

Abstract
Despite their essential role in the process of chromosome segregation in eukaryotes, kinetochore proteins are highly diverse across species, being lost, duplicated, created, or diversified during evolution. Based on comparative genomics, the duplication of the inner kinetochore proteins CenH3 and Cenp-C, which are interdependent in their roles of stablishing centromere identity and function, can be said to be rare in animals. Surprisingly, the Drosophila CenH3 homolog Cid underwent four independent duplication events during evolution. Particularly interesting are the highly diverged and subfunctionalized Cid1 and Cid5 paralogs of the Drosophila subgenus, which show that over one thousand Drosophila species may encode two Cid genes, making those with a single copy a minority. Given that CenH3 and Cenp-C likely co-evolve as a functional unit, we investigated the molecular evolution of Cenp-C in species of Drosophila. We report yet another Cid duplication within the Drosophila subgenus and show that not only Cid, but also Cenp-C is duplicated in the entire subgenus. The Cenp-C paralogs, which we named Cenp-C1 and Cenp-C2, are highly divergent. The retention of key motifs involved in centromere localization and function by both Cenp-C1 and Cenp-C2 makes neofunctionalization unlikely. In contrast, the alternate conservation of some functional motifs between the proteins is indicative of subfunctionalization. Interestingly, both Cid5 and Cenp-C2 are male germline-biased and evolved adaptively. Our findings point towards a specific inner kinetochore composition in a specific context (i.e., spermatogenesis), which could prove valuable for the understanding of how the extensive kinetochore diversity is related to essential cellular functions.
Introduction
During eukaryotic cell division, accurate chromosome segregation requires the interaction of chromosomes with the microtubules from the spindle apparatus. This interaction is mediated by the kinetochore, a multiprotein structure that is hierarchically assembled onto centromeres. Upstream in the assembly of the kinetochore are CenH3 and Cenp-C, two interdependent proteins in their roles of establishing centromere identity and function. CenH3 is the histone H3 variant found in centromeric nucleosomes and, therefore, considered the centromere epigenetic marker (Dalal et al. 2007). During kinetochore assembly, Cenp-C binds to CenH3 and recruits other kinetochore proteins (Przewloka et al. 2011; Liu et al. 2016). CenH3 and Cenp-C are fundamentally interdependent because the centromeric localization of one depends on the centromeric localization of the other (Erhardt et al. 2008; Orr and Sunkel 2011). This interdependence is also illustrated by the fact that both CenH3 and Cenp-C have similar phylogenetic profiles (i.e., they have similar patterns of presence and absence across the eukaryotic evolutionary tree) and likely co-evolve as a functional unit (van Hooff et al. 2017). One interesting case is that seen in insects, where CenH3 was lost independently five times, and in all these cases Cenp-C was also lost (Drinnenberg et al. 2014).
Despite the essentiality of centromeres, both centromeric DNA (CenDNA) and proteins are remarkably diverse (Henikoff et al. 2000; Talbert et al. 2004; Plohl et al. 2008). This rapid evolution despite the expectation of constraint is referred to as the “centromere paradox” (Henikoff et al. 2001). This paradox may be explained by the centromere drive hypothesis, which proposes that genetic conflicts during female meiosis drive centromere evolution (Henikoff et al. 2001; Dawe and Henikoff 2006).
In the female meiosis of animals and plants, the meiotic spindle fibers are asymmetric in a way that one pole will originate a polar body and the other will give rise to the oocyte. As a result, there is potential for non-mendelian (biased) inheritance if a pair of homologous chromosomes have kinetochores that interact unequally with the spindle fibers (Ross and Malik 2014). The heterogeneity in kinetochore function between homologs is a result of differences in abundance of centromeric DNA sequences. One homolog may have a ‘strong’ centromere, which has an expanded cenDNA that recruits more kinetochore proteins and delivers its chromosome into the oocyte at > 50% frequency, or a ‘weak’ centromere, which has a contracted cenDNA that in turn recruits less kinetochore proteins and delivers its chromosome into the oocyte at < 50% frequency (Iwata-Otsubo et al. 2017). However, the spread of expanding centromeres throughout a population might be accompanied by deleterious effects, such as increased male sterility or a skewed sex ratio (Fishman and Saunders 2008; Rutkowska and Badyaev 2008; Malik and Henikoff 2009). The centromere drive hypothesis proposes that changes in CenH3 and Cenp-C related to more ‘flexible’ DNA-binding preferences are expected to counteract the transmission advantage gained by expanded centromeres and diminish the associated deleterious effects, thus restoring meiotic parity for both homologs (Henikoff et al. 2001; Dawe and Henikoff 2006).
