

ABSTRACT
Ploidy varies considerably in nature. Yet, our understanding of the impact of ploidy on adaptation is incomplete. Many microbial evolution experiments characterize adaptation in haploid organisms, but few focus on diploid organisms. Here, we perform a 4,000-generation evolution experiment using diploid strains of the yeast Saccharomyces cerevisiae. We show that the rate of adaptation and spectrum of beneficial mutations are influenced by ploidy. Haldane’s sieve effectively restricts access to beneficial mutations in diploid populations, leading to a slower rate of adaptation and a spectrum of beneficial mutations shifted towards dominant mutations. Genomic position also plays an important role, as the prevalence of homozygous mutations is largely dependent on their proximity to a recombination hotspot. Our results demonstrate key aspects of diploid adaptation that have previously been understudied and provide support for several proposed theories.
INTRODUCTION
Understanding the impact of ploidy on adaptation is a central challenge in evolutionary biology. Ploidy varies considerably in the natural world from bacteria that are mostly haploid to some plants that can exist as decaploid1. In addition, all sexual organisms alternate between ploidy states through gamete fusion and meiosis2. Despite its importance, we have an incomplete picture of how ploidy impacts the rate of adaptation and spectrum of beneficial mutations during adaptive evolution. In principle, how ploidy impacts adaptation depends largely on assumptions regarding the dominance of new beneficial mutations. If new beneficial mutations are mostly dominant, then diploids, with twice the mutational target size as haploids, will be twice as likely to acquire beneficial mutations, and will have greater evolutionary potential. Alternatively, if beneficial mutations are recessive, then haploids will have access to new beneficial mutations that have no selective advantage in diploid populations3. This filtering of recessive beneficial mutations in diploids was first proposed by Haldane4 and thus is commonly known as Haldane’s sieve. It is likely that Haldane’s sieve is not the only factor affecting the impact of ploidy on adaptation and that other factors, such as deleterious load and physiological differences between haploids and diploids, also contribute to differences in adaptive potential. If deleterious mutations are mostly recessive, negative phenotypes will be largely masked in diploids. However, if deleterious mutations are dominant (e.g. haploinsufficient mutations), diploids would be negatively impacted by having twice the deleterious load as haploids. In addition, it has been shown in yeast that different ploidy states experience differential regulation of some genes5, which may further impact the mutations that are selectively accessible in haploids and diploids.
The budding yeast, Saccharomyces cerevisiae, can be stably propagated asexually in both haploid and diploid states. This provides an ideal system for studying the effect of ploidy on adaptation. Early experimental results using yeast seemed to demonstrate that diploids evolved faster6. Paquin and Adams looked at rates of adaptation indirectly by monitoring the purging of neutral markers as a proxy for the frequency of selective sweeps and found that diploids fixed mutations 60% faster than haploids, and that this rate did not slow over time6. However, despite this early work, most recent studies find that haploid yeast adapt more quickly compared to diploids, and that this result holds across many strain backgrounds and many environments7–10. Yet, why haploids adapt more quickly is unclear.
Extensive sequencing of laboratory-evolved populations has focused almost exclusively on the spectrum of beneficial mutations in bacterial and haploid yeast populations, rather than in diploids11–14. Collectively, this work shows that the spectrum of beneficial mutations in haploid populations is skewed towards loss-of-function mutations. While there is less information regarding the spectrum of beneficial mutations in higher ploidy populations, one recent effort used whole-genome sequencing and gene expression analysis to explore adaptation of polyploid yeast (including diploids) to Raffinose media and observed a broader spectrum of beneficial mutations in tetraploids15. Some work in diploids implicates Haldane’s sieve as a filter for recessive beneficial mutations. For example, Gerstein et al. constructed heterozygous diploids from a collection of nystatin-resistant haploid yeast, and found that all evolved nystatin-resistant mutations were recessive16. Recent theory also suggests that beneficial mutations in diploids may be overdominant (more beneficial as heterozygotes than homozygotes)17 and there is some experimental evidence suggesting this is the case10,18. Based on these predictions, there is a need to determine how diploidy changes the spectrum of beneficial mutations and the dynamics of adaptation.
Here we measure the rate of adaptation for 48 replicate diploid populations through 4,000 generations and compare these results to previously evolved haploid populations13. We sequence two clones each from 24 populations after 2,000 generations. For two populations we perform whole-genome whole-population time-course sequencing to determine the dynamics of adaptation in diploids and the effect of diploidy on the spectrum of fixed heterozygous and homozygous mutations. We show that diploids adapt more slowly than haploids, have a unique spectrum of beneficial mutations, and that the prevalence of homozygous mutations depends on their genomic position. In addition, we validate haploid-specific, diploid-specific, and shared mutational targets by reconstruction.
