 1951), extensive fate mapping and gene expression analysis has resulted in the identification of four distinct embryonic regions corresponding to four different neural maturation states (reviewed in Gouti et al., 2015 and Henrique et al., 2015; summarized in Fig 1A). The most caudal region is the caudal lateral epiblast and node-streak border region containing bipotent neuromesodermal progenitors (NMPs) cells that contribute to both neural and mesodermal tissues (region 1; Brown and Storey, 2000; Cambray and Wilson, 2007; Tzouanacou et al., 2009; reviewed in Henrique et al., 2015). NMPs are defined molecularly by the co-expression of two key transcription factors, the pan-neural marker Sox2 and the mesodermal marker T/Bra, although NMPs also transcribe the pre-neural identity marker Nkx¡.2 (also known as Sax1; Delfino-Machin et al., 2005; Gouti et al., 2015; Gouti et al., 2017). Immediately rostral to the NMP domain is the pre-neural tube (PNT; Gouti et al., 2015; Henrique et al., 2015). Cells in the PNT downregulate T/Bra but continue to express Sox2 (Delfino-Machin et al., 2005; Gouti et al., 2015). PNT can be further subdivided into a caudal PNT that continues to express Nkx1.2 (region 2) and the rostral PNT which downregulates Nkx1.2 and activates Pax6 transcription (region 3; Bel-Vialar et al., 2007; Bertrand et al., 2000; Delfino-Machin et al., 2005; Sasai et al., 2014; Spann et al., 1994). Finally, rostral to the PNT and situated adjacent to the developing somites is the neural tube (NT; region 4; Gouti et al., 2015; Henrique et al., 2015). NT cells are Nkx1.2’-negative and Pax6-positive and begin to transcribe the neural differentiation genes Ngn1/2 and NeuroM(Diez del Corral et al., 2003). Thus, from caudal-to-rostral, four spatially distinct populations can be identified that correspond to four maturation states (summarized in Fig 1A).

(A) Schematic representation of the caudal end of HH10 chicken embryo showing primary subdivisions (central diagram; adapted from Olivera-Martinez and Storey, 2007), and expression domains of key transcription and signaling factors (left and right of diagram, respectively). (B) Cdx4 is transcribed in a dynamic dorsal-ventral (DV) gradient along the rostro-caudal (RC) axis of embryos (HH11). Red lines indicate position of transverse sections shown on right. (C) Distribution of PAX7 (dorsal), PAX6 (dorsal-to-intermediate), and NKX6.1 (ventral) proteins relative to Cdx4 transcription domain. (D) Cdx4 and Pax6 transcription domains overlap in the rostral pre-neural tube (PNT) at stage HH 11-11+ (ISH; Cdx4 expression in purple in Da, and red in Db and Dc; Pax6 expression in purple in Db and Dd). Arrow shows position of somite 13. (E) Graphical representation of the experimental approach used throughout this work. The PNT of HH10-11 stage embryos were electroporated on the left side with appropriate constructs carrying a GFP-reporter gene. Embryos were processed for analysis 8 (HH 12-13) or 24 (HH16-16+) hours post electroporation (hpe), when electroporated cells are localized to the rostral portion of the PNT (HH 12-13) or the caudal portion of the NT (thoracic level; HH16-16+), respectively. Control experiments demonstrate that electroporation alone has not affect gene transcription, and that overexpression of electroporated constructs is long lasting (Fig S1 for all experiments). Arrowhead shows the position the last somite formed at the time of electroporation. (F) CDX4 does not regulate DV patterning in the neural tube. Ectopic Cdx4 did not change the distribution of PAX7 or NKX6.1 proteins (n=6/6 for both), but caused ectopic PAX6 accumulation outside its normal domain (arrowheads, n=6/6). Marker proteins are in red and electroporated cells are in green (nuclear GFP tag). Embryos were electroporated at HH10-11 and analyzed 24 hpe (HH16). (E) Summary of results. Scale bar is 200μm for whole mount and 40μm for transverse sections.
Recently, single cell transcriptome analysis of in vitro differentiating NMPs has confirmed and expanded the known transcriptional signatures observed throughout NMPs initial cell fate choice decision, allowing the more accurate assignment of gene activities to particular specification states (Gouti et al., 2014; Gouti et al., 2017). For example, CDX transcription factors have been implicated in NMP maintenance, and also axial patterning and elongation (Amin et al., 2016), however the separation of these activities has been challenging to dissect due to the multiple and partially redundant activities of the three functionally similar CDX proteins (CDX1, CDX2 and CDX4; van Rooijen et al., 2012). By studying the differentiation of NMPs derived from mouse Embryonic Stem Cells (mESC) lacking individual or different combination of Cdx genes, CDX proteins were shown to regulate the temporal maintenance of T/Bra (Gouti et al., 2017). By maintaining or down regulating T/Bra transcription, Cdx genes regulate the fate decision of NMP cell to become either mesoderm or neural tissues (Gouti et al., 2017). In addition to the NMPs, Cdx are also transcribed in NMP descendants in the PNT and NT (Gaunt et al., 2005; Marom et al., 1997; Fig 1B), where their function remains largely unknown.
The transition from NMP to pre-neural to neural transcriptional states is under the control of three signaling factors: FGF, Wnt and Retinoic Acid (RA; Diez del Corral et al., 2003; Olivera-Martinez and Storey, 2007). At the caudal end of the embryo, FGF8 and Wnts (Wnt3a and Wnt8c) are transcribed in a caudal to rostral gradient that promotes potency by maintaining T/Bra, Sox2 and Nkx1.2 expression while simultaneously preventing Pax6 transcription (Bertrand et al., 2000; Delfino-Machin et al., 2005; Diez del Corral et al., 2003; Olivera-Martinez et al., 2012). FGF also maintains tissue proliferation by limiting precocious cell cycle exit (Akai et al., 2005). In contrast, RA secreted from somites establishes a rostral to caudal signaling gradient that promotes differentiation: first by promoting transcription of neural identity genes Pax6 (Diez del Corral et al., 2003; Novitch et al., 2001; Pituello et al., 1999), and subsequently, by promoting transcription of downstream neurogenic genes Ngn1/2 and NeuroM (Diez del Corral et al., 2003). By inducing Pax6 and Ngn2, RA induces cells to exit the proliferation program (Bel-Vialar et al., 2007; Lacomme et al., 2012). The signaling activities of FGF/Wnt and RA are segregated to opposite caudal and rostral regions of the nascent spinal cord through positive and negative interactions: caudally, high FGF directly prevents RA synthesis and stimulates its degradation, while rostrally, low FGF indirectly promotes RA production through a Wnt8c-dependent mechanism (Boulet and Capecchi, 2012; Olivera-Martinez et al., 2012; Olivera-Martinez and Storey, 2007; Sakai et al., 2001; White et al., 2007). In turn, RA inhibits Fgf8 transcription rostrally, creating a zone where cells can exit the cell cycle and differentiate (Diez del Corral et al., 2003; Kumar and Duester, 2014). These interactions have been proposed to function as the signaling switch that drives the transition of cellular states in the caudal neural tube (Diez del Corral and Storey, 2004; Olivera-Martinez and Storey, 2007). While the signal interactions regulating the transition from NMP to pre-neural to neural states have been extensively investigated, the underlying transcription factor network driving the cell transitions are incompletely understood.