The kinetochore is highly diverse across species, with proteins being lost, duplicated, created, or diversified during evolution (van Hooff et al. 2017). Given that data directly supporting a correlation between the evolution of cenDNA, CenH3 and Cenp-C are still absent, it is not known if and how such structural divergence is related to centromere drive suppression. However, the subfunctionalization of CenH3 paralogs in some lineages of Drosophila has been hypothesized to be linked to centromere drive suppression. Kursel and Malik (2017) have recently reported that the Drosophila CenH3 homolog Cid underwent four independent duplication events during evolution, and some Cid paralogs are primarily expressed in the male germline and evolve under positive selection (Kursel and Malik 2017). These duplications could have allowed the rapid evolution of centromeric proteins without compromising their essential function by separating functions with divergent fitness optima. The existence of germline-biased CenH3 duplicates (which do not interfere with essential mitotic functions) in genetically tractable organisms provides an opportunity to study the functional consequences of the genetic variation for kinetochore-related processes.
Given the interdependence between CenH3 and Cenp-C, we decided to further analyze the molecular evolution of the Cid and Cenp-C genes in Drosophila species. Here, we report a novel Cid duplication within the Drosophila subgenus and show that not only Cid, but also Cenp-C is duplicated in the entire Drosophila subgenus. The Cid and Cenp-C paralogs likely subfunctionalized, as some motifs are alternatively conserved between the paralogs. Interestingly, both the Cid and Cenp-C duplications generated copies that are male-biased and evolve under positive selection. Our findings point towards a specific kinetochore composition in a specific context (i.e., the male germline), which could prove valuable for the understanding of how the extensive kinetochore diversity is related to essential cellular functions.
Results and Discussion
Cid1 was replaced by a new paralog in a clade within the Drosophila subgenus
Duplicate Cid genes exist in D. eugracilis (Cid1, Cid2) and in the D. montium subgroup (Cid1, Cid3, Cid4), both within the Sophophora subgenus, and in the entire Drosophila subgenus (Cid1, Cid5). In all analyzed species from the Drosophila subgenus, Cid1 is flanked by the cbc and bbc genes, and Cid5 is flanked by the Kr and CG6907 genes (Kursel and Malik 2017). As expected, we found two Cid genes while looking for the orthologs of Cid1 and Cid5 in the assembled genomes of two cactophilic species from the Drosophila subgenus, D. buzzatii and D. seriema (repleta group). Surprisingly, while one of the genes is present in the expected locus of Cid5, the other one is located in an entirely different locus, flanked by the CG14341 and IntS14 genes. We named this new paralog as Cid6.
By investigating the Cid1 locus of D. buzzatii, we found a myriad of transposable elements (TEs) surrounding a 116-bp fragment of the original Cid1 gene (fig. 1, upper panel). Due to fragmentary genome assembly, the Cid1 locus of D. seriema could not be identified. Both Cid5 and Cid6 of D. buzzatii and D. seriema share ∼40% amino acid identity but, in contrast, Cid6 of each species and Cid1 of the closely related D. mojavensis are much more similar, sharing ∼80% identity. Fluorescent in situ hybridizations on polytene chromosomes showed that Cid6 is distal (in relation to the chromocenter) in the Muller element B of D. buzzatii and D. seriema, and that Cid1 is proximal in the Muller element C of D. mojavensis and the outgroup D. virilis (fig. 1, lower panel). Therefore, we inferred that Cid1 was degenerated by several TE insertions after the origin of Cid6 by an inter-chromosomal duplication of Cid1 in the lineage that gave rise to D. buzzatii and D. seriema. The time of divergence between D. buzzatii and D. seriema has been estimated at ∼4.6 mya, and the divergence between them and the closely related D. mojavensis has been estimated at ∼11.3 mya (Oliveira et al. 2012). Therefore, the Cid1 duplication that gave rise to Cid6 happened between ∼4.6 and 11.3 mya.