RESULTS
Most of the fitness advantage of mutations in evolved haploids is recessive
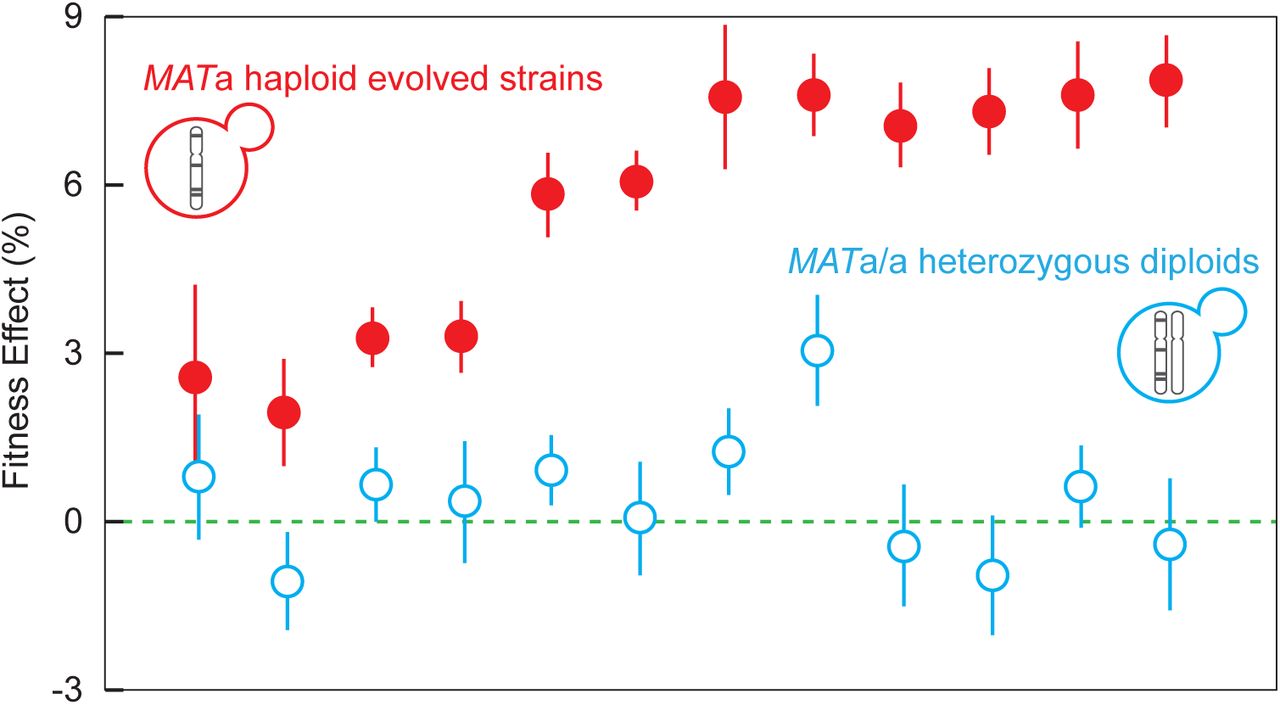
Previously, we determined the spectrum of mutations in haploid populations evolved for 1,000 generations13. We observe a large number of nonsense and frameshift mutations, suggesting that adaptation in haploids may be driven largely by loss-of-function mutations. Many of these mutations are likely to be recessive given that only 3% of gene deletions are haploinsufficient or haploproficient19. To test this hypothesis, we isolated twelve individual clones containing diverse mutations from ten evolved haploid populations and crossed them to the MATα version of the ancestor to generate diploids heterozygous for all of the evolved mutations. To control for effects of mating type, the heterozygous diploids were converted to MATa/a. Fitness of the haploid evolved strains ranged from 2% to 8% (Fig. 1). Heterozygous diploid fitness, however, was only significantly different from zero in one of twelve populations. Interestingly, this one haploid population with partially dominant fitness effects contains epistatically-interacting alleles of KEL1 and HSL7, large effect driver mutations that were not common targets of selection20. Outside of this one example, beneficial mutations in haploids are recessive. Based on this result, we expect Haldane’s sieve to filter out most of these mutations were they to occur in a diploid background, leading to a slower rate of adaptation in diploids and a spectrum of beneficial mutations that is shifted towards dominant gain-of-function mutations.

Filled red circles indicate fitness of twelve evolved MATa clones from the haploid experiment14. Open blue circles indicate the fitness of the same twelve strains as MATa/a diploids where all of the evolved mutations are heterozygous. Error bars are standard error between 7 replicates. Haploid fitness data are from Ref. 20. In order from left to right, the data points correspond to populations BYS1-E03-745, RMS1-G02-545, BYS2-C06-1000, BYS1-A08-545, BYS2-D06-910, BYS1-A09-1000, BYS-D08-1000, BYS2-E01-745, RMS1-G02-825, RMS1-H08-585, RMS1-H08-585, and RMS1-D12-910 in Refs. 13,20.
Diploids evolve more slowly than haploids
To test our hypothesis that the rate of adaptation and mutational spectrum differs between ploidies, we evolved 48 diploid populations in rich glucose medium (YPD) for 4,000 generations under conditions identical to those used for the haploid experiment13. The fitness of each population was measured approximately every 250 generations using a competitive growth rate assay. On average, diploid populations increased in fitness by only 5.8% over 4,000 generations, whereas haploid populations increased in fitness by 8.5% over 1,000 generations (Fig. 2). As expected given the stochastic nature of evolution, we observe a large variation in fitness gains and trajectories across replicate populations. Diploids increase in fitness by between 2% and 11% over 4,000 generations, whereas haploids increase in fitness between 5% and 12% over 1,000 generations (Fig. 2). In order to predict future rates of adaptation, we fit the averaged data to both a power law (which has been shown to accurately model haploid evolution21) and a linear fit. The haploid data are described better by a power law fit than by a linear fit (p<0.001, F-test) but the diploid data are not (p=1, F-test).

Over the course of a 4,000 generation evolution experiment, diploids adapt more slowly than haploids. Fitness effects for haploids and diploids are plotted as averages represented by red circles and blue squares, respectively. Light red and light blue traces indicate individual trajectories of haploid and diploid populations, respectively. The red dotted line represent the fit of the haploid average to the power law equation, y = (bx + 1)a −1. The blue dotted line represents the linear fit of the diploid average data. Haploid data are from Ref. 13.