FGF, Wnt and RA signals are known regulators of Cdx transcription (Deschamps and van Nes, 2005; Lohnes, 2003), making CDX transcription factors good candidates to regulate PNT cell maturation. In chicken embryos, Cdx genes are transcribed in nested domains, at levels that are high in NMPs and low in the NT (Marom et al., 1997). These high-to-low levels of transcription have also been observed in differentiating NMPs in vitro (Gouti et al., 2017). In the spinal cord, CDX factors are essential for tissue specification and rostro-caudal patterning (Deschamps et al., 1999; Nordstrom et al., 2006; Shimizu et al., 2006; Skromne et al., 2007; van den Akker et al., 2002), controlling the initial specification of post-occipital tissues (van Rooijen et al., 2012), and the subsequent patterned transcription of Hox expression domains (Deschamps et al., 1999; Hayward et al., 2015). Thus, Cdx genes are attractive candidates to integrate multiple signals into coherent cell maturation states.
Here we show that chicken CDX4, the only CDX present in the chick PNT and NT during early segmentation stages (HH10-16; Marom et al., 1997), controls the progression of PNT cells towards more mature states without promoting their terminal differentiation. In the PNT, transient CDX4 results in Nkx1.2 downregulation and Pax6 activation, which drives cells with recently acquired neural identity (Sox2+, Nkx1.2+, Pax6-) toward a more restricted neural progenitor state (Sox2+, Nkx1.2-, Pax6+). Significantly, Pax6 activation by CDX4 is dependent on RA secreted by somites, which restricts the maturation of cells to the rostral PNT. Furthermore, we show that CDX4 prevents Ngn2 transcription even in the presence of the Ngn2- activator PAX6, thus preventing the premature cell’s terminal differentiation. Our results support a model in which CDX4 is an integral component of a gene regulatory network that functions to simultaneously reduce the potency and increase the differentiation state of cells. We propose that this gene regulatory network operates under the control of previously described signaling network involving FGF, Wnt, and RA.
RESULTS
Cdx4 is transcribed in the caudal neural tube where it regulates Pax6 transcription
CDX4 neural function in chicken embryos was first analyzed by correlating its transcription domain to distinct progenitor cell maturation zones of the caudal neuroectoderm (Fig 1A; Olivera-Martinez and Storey, 2007). As previously reported in whole chick embryos (HH10-12; Morales et al., 1996; Marom et al., 1997), Cdx4 is transcribed in the pre-neural tube (PNT) and nascent neural tube (NT) in a high caudal to low rostral gradient (Fig 1B). However, transverse sections also revealed that Cdx4 is transcribed in a highly dynamic dorsal-to-ventral (DV) gradient: caudally, Cdx4 transcription was ubiquitous throughout the medio-lateral extent of the PNT (dorsal-ventral extent in the NT), whereas rostrally, Cdx4 transcription was progressively excluded from ventral regions as well as the roof plate (Fig 1B, transverse sections). Due to the lack of chicken specific CDX4 antibody, we were unable to examine the CDX4 protein profile in the neural tube. However, a similar dorsally restricted expression profile has been reported for Cdx4 in mouse embryos (Gaunt et al., 2005), suggesting evolutionary conserved transcriptional mechanisms and a potential function for CDX4 in the specification of DV neural cell identities.
To test the role of CDX4 in DV specification, we analyzed Cdx4 transcriptional domain relative to various DV identity markers including the dorsal cell marker Pax7 (Briscoe et al., 2000; Diez del Corral et al., 2003), the dorsal-to-intermediate cell marker Pax6 (Briscoe et al., 2000; Novitch et al., 2003), and the ventral cell marker Nkx6.1 (Briscoe et al., 2000; Diez del Corral et al., 2003; Novitch et al., 2003). At HH11, we observed a correlation between the transcriptional domain of Cdx4 and some of these markers. For example, in the caudal NT, PAX7 domain was nested within, and NKX6.1 domain was complementary to Cdx4 domain of transcription (Fig 1B, C). However, more rostrally in the NT, we saw a loss of correlation between D/V markers and Cdx4 domain of transcription; PAX7 domain was broader than, and NKX6.1 domain no longer complemented Cdx4 transcription domain (Fig 1B, C). The only correlation we observed was between Cdx4 and Pax6, with levels of Cdx4 transcript decaying as levels of Pax6 transcript and PAX6 protein increased in a caudal to rostral direction (Fig 1C, D).
To formally test Cdx4 involvement in DV cell fate specification, we artificially maintained high levels of Cdx4 in the NT in a domain where Cdx4 would normally be downregulated. We reasoned that if CDX4 regulates DV cell specification, increasing Cdx4 levels would result in a change in the localization of DV marker genes. We overexpressed CDX4 by electroporating wild type Cdx4 in the PNT of stage HH10-11 embryos, a region that transcribes endogenous Cdx4, and analyzed the protein distribution of PAX7, PAX6, and NKX6.1 24-hours post-electroporation (hpe; HH16-17), at a time when electroporated cells have become part of the NT and no longer transcribe endogenous Cdx4 (Fig 1B, E). While artificially maintained high levels of Cdx4 expression did not change NKX6.1 and PAX7 protein distribution (Fig 1F; n=6/6 for both conditions), ectopic Cdx4 caused production of PAX6 protein outside its normal domain, both ventrally and dorsally (Fig 1F; n=6/6). In this and all other experiments, electroporation of a control reporter GFP vector had no effect on target gene transcription and protein distribution (Fig S1). Together, these results suggest that CDX4 is not a general regulator of DV identity markers, but instead, a specific regulator of Pax6 transcription (Fig 1G).
CDX4 regulates Pax6 transcription during neural progenitor cell maturation
In addition to its function in DV cell specification, PAX6 promotes the maturation of neural progenitor cells in the PNT (Bel-Vialar et al., 2007). Given that our results do not support a function for CDX4 in global DV cell specification (Fig 1), we hypothesized that CDX4 might regulate Pax6 transcription during PNT cell maturation. To test this hypothesis, we asked whether the presence of CDX4 was sufficient to change Pax6 transcription in the rostral PNT, a region where Pax6 transcription initiates. Embryos were electroporated in the PNT with different Cdx4 constructs (HH10-11), grown for 8 hours only (HH12-13), and analyzed by in situ hybridization for premature Pax6 activation. Two constructs were used in this assay, a wild type and a constitutive active version of CDX4 that phenocopies CDX functions in Hox gene transcription assays (VP16CDX4; Bel-Vialar et al., 2002; Faas and Isaacs, 2009). In these short incubation experiments, VP16CDX4 was able to induce Pax6 transcription more caudally and at higher levels than CDX4 (Fig 2A; n=4/6 by ISH. Fig 2B; n=3/4 by IHC), while CDX4 could induce Pax6 after long incubation periods (24 hpe; Fig 1F). These results suggest that CDX4 has the potential to regulate Pax6 transcription in the rostral PNT and caudal NT.
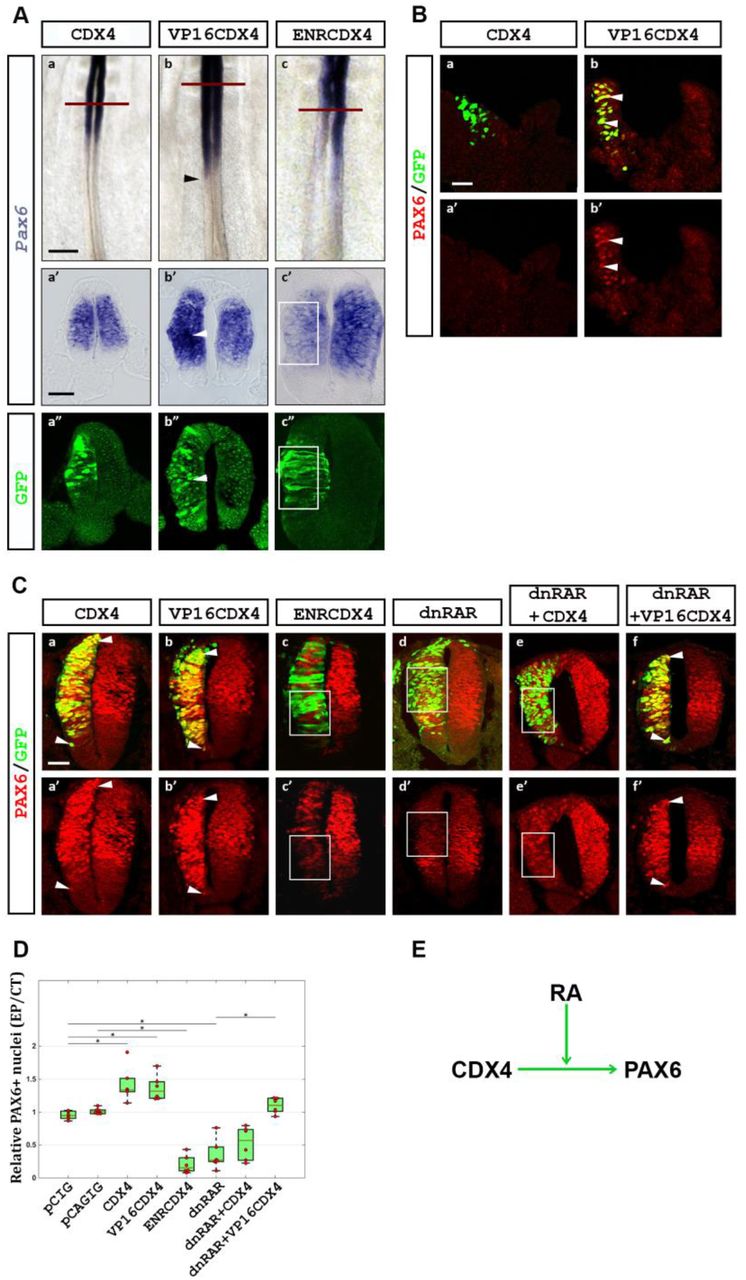
(A) CDX4 regulates Pax6 transcription in the rostral PNT. In the PNT, Cdx4 has no effect (a, a’; n=6/6), VP16Cdx4 induces (arrowheads in b, b’; n=4/6), and EnRCdx4 downregulates (c, c’, box; n=6/6) Pax6 transcription (purple signal by ISH; green GFP tag labeled by IHC). Embryos were electroporated at HH10-11 and analyzed 8 hpe (HH12-13). (B) Similarly, ectopic VP16Cdx4 (n=3/4) but not Cdx4 (n=0/4), causes ectopic PAX6 protein accumulation (arrowheads). (C) CDX4 requires Retinoic Acid (RA) to activate Pax6 transcription in rostral regions. In the NT, both Cdx4 and VP16Cdx4 overexpression result in ectopic PAX6 protein accumulation (a, a’, b, b’; arrowheads; n=6/6 for both), whereas EnRCdx4 overexpression causes the loss of PAX6 (c, c’; box; n=6/6). Inhibition of RA signaling using a dominant negative RA receptor (dnRAR) causes the loss of PAX6 (d, d’; box; n=6/6). In the absence of RA signaling, Cdx4 overexpression is unable to induce ectopic PAX6 (e, e’; box; n=6/6). Under similar conditions, VP16Cdx4 overexpression induces ectopic PAX6 (f, f; arrowheads; n=6/6). Embryos were electroporated at HH10-11 and analyzed 24 hpe (HH16-17). (D) Quantification of PAX6 positive cells after experiments shown in C. Box-scatter plot representing ratio of PAX6 positive cells on electroporated side to that on the contralateral control side (as per Karaz et al., 2016). Cells were counted using ImageJ. Significance is shown with a bar and a star (two tailed t-test analysis, p<0.05). (E) Summary of results. Scale bar is 200μm for whole mount and 40μm for transverse sections.