The spectrum of beneficial mutations is different between diploids and haploids
Having confirmed that diploids adapt more slowly than haploids, we next determined the spectrum of mutations in diploids by sequencing two clones each from 24 diploid populations from generation 2,000. Each clone was sequenced to an average depth of ~30x coverage and we called mutations only if they were present in both clones. We classified each mutation as heterozygous or homozygous. We identify 381 mutations across the 24 populations. Heterozygous mutations outnumber homozygous mutations 9:1. The number of mutations per population ranged from 5 in population B04 to 27 in population G08. Of the 381 mutations, 118 are intergenic, 56 are synonymous, 175 are missense, and 31 are nonsense or frameshift (Table S1). Comparing this to the set of haploid mutations, we see that diploids are slightly enriched for intergenic and synonymous mutations but this is not significant by Chi-square analysis (295 intergenic, 101 synonymous, 492 missense, 118 nonsense/frameshift, p=0.025). Homozygous mutations are overrepresented on the right arm of Chr. XII (p<0.001, Chi-square). Excluding Chr. XII, the genome-wide distribution of homozygous and heterozygous mutations is not significantly different from random (p=0.06, Chi-square, Fig. 3).

Sequencing data for 381 diploid mutations show an overrepresentation of homozygous mutations on the right arm of Chr. XII (p<0.001, Chi sq.). Chromosomes are represented as grey bars. Homozygous mutations are represented by red lines sticking out of the top of chromosomes. Heterozygous mutations are represented by blue lines sticking out of the bottom of chromosomes. Mutations that were seen as both homozygous and heterozygous in paired samples are shown as green lines in the middle of chromosomes.
Of the mutations in our diploid populations, we identified putative targets of selection as those genes in which mutations were seen at least twice (Table 1). We observe interesting differences in the biological processes that are common targets of selection in diploids. Genes involved in the Ras pathway and the mating pathway are common targets of selection in haploid populations13. However, no mutations in either of these two pathways were detected in the evolved diploids. Cell wall biogenesis/assembly and cytokinesis are shared targets of selection in both haploid and diploid populations. However, even within these biological processes, there are both shared (e.g. KRE6) and ploidy-specific target genes, such as the haploid-specific GAS1 and KRE5, and the diploid-specific CTS1. Furthermore, the most common haploid target of selection, IRA1 (a negative regulator of Ras), was not mutated in any of our diploid populations.
Common targets of selection in diploid populations
Evolved alleles show varying degrees of dominance and ploidy-dependence
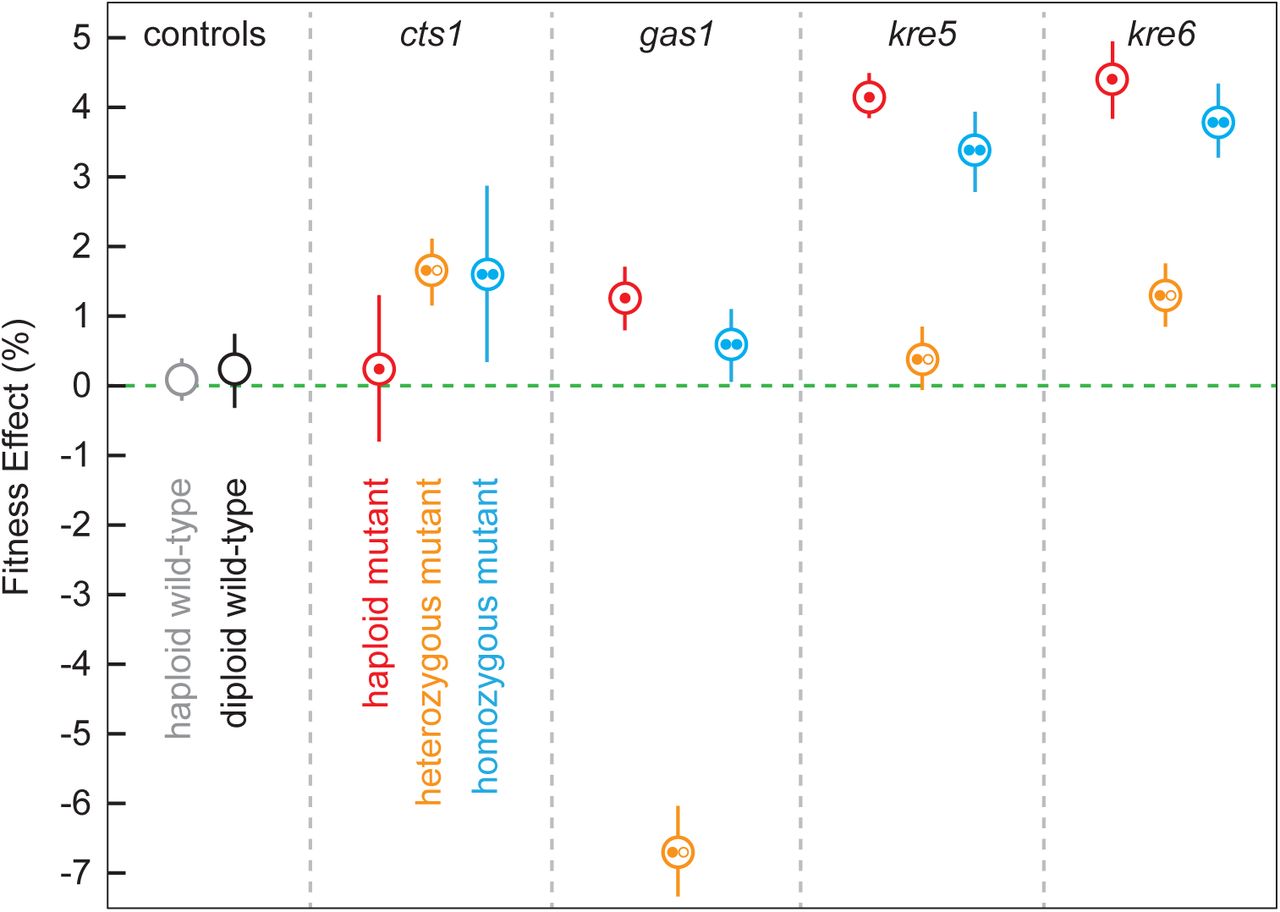
Given that cell wall mutants represent a diverse subset of beneficial mutations, including haploid-specific (GAS1, KRE5), diploid-specific (CTS1), and common (KRE6) beneficial mutational targets, we focused on these four targets of adaptation. We reconstructed these alleles as haploids, heterozygous diploids, and homozygous diploids and performed competitive fitness assays on all constructs to test whether the relative fitness effects of evolved alleles are contingent on the ploidy in which they arose (Fig. 4). Consistent with our classification of these mutations as putative drivers of adaptation, we found that each is beneficial in the background in which it arose. Mutations in the shared target of selection, KRE6 are beneficial as haploids, heterozygous diploids, and