To test if CDX4 is necessary for Pax6 activation in the PNT, we outcompeted endogenous CDX4 by overexpressing a dominant negative form of CDX4 in which the transcription activation domain of the protein was replaced with the transcriptional repressor domain of the Drosophila Engrailed protein (ENRCDX4; Han and Manley, 1993). This chimeric form of CDX4 has been shown to repress transcription of downstream CDX targets (e.g., Hox genes; Bel-Vialar et al., 2002; Isaacs et al., 1998). Overexpression of EnRCdx4 caused Pax6 downregulation in the rostral PNT (8 hpe; Fig 2Ac; n=6/6), indicating that in this region, CDX4 is necessary for Pax6 transcription.
CDX4 activation of Pax6 in the PNT is dependent on Retinoic Acid signaling
Transcription of Pax6 is restricted to the rostral PNT despite that Cdx4 is transcribed in both caudal and rostral PNT regions (Fig 1D) and, upon overexpression, CDX4 can induce Pax6 transcription ectopically (Fig 1F, 2A). To investigate the possible mechanisms that restrict Pax6 transcription to the rostral PNT, we turned our attention to Retinoic Acid (RA). Somite-derived RA regulates spinal cord neurogenesis by activating numerous target genes in the rostral PNT, including Pax6 (Novitch et al., 2003; Pituello et al., 1999). Given that RA and CDX4 interact during zebrafish spinal cord cell specification (Chang et al., 2016; Lee and Skromne, 2014), we hypothesized that RA and CDX4 might also interact during spinal cord maturation. To test this hypothesis, we electroporated PNT with dominant negative RA receptors (dnRAR) to block RA signaling (Novitch et al., 2003), and then analyzed the transcription of Pax6 24-hpe, at a time when electroporated cells would be undergoing maturation. As previously shown (Novitch et al., 2003), overexpression of dnRAR blocked RA signaling and caused Pax6 down regulation (Fig 2Cd, D), even as dnRAR enhanced Cdx4 transcription (Fig S2). To test if induction of Pax6 by CDX4 is RA-dependent, we co-electroporated different Cdx4 constructs together with dnRAR. In RA-deficient cells, CDX4 was unable to induce Pax6 (Fig 2Ce, D; n=6/6), despite its ability to do so in RA-responsive cells (Fig 1D). Significantly, however, VP16CDX4 was able to induce Pax6 transcription even in the absence of RA (Fig 2Cb, Cf, D; n=6/6). Together, these results suggest that Pax6 activation by CDX4 is dependent on RA signaling, and illuminates a mechanism for the restricted transcription of Pax6 to the rostral portion of the PNT (Fig 2E).
CDX4 inhibits PAX6-dependent activation of Ngn2 in the NT
PAX6 is present in both the rostral PNT and the NT, but it only activates neural differentiation genes in the NT (Bel-Vialar et al., 2007). Then, what prevents PAX6 from prematurely activating neural differentiation genes in the PNT? To address this question we analyzed the transcription of the neural differentiation gene Ngn2, a downstream target of PAX6 (Scardigli et al., 2003). Ngn2 transcription domain is nested within that of Pax6 and lays immediately rostral to that of Cdx4 (Fig 1B; Fig 3A), raising the possibility that CDX4 activity is incompatible with Ngn2 transcription. To test this possibility, we overexpressed Cdx4, VP16Cdx4 and EnRCdx4 in HH10-11 embryos and analyzed their effect on NGN2 distribution at HH16-17 (24 hpe). As expected, ENRCDX4 caused a loss of NGN2 (24 hpe; Fig 3Bc, 3C; n=6/6), as this construct also reduced the levels of PAX6 (24 hpe; Fig 2Cc), and PAX6 is required for Ngn2 transcription (Scardigli et al., 2003). Surprisingly, CDX4 and VP16CDX4 also caused the loss of NGN2 (24 hpe; Fig 3Ba-b, 3C; n=6/6), under conditions that resulted in ectopic PAX6 (Fig 2Ca-b), suggesting that CDX4 represses Ngn2. This result was consistent with Cdx4 and Ngn2 complementary expression domains (Figs 1B, 3A). To confirm that CDX4 represses Ngn2 in the presence of PAX6, we repeated the experiment by simultaneously co-expressing Cdx4 and Pax6. While PAX6 on its own was able to ectopically activate Ngn2 (Fig 3Bd, C; n=6/6; Bel-Vialar et al., 2007), it was unable do so in the presence of CDX4 (Fig 3Be, C; n=6/6). Taken together, these results suggest that CDX4 in the PNT promotes Pax6 and prevents Ngn2 transcription (Fig 3D).

(A) Ngn2 expression in the NT of wild type HH11 embryos. Expression is first observed caudally at the site of NT closure. (B) Cdx4 and VP16Cdx4 overexpression results in the loss of NGN2 (a, a’, b, b’; boxes; n=6/6 for both conditions), despite both inducing the Ngn2-activator Pax6 (Fig 2A, C). EnRCdx4 overexpression also causes the loss of NGN2 (c, c’; box; n=6/6), similar to its effect on PAX6 (Fig 2Cc). Pax6 overexpression results in ectopic NGN2 (d, d’; arrowhead; n=6/6), but not in the presence of Cdx4 (e, e’; box; n=6/6). Embryos were electroporated at HH10-11 and analyzed 24 hpe (HH16-17). (C) Quantification of NGN2 positive cells after experiments shown in B. Box-scatter plot representing ratio of NGN2 positive cells on electroporated side versus contralateral control side. Cells were counted using ImageJ. Significance is shown with a bar and a star (two tailed t-test analysis, p<0.05). (D) Figure summarizing CDX4-PAX6-NGN2 interactions. Scale bar is 200μm for whole mount and 40μm for transverse sections.
CDX4 inhibits Nkx1.2 expression in early neural progenitor cells
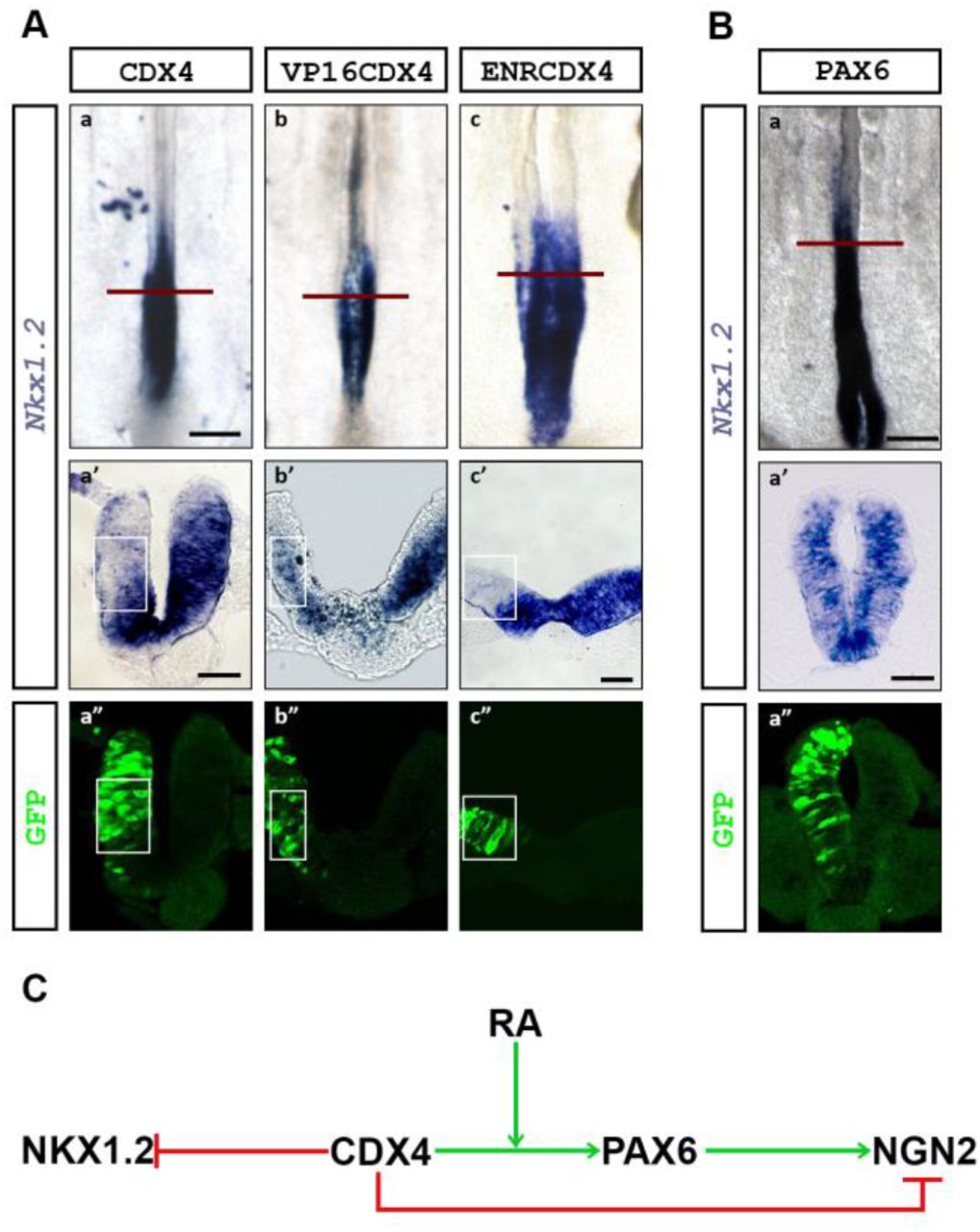
Cdx4 transcription domain in the PNT includes the caudal region that contains T/Bra-, Sox2+ and Nkx1.2+ early neural progenitors. This observation prompted us to ask whether CDX4 also regulates aspects of early PNT maturation. To address this question, we electroporated the caudal PNT with different Cdx4 constructs at HH10-11 and, after growing the embryos for 8 hpe to HH12-13, we analyzed the transcription of the early PNT marker Nkx1.2 (Delfino-Machin et al., 2005; Gouti et al., 2017; Gouti et al., 2015; Sasai et al., 2014). Overexpression of Cdx4 and VP16Cdx4 caused downregulation of Nkx1.2 transcription (Fig 4A; n=6/6 for both conditions), suggesting that CDX4 is a negative regulator of Nkx1.2. Unexpectedly, however, EnRCdx4 overexpression also caused Nkx1.2 downregulation (Fig 4A; n=6/6), suggesting that CDX4 is also necessary for Nkx1.2 transcription. We interpret these results to indicate that Nkx1.2 is under both positive and negative CDX4 regulation, with high levels of CDX4 repressing Nkx1.2 (Fig 4C).

(A) Overexpression of Cdx4 (a, a’), VP16Cdx4 (b, b’), and EnRCdx4 (c, c’) downregulate Nkx1.2 transcription (boxed region; n=6/6 for all conditions). (B) Overexpression of Pax6 has no effect on Nkx1.2 transcription (n=6/6). Embryos were electroporated in the caudal PNT at HH10-11 and analyzed 8-hpe (HH12-13). (C) Figure summarizing CDX4-NKX1.2 interactions. Scale bar is 200μm for whole mount and 40μm for transverse sections.
One possible mechanism to explain the loss of Nkx1.2 transcription after Cdx4 overexpression is that CDX4 activates neural differentiation genes that then repress Nkx1.2. Such a candidate could be Pax6, as its expression domain and that of Nkx1.2 are mutually exclusive (summarized in Fig 1A), its transcription is induced by CDX4 (Fig 2), and its overexpression induces neural differentiation (Bel-Vialar et al., 2007). To test this possibility, we repeated these experiments overexpressing Pax6 and analyzing Nkx1.2 transcript at HH12-13. Under these conditions, PAX6 did not change Nkx1.2 transcription (Fig 4B; n=6/6). While this result does not rule out indirect mechanisms for the regulation of Nkx1.2 by CDX4, the result suggests that PAX6 is not a Nkx1.2 repressor (Fig 4C).
NKX1.2 and PAX6 interactions result in the segregation of their expression domains to different regions of the PNT
Nkx1.2 and Pax6 transcription domains are mutually exclusive, but both span Cdx4 transcription domain (summarized in Fig 1A), suggesting the possibility of cross-regulatory interactions between these genes. To test for possible cross-regulatory interactions between Nkx1.2 and Pax6, and between these two genes and Cdx4, we analyzed the expression of these genes after their overexpression in the caudal PNT at HH10-11. To analyze NKX1.2 function, we overexpressed the mouse version of Nkx1.2 (mNkx1.2), which acts as a repressor in mouse cell lines (Tamashiro et al., 2012) and chicken embryos (Sasai et al., 2014). Consistent with previous report (Sasai et al., 2014), overexpression of mNkx1.2 represses Pax6 at HH12 (8 hpe; Fig 5Ac, n=6/6). In addition, mNKX1.2 also repressed cNkx1.2 transcription (Fig 5Aa, n=6/6), suggesting negative autoregulation. However, mNkx1.2 overexpression had no effect on Cdx4 transcription (Fig 5Ab; n=6/6). Using the same strategy, we analyzed PAX6 activity on Cdx4. In this experiment, overexpression of Pax6 downregulated and dominant-negative EnRPax6 upregulated Cdx4 transcription (Fig 5B; n=6/6 for all conditions), suggest that PAX6 functions to represses Cdx4. Together, these results providing a mechanism to explain the segregation of Nkx1.2 and Pax6 transcriptional domains to caudal and rostral PNT, respectively, and the downregulation of Cdx4 in the caudal NT (Fig 5C; see model below).