homozygous diploids, though the benefit as a heterozygote is less than that of a homozygote, showing that it is partially dominant (coefficient of dominance, h=0.34). The diploid-specific target of selection, CTS1, is equally beneficial as a heterozygote or a homozygote (h≈1), however this mutation is neutral in a haploid background. Mutations in the haploid-specific targets, GAS1 and KRE5, are beneficial in haploids and homozygous diploids. In the case of KRE5, the heterozygous diploid fitness is not statistically different from zero (h=0.10, Fig. 4). In the case of GAS1, the heterozygote is strongly deleterious.

Average fitness effects of haploid (single filled circle in red), heterozygous diploid (one filled circle and one open circle in yellow) and homozygous diploid (two filled circles in blue) cell wall mutants are compared to wild-type haploids (light grey empty circle) and wild-type diploids (dark grey empty circle). The corresponding gene mutated in each is listed at the top. The cts1 mutation is from the F05 population of our diploid data (Table S1). The gas1, kre5, and kre6 mutations are from our haploid populations BYS2-D06, RMB2-B10, and RMS1-H08, respectively14. Each point is an average of seven replicates, or six replicates for the wild-type points. Error bars are the standard error of these averages.
Gene conversion occurs more rapidly for CTS1 than ACE2
Interestingly, although our reconstruction experiments show no significant fitness difference between a heterozygous and homozygous CTS1 mutation, all six CTS1 mutations are homozygous in the diploid populations. Another common diploid-specific target, ACE2, is observed as a homozygous mutant in two of the four populations in which it arose. Both ACE2 and CTS1 are located on Chr. XII. Two possible mechanisms could explain this observation: gene conversion or chromosome loss. To verify that our strains have two copies of Chr. XII, we measured read coverage in our sequencing data and found that coverage across Chr. XII was consistent with coverage across the rest of the genome in all samples containing a CTS1 or ACE2 mutant. We also performed tetrad dissections of CTS1 mutant strains and found four-spore viable tetrads, which is unlikely if all or part of one copy of Chr. XII is missing. These homozygous mutations therefore likely arise from gene conversion events.
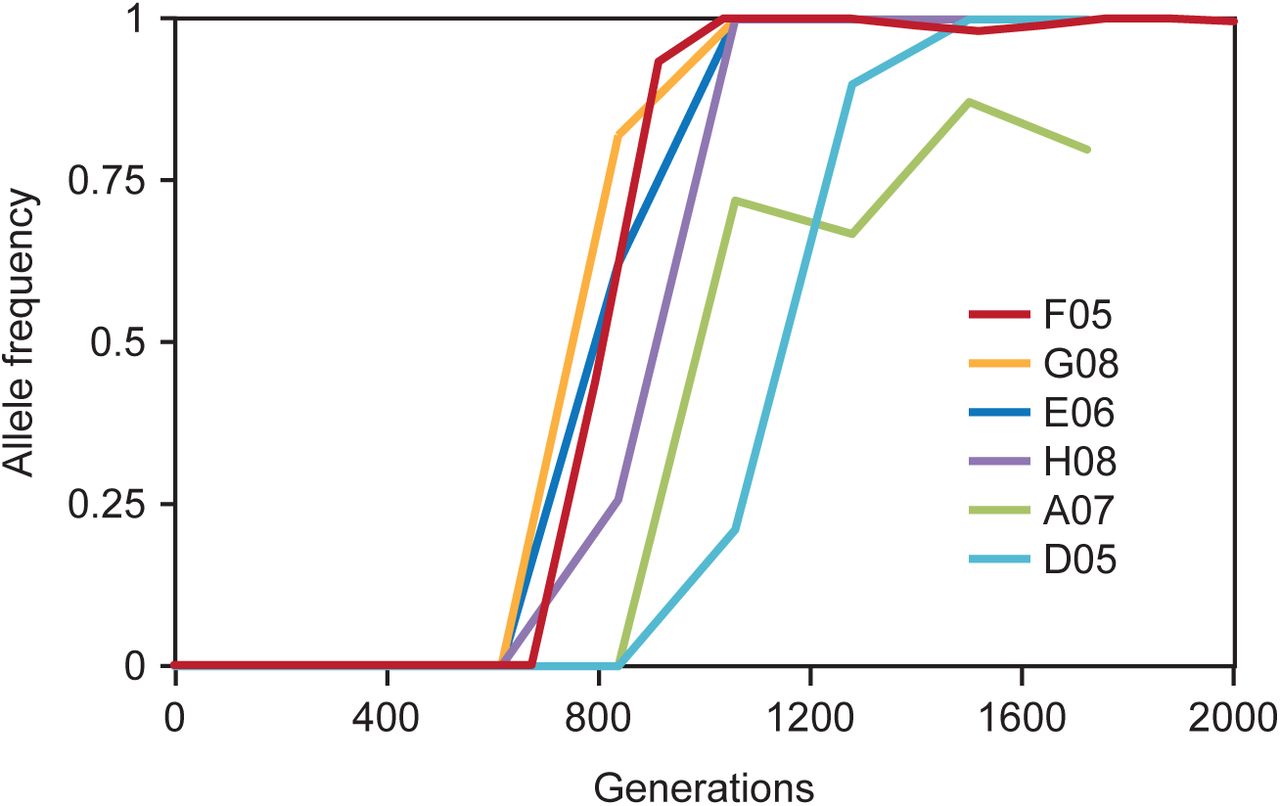
With this in mind, we investigated the dynamics of genome sequence evolution by performing whole-genome whole-population time-course sequencing on two populations, one with a homozygous CTS1 mutation, and one with a homozygous ACE2 mutation. In population A05, the ace2 allele establishes around generation 900, rises to a frequency of ~0.5 by generation 1,100, and remains there until around generation 1,400, when it rises to a frequency of ~1, fixing in the population by generation 1,600 (Fig. 5A). In population F05, the cts1 allele establishes around generation 700, rises to a frequency of ~0.5 around generation 800, and continues without pausing to reach a frequency of ~1 around generation 1,000 (Fig. 5B). In this case, the mutant cts1 homozygote establishes before the heterozygote fixes. In addition to CTS1 and ACE2 mutations, we observe other mutations in these populations, including a few heterozygosities that were present in the ancestral strain. We can use the information from the dynamics of these additional mutations to inform our theory of gene conversion leading to homozygosity. The A05 population contains three non-ACE2 variants, two of which sweep as heterozygotes. The third is an existing polymorphism in an LTR that was heterozygous in the ancestor and is approximately 