(A) NKX1.2 negatively regulates the transcription of its own gene and of Pax6, but not Cdx4. Overexpression of mNkx1.2 downregulates cNL·1.2 (a, a’) and Pax6 (c, c’), without altering Cdx4 transcription (a’, c’; boxed regions; n=6/6 for all conditions). (B) PAX6 represses Cdx4. Ectopic Pax6 downregulates (a, a’; boxed region), and EnRPax6 upregulates (b, b’; arrowhead) Cdx4 transcription (n=6/6 for both conditions). Embryos were electroporated at HH10-11 and analyzed 8hpe (HH12-13). (C) Figure summarizing NKX1.2-CDX4-PAX6 interactions. Scale bar is 200μm for whole mount and 40μm for transverse sections.
DISCUSSION
A gene regulatory network controlling spinal cord neuronal maturation
CDX4 promotes loss of potency in the caudal pre-neural tube
Previous work has shown that CDX are key in the establishment and subsequent differentiation of NMPs into neural and mesodermal precursors by balancing the activity of Wnt3a, Fgf8 and RA signaling (Amin et al., 2016; Chawengsaksophak et al., 2004; Gouti et al., 2017). Mouse embryos deficient for all Cdx genes fail to develop post-occipital structures due to the premature differentiation of NMP cell (Amin et al., 2016; van Rooijen et al., 2012). The primary cause for this premature differentiation is the premature activation of the RA pathway, which in cell culture conditions causes NMPs to follow a neural fate by maintaining Sox2 and Nkx2.1, and repressing T/Bra transcription (Gouti et al., 2017).
Here we show important additional functions for CDX4, the only CDX member present in chick PNT post HH12 (Marom et al., 1997), in the progressive maturation of spinal cord neuronal progenitors. As NMPs’ descendants acquire neural identity (from T/Bra+, Sox2+, Nkx1.2+ to T/Bra-, Sox2 +, Nkx1.2+), CDX4 promote their further maturation by repressing Nkx1.2 transcription (Fig 4). Control of Nkx1.2 transcription is tightly balanced by Wnt and FGF signaling (Bertrand et al., 2000; Tamashiro et al., 2012), signals that are regulated by CDX (Chawengsaksophak et al., 2004; Gouti et al., 2017; Savory et al., 2009; van Rooijen et al., 2012). We speculate that increasing or decreasing CDX4 levels could cause an imbalance in Wnt and FGF that could lead to a loss of Nkx1.2 transcription. Given that NKX1.2 inhibits floor plate cell specification by repressing Pax6 (Sasai et al., 2014), we propose that CDX4 downregulation of Nkx1.2 is one of the first steps in PNT cell maturation.
CDX4 promotes neural cell determination in the rostral pre-neural tube
Progression of cells from caudal to rostral PNT is marked by the acquisition of neural determination markers. CDX4 promotes new maturation states by directing Pax6 activation, a factor involved in neural progenitor maturation (Bel-Vialar et al., 2007). CDX factors have been observed to increase Pax6 transcription in embryoid bodies (McKinney-Freeman et al., 2008). We propose that in the PNT, CDX4 regulation of Pax6 occurs via two distinct mechanism (Fig 6): by the indirect down regulation of the Pax6 repressor NKX1.2 (Fig 5), and by the direct activation of Pax6 transcription (Fig 2). Importantly, CDX4 activation of Pax6 is dependent on RA (Fig 2), which is secreted from somites (Molotkova et al., 2005; Olivera-Martinez and Storey, 2007). This spatial distribution of RA restricts the Pax6 inducing activity of CDX4 to the rostral PNT. RA/CDX regulation of Pax6 is likely to be evolutionarily conserved across vertebrates, as in all species examined the second intron of Pax6 contains an ultraconserved non-coding region that harbors both RA response elements (RAREs; Cunningham et al., 2016) and CDX4 binding sites (Paik et al., 2013 and Fig S3). In addition to DNA binding, RA has been implicated in opening up the Pax6 locus by antagonizing FGF signaling (Patel et al., 2013). In this scenario, RA could function to provide locus accessibility to CDX4 and other factors. Thus, in the PNT, RA provides the context in which CDX4 can further promote neural progenitor cell maturation.

Gene regulatory network of the genetic interactions identified in figures 1-5, superimposed to the FGF-Wnt8C-RA signaling network shown by others to regulate cell transitions states during spinal cord neuronal cell maturation (Olivera-Martinez and Storey, 2007). Network map was generated using Biotapestry (Longabaugh et al., 2005). In this model, CDX4 is at the core of the gene regulatory network that coordinates upstream signaling information into downstream transcriptional response.
CDX4 prevents premature neural cell differentiation
PAX6 induces neural cell differentiation (Bel-Vialar et al., 2007), and yet, despite Pax6 being induced by CDX4 in the rostral PNT (Fig 2), neural cell differentiation does not begin until after the NT has formed and Cdx4 has been down regulated (Fig 1). Two mechanisms by which PAX6 promotes differentiation is by downregulating Cdx4 (Fig. 5B) and by activating Ngn2 (Bel-Vialar et al., 2007; Scardigli et al., 2003), a gene that promotes cell cycle exit and further differentiation (Lacomme et al., 2012). Our data shows that CDX4 represses Ngn2 transcription even in the presence of PAX6 (Fig 3), thus priming but delaying spinal cord terminal cell differentiation. Along the caudal-to-rostral axis of the neural tube, CDX4 transcription is gradually restricted to the dorsal neural tube (Fig 1), at a time when Ngn2 transcription initiates ventrally (Fig 3). At the moment, it is unclear how CDX4 represses Ngn2, as CDX are known transcriptional activators (Isaacs et al., 1998). In sum, by regulating the activation of specification, determination and differentiation genes, CDX4 controls the transition of neural cells from one state to the next during the early maturation of the spinal cord. The regulation of cell transitions by CDX proteins may be a general property of this family of transcription factors, as CDX family members have also been described to control maturation of multipotent cell precursors in intestinal (Hryniuk et al., 2012; Saad et al., 2011) and hematopoietic (McKinney-Freeman et al., 2008; Wang et al., 2008) tissues.
A model of spinal cord neuronal maturation that integrates transcription and signaling networks
In current models of spinal cord development, cells progressively lose potency and acquire neural characteristics under the control of FGF, Wnt and RA signaling (Diez del Corral and Storey, 2004; Gouti et al., 2014). Mutual interactions among these signaling factors restrict the activity of respective pathways to specific domains within the caudal and rostral PNT to direct cell fate decisions. In the caudal end, high levels of FGF promote Wnt transcription while repressing RA pathway activity through a variety of mechanisms (Boulet and Capecchi, 2012; Olivera-Martinez et al., 2012; Sakai et al., 2001; White et al., 2007). In turn, WNT8c promotes RA synthesis in anterior pre-somitic mesoderm and somites, away from the caudal domain of FGF activity. RA secreted from these anterior sources represses FGF synthesis, helping establish and refine the high-caudal to low-rostral gradients of FGF, and indirectly, Wnts (Diez del Corral et al., 2003; Kumar and Duester, 2014). This cross-repressive activities of FGF/Wnts and RA create a caudal-to-rostral gradient of potency signals and a rostral-to-caudal gradient of pro-differentiation signaling that promote the gradual maturation of spinal cord cells (Fig 1, 2). Molecularly, FGF and Wnt maintain NMPs cells by promoting the transcription of multipotency genes T/Bra, Sox2 and Nkx1.2, while simultaneously repressing the differentiation genes Pax6, and Ngn1/2. In contrast, RA promotes differentiation by repressing T/Bra and Nkx1.2 and inducing Pax6 and Ngn1/2 transcription. Thus, the switch from NMP to pre-neural to neurogenic identities is the response of cells to change in extracellular signals (Diez del Corral et al., 2003).
We have expanded the model of spinal cord neurogenesis by integrating signaling and transcription network models (Fig 6). The FGF-Wnt-RA network model provides a series of interactions that result in the spatiotemporal separation of regulatory inputs without providing intracellular mechanisms for the specification and separation of cells states, whereas the transcription factor network provides a molecular mechanism for the specification of different cellular states, but lacks the inputs necessary to drive the system forward. CDX4, at the core of the transcription factor network, provides an integration point for the inputs to regulate effector genes, as Cdx4 transcription is directly regulated by FGF, Wnt and RA (Chang et al., 2016; Keenan et al., 2006; Lee and Skromne, 2014; Nordstrom et al., 2006; Tamashiro et al., 2012). FGF and Wnt promote potency by directly activating Nkx1.2 (Diez del Corral et al., 2003; Tamashiro et al., 2012), but also initiate the loss of potency by sustaining Cdx4 transcription that indirectly represses Nkx1.2 (Fig 4B). A similar “dual-activity” phenomenon is observed in the regulation of Pax6, with FGF both repressing (Bertrand et al., 2000) and activating (via CDX4; Fig 2B) Pax6 transcription. While the mechanism by which CDX4 antagonizes FGF activity at the Pax6 locus is unknown, it may involve a change in Pax6 chromatin state. FGF signaling has been shown to cause the translocation of the Pax6 locus to the nuclear boundary associated with inactive chromatin (Patel et al., 2013). CDX4 could antagonize this activity, as CDX family members have been associated with the clearance of repressive histone modifications in other loci (e. g., Hox; Mazzoni et al., 2013). Regardless of the mechanism, we observe that for two genes, Nkx1.2 and Pax6, CDX4 antagonizes FGF and synergizes with RA. We propose this FGF-Cdx/RA antagonism provide a time delay mechanism to separating early, intermediate and late states of cell differentiation. Experiments are under way to test the interactions between the signaling and transcription factors discussed in this model.
CDX and the coordinated control of spinal cord neuronal maturation, patterning and growth
In addition to regulating spinal cord neuronal maturation, CDX factors are key regulators of axial patterning and elongation. In the context of patterning, CDX4 work together with FGF (and Wnts) to activate transcription of branchial and thoracic Hox genes (Bel-Vialar et al., 2002; Marletaz et al., 2015; Nordstrom et al., 2006; Shimizu et al., 2006; Skromne et al., 2007) and antagonizes RA’s ability to induce hindbrain Hox genes (Lee and Skromne, 2014; Marletaz et al., 2015; Skromne et al., 2007). Significantly this interaction is in contrast to the CDX4-FGF antagonism and CDX4-RA cooperation that we observed during spinal cord neuronal maturation (Fig 6). The molecular mechanism underlying this context-dependent switch in CDX4 activities is currently unknown. However, CDX4 involvement in both processes is significant as it provides a mechanism for coordinating the maturation and anterior-posterior identity specification of spinal cord neurons.
CDX role in vertebrate body extension involves maintaining progenitor population via two distinct mechanisms. Early in spinal cord development CDX cooperate with T/BRA to promote FGF and Wnt signaling cascades and sustain NMP proliferation (Amin et al., 2016; Gouti et al., 2017), whereas, later in development, CDX activate Hox13 genes involved in axial termination (van de Ven et al., 2011; Young et al., 2009). Mutations in mouse that inactive Cdx or prematurely activate Hox13 impairs elongation and morphogenesis of the spinal cord neuroepithelium, which results in irregular or duplicated neural structures (van de Ven et al., 2011). These neural tube defects are similar to those observed in mutants in the mesoderm specification genes T/Bra and Tbx6 (Chapman and Papaioannou, 1998; Yamaguchi et al., 1999), leading to the proposal that caudal spinal cord defects associated with the loss of CDX arise through defects in the specification of NMP descendent (van de Ven et al., 2011). In light of our results, however, the neural tube abnormalities associated with CDX loss could also be explained, at least in part, to defects in spinal cord neuronal maturation. Future work will need to determine the contextual contribution of CDX in coordinating spinal cord cell maturation, differentiation and axial identity specification.
MATERIALS AND METHODS
Chicken embryo incubation and harvesting
Fertilized broiler chicken eggs (Morris Hatchery, Inc.; Miami, FL) were incubated at 38.2° C in a humid chamber until reaching the appropriate stage of development. The embryos were staged according to Hamburger and Hamilton normal table of development (Hamburger and Hamilton, 1951). Embryos post-electroporation were incubated until stipulated time for further analysis.