387 kb away from ACE2 on Chr. XII. When the ace2 allele fixes in this population, the LTR also loses heterozygosity (Fig. 5A). The F05 population contains 11 non-CTS1 mutations, including seven heterozygous mutations, two homozygous mutations, and two positions that were heterozygous in the ancestor, but lost heterozygosity during the evolution of this population. Among these is a mutation in DUS4, which sweeps to fixation with the cts1 allele, and the previously described heterozygosity in an LTR which loses heterozygosity at the same time as CTS1 (Fig. 5B). In this case, the LTR is approximately 84 kb away from CTS1.

a, Time-course sequencing of population F05 containing a cts1 mutation shows multiple mutations fixing as a cohort with cts1. The cts1 mutant fixes in the population quickly, without a pause around 0.5 signifying heterozygote fixation. Mutations are shown with the chromosomes on which they occur. Dashed lines represent intergenic mutations. b, Genotypes of CTS1 at three time points are shown as homozygous wild type (two open circles), heterozygous (one filled and one open circle), and homozygous mutant (two filled circles). Homozygous mutants appear before homozygous wild type is eliminated. c, Time-course sequencing of population A05 containing an ace2 mutation. There is a decrease in slope around an allele frequency of 0.5, suggesting heterozygote fixation before homozygous mutant establishment. Mutations are shown with the chromosomes on which they occur. d, Genotypes of ACE2 at three time points are shown as homozygous wild type (two open circles), heterozygous (one filled and one open circle), and homozygous mutant (two filled circles). Homozygous mutants only appear after the homozygous wild type is eliminated by the sweep of the heterozygous mutant.
We hypothesize that the length of the pause of the ace2 allele at a frequency of 0.5 is the waiting time between the heterozygote fixing in the population and the gene conversion event. To test this, we performed Sanger sequencing of clones from both populations before, during, and after the sweep, corresponding to allele frequencies of 0, ~0.5 and 1. For ACE2, 19/20 clones at an allele frequency of ~0.5 are heterozygous, consistent with a full sweep of the heterozygote before gene conversion (Fig. 5C). For CTS1 at an allele frequency of ~0.5, we observe a mix of heterozygous, homozygous mutant, and homozygous ancestral clones, meaning that the heterozygote did not fix in the population before the gene conversion event, as we saw for the ace2 allele in population A05 (Fig. 5D). This suggests that the cts1 allele gene converts very quickly compared to the ace2 allele.
While the F05 allele of CTS1 is a nonsense mutation (likely to be a loss-of-function), the other five CTS1 mutant alleles seen in our diploids are missense mutations (possibly alteration-of-function). Even so, all six show similar dynamics of adaptation, with similar rates of gene conversion (Figure S1). Based on this, we hypothesize that the difference in gene conversion rates between CTS1 and ACE2 mutations is likely not due to the effects of individual mutations, but rather is due to the location of the CTS1 and ACE2 genes themselves.

a, The average rate of adaptation for haploids (red) and diploids (blue). The haploid rate decreases over time, while the diploid rate remains constant. The red dotted line is the extrapolation of the haploid rate of adaptation based on the power law. b, Average adaptation relative to ancestor for haploids (red), and diploids (blue), with predictions out to 16,000 generations using power law for haploid data (red dotted line) and linear fit for diploid data (blue dotted line).
DISCUSSION
We show that our diploid populations adapt more slowly than haploids because recessive beneficial mutations are not selectively accessible to diploids. We have used our power law and linear fits to predict future rates of adaptation for our evolving haploids and diploids, respectively. In our model, the rate of haploid adaptation starts off higher but decreases over time following the power law, while the diploid rate of adaptation is lower but constant, following a linear fit. Therefore, the model predicts that around generation 3,500, the haploid and diploid rates of adaptation are equal, but past this point, the diploid rate is faster than the haploid rate (Fig. S1A). Additionally, our model predicts that by generation 15,000, diploids will have achieved more total adaptation relative to their ancestor than haploids (Fig. S1B). The declining rate of adaptation in haploids may be due to the exhaustion of available beneficial recessive mutations.
Through our comparison of the mutations gained during haploid and diploid adaptation, we observe that both the mating pathway and the negative regulation of Ras were prominent targets of adaptation in haploids, but were not targets of selection in diploids. This makes sense for the mating pathway, as it is repressed in diploids (and therefore sterile mutations are not selectively advantageous). As to why negative regulation of Ras is not a target of adaptation in diploid populations, there are two possibilities. The first is that all or most of the spectrum of beneficial mutations that can be made to genes involved in the negative regulation of Ras are recessive in their fitness effects, and thus unlikely to pass through Haldane’s sieve. The other possibility is that the Ras pathway is regulated differently in haploids and diploids, such that some genes are not functionally redundant in haploids and diploids. Some yeast genes are known to have ploidy-specific regulation5. Interestingly, CTS1 is included among these differentially regulated genes, increasing in expression with ploidy. This physiological difference between haploids and diploids may explain why mutations to CTS1 are diploid-specific.
By examining our diploid evolved mutations, we found that Haldane’s sieve is effective at filtering out recessive beneficial mutations from evolving diploid populations, but it is not the only factor affecting the rate of adaptation and spectrum of beneficial mutations in diploids. We also see that genomic position is an important factor, largely due to varying rates of recombination throughout the genome. This is particularly visible on the right arm of Chr. XII, which contains the rDNA locus, a known recombination hotspot in yeast22. We find that homozygous mutations are rare (only 10% of diploid mutations) but are largely concentrated on the right arm of Chr. XII (Fig. 3), particularly in the CTS1 and ACE2 genes (Fig. S2). This implies that the ability for beneficial diploid mutations to become homozygous, and thus to escape from Haldane’s sieve will depend strongly on local rates of gene conversion. Our results here further validate the point made by Mandegar and Otto that mitotic recombination is an important factor in the spread of beneficial alleles in evolving asexual populations23.
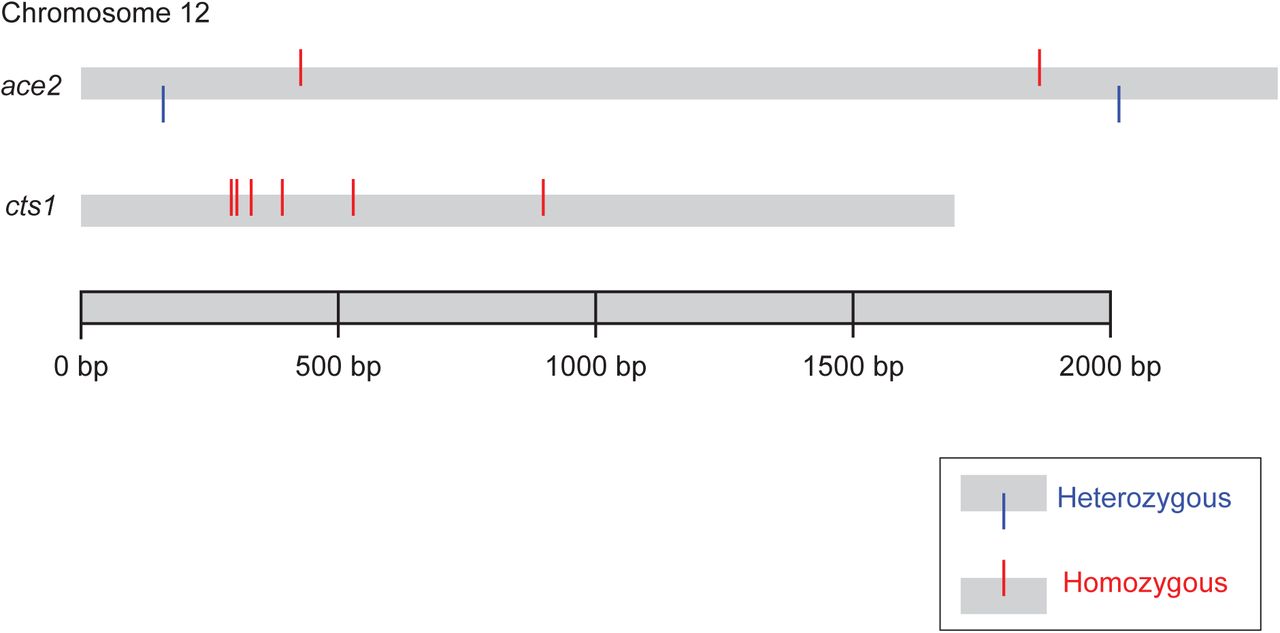
The ACE2 locus on Chr. XII is the site of two homozygous and two heterozygous mutations. The CTS1 locus on Chr. XII is the site of six homozygous mutations. Homozygous mutations are represented by red lines sticking out of the top of chromosomes. Heterozygous mutations are represented by blue lines sticking out of the bottom of chromosomes.