DNA constructs and chicken in ovo electroporation
Gene overexpression studies were done using standard cloning and electroporation techniques. To achieve high level of gene expression and to track electroporated cells, gene of interest was cloned either into pCIG or pCAGIG vector (Matsuda and Cepko, 2004; Megason and McMahon, 2002). These vectors use the chicken Actin promoter to drive high gene expression levels, and carry a GFP gene as a reporter for transcription. Genes of interest were either cloned into vectors in the laboratory (Cdx4, VP16Cdx4, EnRCdx4, mNkx1.2; for details see supplementary material), or obtained already in the appropriate vector from other laboratories (Pax6-pCIG and EnRPax6-pCIG were kindly provided by Dr. Francois Medevielle (Bel-Vialar et al., 2007); and mNkx1.2-pEF2 was kindly provided by Dr. Yusuke Marikawa (Tamashiro et al., 2012). Plasmids for electroporation were purified using QIAGEN maxi-prep kit, and diluted to a final concentration of 0.5 μg/μl in 1X PBS, with 50ng/ml Fast Green dye to aid in the visualization of the cocktail mix during the procedure. Neural tube of chicken embryos stage HH10-11 were injected with the DNA cocktail mix and immediately electroporated unilaterally following standard protocols (Itasaki et al., 1999; Nakamura and Funahashi, 2001). Only normal-looking embryos with good electroporation in the desired region (e.g., neural tube, pre-neural tube, or caudal neural plate depending on experimental requirements) were selected for further processing by in situ hybridization or immunohistochemistry. Analysis was focused on same axial level in all stage: PNT for stage HH12-13 (prospective thoracic level; Liu et al., 2001), and NT for stage HH16-17 (thoracic level between somites 20-25; Evans, 2003).
In situ hybridization
Analysis of gene transcription by in situ hybridization was done using digoxigenin (DIG)-labeled antisense RNA probes synthesized and hybridized using standard protocol (Wilkinson and Nieto, 1993). Briefly, embryos were harvested at the appropriate stage and fixed with 4% paraformaldehyde diluted in 1x PBS at 4° C overnight, before processing for in situ hybridization. After a series of washes, embryos were exposed overnight in hybridization solution to DIG-labeled antisense RNA probes against Pax6, Nkx1.2, T/Bra, or Cdx4. mRNA expression was detected using an Alkaline Phosphatase coupled Anti-DIG antibody (Roche) and developing embryos with nitro-blue tetrazolium salt (NBT, Thermo Scientific) and 5-bromo-4-chloro-3-indolyl-phosphate (BCIP, Biosynth) at room temperature until dark purple precipitate deposited revealing the areas of gene transcription. For double in situ hybridization, one of the probes was labeled with FITC and developed with Fast Red (Sigma-Aldrich). Post-development, embryos were washed with 1x TBST and then fixed in 4% PFA.
Cryo-sectioning and Immunohistochemistry
Embryos harvested for immunohistochemistry (IHC) analysis were fixed with 4 % PFA for 3 hours at room temperature. Embryos were then embedded in Shandon M1 embedding matrix media (Thermo Scientific) and quickly frozen over dry ice. Mounted embryos were sectioned on Leica CM1850 cryostat and consecutive 20 μm thick sections were collected on positive-charged glass slides (Globe scientific). Antibody staining was performed following standard protocols on slides stacked in Shandon Sequenza slide rack (Thermo Scientific) and supported by Shandon cover plates.
Primary antibodies against anti-mouse PAX6, PAX7 and NKX6.1 were obtained from development Studies Hybridoma Bank. Anti-chicken NGN2 antibody was a kind gift from Dr. Bennett Novitch (Skaggs et al., 2011). Rabbit polyclonal antibody against GFP Tag was obtained from AnaSpec Inc. Goat anti-mouse Alexa Flour 488, Alexa Flour 556 and goat anti-guinea pig Alexa Flour 568 secondary antibodies (Invitrogen) were used for detecting primary antibodies. Sections were covered with DAPI-containing mounting media (Vectashield) and a cover slip, and sealed with nail polish.
Microscopy
Whole embryo images were taken on Zeiss V20 Stereo microscope with an AxioCam MRc digital color camera (Carl Zeiss). Images of transverse section of neural tube were taken on AXIO Examiner Z1 compound microscope with an AxioCam MRc color camera (Carl Zeiss), or on a Leica SP5 confocal microscope (Leica). Confocal images, thickness 2.304 μm, were processed with ImageJ (Schneider et al., 2012). Images were processed for figures using Adobe Photoshop (CC2017, Adobe) for size and resolution adjustment, and for figure preparation.
Quantification of IHC data
To quantify changes in the levels of candidate proteins after electroporation, cells positive for PAX6 or NGN2 were counted on both electroporated and control sides at the same dorsal-ventral position, and their relative ratio was determined. Images were processed with ImageJ IHC toolbox plugin (Shu et al., 2013) before cell counting to select for cells above threshold level as determined by the program algorithm. A total of 6 embryos per conditions were used for determining significance. Significance of difference between mean values of compared pairs was evaluated using two-tailed t-test (Microsoft Excel). Data for each condition was graphed into a box-plus-scatter plot using MATLAB (2014b, The MathWorks Inc., Natick, MA, 2014).
AUTHOR CONTRIBUTIONS
P.J. and I.S. designed the experiments. P.J. performed the experiments. A. J. D. provided intellectual contributions towards designing and troubleshooting experiments. P.J. and I.S. analyzed the results. P.J., A.J.D and I.S. wrote the manuscript.
COMPETING INTERESTS
No competing interest declared.
FUNDING
P. J. was supported by Sigma XI GIAR, and the University of Miami College of Art and Science Dean’s summer and dissertation grants. I. S. was supported by University of Miami College of Arts and Sciences and the Neuroscience Program, and by the National Science Foundation (IOS-090449 and IOS-1755386). DSHB was created by the NICHD of the NIH and maintained by The University of Iowa, Department of Biology.
ACKNOWLEDGEMENTS
We thank Dr. Ann Foley, Dr. Pantelis Tsoulfas and two anonymous reviewers for their expert insights in improving the quality of the original manuscript, and members of the Skromne lab for intellectual discussion, particularly Dr. S. Bandopadhyay. We also thank Dr. K. G. Story (U Dundee, UK), Dr. M. Gouldin (Salk Institute, USA). Dr. F. Medeville (CBI, France), Dr. S. Mackem (NCI, USA), Dr. Y. Marikawa (U Hawaii, USA), Dr. A. V. Morales (Cajal Institute, Spain) and Dr. B. Novitch (UCLA, USA) for generously providing essential constructs and antibodies.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}