We reconstructed evolved haploid-specific, diploid-specific, and shared alleles in isolation as haploids, heterozygous diploids, and homozygous diploids, and found that the degree of heterozygosity differs between the alleles. Our reconstructed allele of CTS1 shows a surprisingly high degree of dominance (h≈1), yet all six of the cts1 alleles present in our diploid populations rapidly gene converted and fixed as homozygotes. One possible explanation is that the homozygote does have an advantage over the heterozygote, but that we cannot detect this difference with flow cytometry-based fitness assays. One complication is that homozygous mutations in CTS1 result in a cell aggregation phenotype. This aggregation phenotype has previously been shown to occur during yeast laboratory evolution24–27. Our evolved CTS1-mutant diploid populations and the reconstructed cts1 haploid and homozygous diploid strains form aggregates, but the reconstructed heterozygous mutant does not (Fig. S3). It is possible that this phenotype complicates fluorescence-based fitness measurements of these strains.

The reference is the common ancestor of all strains shown here. The top row shows reconstructed single-mutant strains for the F05-cts1 allele. The middle row shows the six evolved populations which contain cts1 mutations. The bottom row shows the four evolved populations which contain ace2 mutations. Each image was taken at 400x magnification.
Furthermore, from the dynamics of adaptation of all six CTS1-mutant populations (Fig. 5A, Fig. S4), we have shown that CTS1 mutants become homozygous both very frequently and quickly. Additionally, from the dynamics of the F05 population containing our CTS1 mutation of interest, we see a mutant dus4 allele that travels to fixation with the cts1 allele, and one ancestral heterozygosity in a long terminal repeat that loses heterozygosity at the same time (Fig. 5A). All three of these genetic loci are on Chr. XII downstream from the yeast rDNA locus. We propose that a single recombination event caused a loss of heterozygosity for all three of these loci, establishing the cts1 homozygous mutant. Our dynamics of adaptation also show the loss of heterozygosity of another ancestral heterozygous long terminal repeat paired with the rise of the ace2 allele to fixation, suggesting that a similar gene conversion may have established the ace2 homozygote. In this same population, there may be another case of gene conversion where the loss of heterozygosity in IOC4 co-occurs with the fixation of an FSH2 mutant. Both of these loci are on Chr. XIII (Fig. 5C). Through these examples and the concentration of homozygous mutations and lack of heterozygous mutations on the right arm of Chr. XII, we also propose that when gene conversion events occur, they involve a single point of recombination leading to loss of heterozygosity from the point of origin through the telomere. This is similar to the types of lesions observed due to break-induced replication28.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Sanger sequencing of five additional cts1 mutants shows that the dynamics of adaptation are similar between cts1 mutants. The six total populations are listed on the right.
While gene conversion allows us to explain the location-specific enrichment of homozygous mutations, these are a minority of our putative driver mutations (Table 1) and are therefore not sufficient to explain the fitness gains we see in diploids. There must also be some adaptive heterozygous mutations, which remain heterozygous. It is possible that some of our heterozygous putative driver mutations are maintained as heterozygotes because they are overdominant as suggested by Sellis et al.17,18. Overall, we have shown here that over the course of 4,000 generations of evolution, diploid populations adapt slower than haploid populations, and that diploid populations have a unique spectrum of beneficial mutations compared to haploid populations. In addition, the majority of adaptive diploid mutations are heterozygous and the prevalence of adaptive diploid homozygous mutations depends on the position of mutations in the genome. Collectively this work fills a gap in understanding how ploidy impacts adaptation, and provides empirical support for the hypothesis that diploid populations have restricted access to beneficial mutations.
METHODS
Strain Construction
The strains used in this experiment are derived from the base strain, yGIL432, a haploid yeast strain derived from the W303 background with genotype MATa, ade2-1, CAN1, his3-11, leu2-3,112, trp1-1, URA3, bar1Δ::ADE2, hmlαΔ::LEU2, GPA1::NatMX, ura3Δ::PFUS1-yEVenus. This strain was previously reported as DBY15105 25. A nearly isogenic MATα version of yGIL432 was constructed by introgressing the MATα allele through three backcrosses into the yGIL432 background. This MATα strain, yGIL646, was crossed to yGIL432 to generate the diploid strain yGIL672. Haploid MATa evolved strains used in Figure 1 are previously described in Ref. 13, and their corresponding MATa/a heterozygous diploids are described in Ref. 20. For the reconstruction experiments, the haploid cts1 mutation in the yGIL432 background was generated using Cas9 allele replacement29. Briefly we retargeted the guide RNA expressing plasmid (Addgene #43803) to cts1 using the gRNA sequence 5′ TTCTTCAAAATCTCAACATA 3′. We co-transformed the gRNA plasmid (pGIL083), a constitutive Cas9 plasmid (Addgene #43802), and a plasmid (pGIL089) containing the cts1 mutant allele into our MATa strain, yGIL432. We screened for plasmid retention, PCR screened single colonies for the allele replacement, and cured the plasmids. Allele replacements of gas1, kre5, and kre6 in the yGIL432 were obtained from Matt Remillard (Princeton University), who constructed them using alleles from our haploid populations BYS2-D06, RMB2-B10, and RMS1-H08, respectively. Each of the haploid MATa strains was crossed to yGIL646 to generate heterozygous diploids. The heterozygous diploids were then sporulated, tetrads were dissected, and haploid spores were mating-type tested and genotyped. Appropriate haploid spores were then crossed to each other to create homozygous diploid strains for each mutation.
Long-Term Evolution
To set up the long-term evolution experiment, a single clone yGIL672 was grown to saturation in rich-glucose YPD medium, was diluted 1:10,000, and was used to seed 48 replicate populations in a single 96-well plate. This initial plate was duplicated and then frozen down for future use. The cultures were evolved through 4,000 generations (400 cycles) of growth and dilution in YPD at 30°C. Every 24 hours, the populations were diluted 1:1024 by serial diluting 1:32 (4 μl into 130 μl) x 1:32 (4 μl into 130 μl) into new medium containing ampicillin (100 mg/L) and tetracycline (25 mg/L). All dilutions were performed using the Biomek Liquid Handler equipped with a Pod96. Approximately every 50 generations, plates were mixed with 50 μl of 60% glycerol and archived at −80°C.
Competitive Fitness Assays
Flow cytometry-based competitive fitness assays were performed essentially as described previously13,20,25 Briefly, experimental and reference strains were grown to saturation in separate 96-well plates. If the strains to be tested were coming from the freezer, the strains were passaged once by diluting 1:1024 to re-acclimate the strains to the appropriate medium. Experimental and reference strains were mixed 50:50 using the Biomek Liquid handler and were propagated for 40 generations under identical conditions to the original evolution experiment. In this project we used two reference strains: diploid MATa/a, and diploid MATa/α. These reference strains are derived from the yGIL432 base strain, but contain a constitutive ymCitrine integrated at the ura3 locus.
We used previously collected competitive fitness data from evolved MATa haploid strains13. We performed a competitive fitness assay as described below on previously constructed heterozygous MATa/a diploid strains20. To perform this assay, we constructed a MATa/a diploid version of the fluorescently-labeled reference strain using a plasmid containing LEU2 under a MATa-specific promoter to select for gene conversion at the mating type locus. These data were normalized to their respective references and compiled into Figure 1.
Whole-Genome Sequencing
For sequencing clones, we struck to single colonies on YPD and picked two colonies from each population to sequence. These individuals were grown to saturation in liquid media and total genomic DNA was isolated for each sample. For whole-genome whole-population time-course sequencing, we thawed each population at 18 time points from generation 0 to 2,000, and transferred 10 μl into 5 ml of YPD. We made genomic DNA preparations as above.
We followed a modified version of the Nextera sequencing library preparation protocol30, modified further as described in Ref. 20. We used the Nextera sequencing library preparation kit and protocol to isolate total genomic DNA and add the unique Nextera library barcodes to all 48 samples. We measured the concentration of DNA in each sample using a NanoDrop spectrophotometer and confirmed these values using a Qubit fluorometer. We equalized the DNA concentration of each sample via dilution and mixed all 48 samples into a single pool. We used a BioAnalyzer High-Sensitivity DNA Chip (BioAnalyzer 2100, Agilent) to confirm that the pool contained the appropriate length DNA fragments, and performed gel extraction on the pool to remove short fragments. The pool was run on an Illumina HiSeq 2500 sequencer with 157 nucleotide single-end reads by the Sequencing Core Facility within the Lewis-Sigler Institute for Integrative Genomics at Princeton University. After an initial sequencing run provided us with the number of reads from each sample, we remixed the pool to better represent underrepresented samples and it was resequenced.
Sequencing Analysis Pipeline
The raw sequencing information was first merged from 3 lanes of sequencing via concatentation. This single file was split into 48 files by the barcodes corresponding to each sample using a custom Perl script (barcode_splitter.py) from L. Parsons (Princeton University). Each sample was aligned to the same customized W303 genome based on the S288C genome available on SGD using the Burrows-Wheeler Aligner (BWA, Version 0.7.12), using default parameters except “Disallow an indel within INT bp towards the ends” set to 0 and “Gap open penalty” set to 5, creating both a .bam and .bai file for each. Variants were called using the FreeBayes variant caller (Version 0.9.21-24-g840b412) and merged together into a single spreadsheet. Variants that existed only in paired clones (448 mutations total) were annotated manually using the Integrated Genome Viewer (IGV) and false calls were removed. This fully annotated table (containing 383 mutations) is available in Table S1.
For whole-genome whole-population time-course sequencing, we used the same Nextera sequencing library preparation protocol as described above, with the following changes: Instead of two clones from each of 24 populations, we isolated genomic DNA from whole populations at 18 time points for two populations for a total of 36 samples. During the sequencing analysis, after splitting the reads based on the Nextera barcodes, we used a script (clipper.sh) to remove any Nextera adapter sequences introduced by sequencing short fragments. We used a previously-described set of scripts (allele_counts.pl and composite_scores.pl) to call real mutations13.
Reconstruction Experiments
We used three previously identified mutations, which were identified in evolved haploids (mutations to gas1, kre5, and kre6), and one mutation identified in a diploid population (mutation to cts1). We used CRISPR/Cas9 genome editing to reconstruct our evolved cts1 mutation in our ancestral haploid background. The other three mutations were previously reconstructed as single mutants in our haploid background by Matt Remillard (Princeton University), who generously shared these resources with us. These four haploid MATa strains were crossed to our haploid MATα ancestor to construct heterozygous diploids for each mutation. These heterozygous diploids were sporulated, tetrads were dissected, and haploid spores were mating-type tested and Sanger-sequenced to determine which spores contained the mutation but were of opposite mating type. These haploid spores were then crossed to create diploids homozygous for each mutation. Fitness of each of these twelve total samples was measured via competitive fitness assays across seven replicates along with control haploids and diploids with no mutations, all against the appropriate fluorescent ancestor strains.
ACKNOWLEDGEMENTS
We thank Matthew Remillard (Princeton University) for providing strains. We thank Alex Nguyen and Michael Desai (Harvard University) for providing the plasmid with mating-type-specific selectable markers. We thank Anna Selmecki, Sean Buskirk, Katie Fisher, and Ryan Vignogna for their comments on the manuscript. This work was supported by the Charles E. Kaufman Foundation of The Pittsburgh Foundation.
REFERENCES