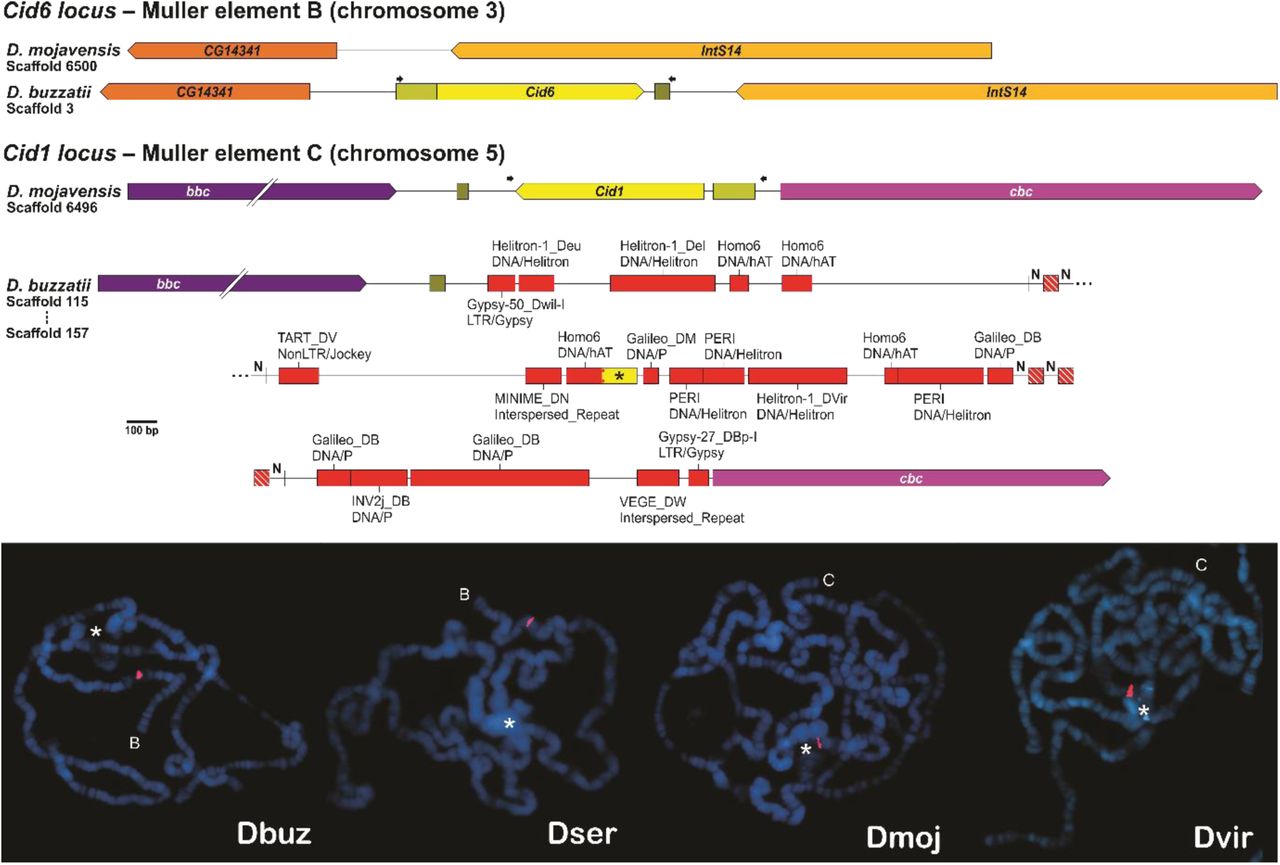
(Upper panel) Comparison between the Cid1 and Cid6 loci of D. buzzatii and the corresponding regions of D. mojavensis. The black asterisk indicates a fragment of Cid1, ‘N’ indicates unidentified nucleotides, red boxes indicate transposable elements, and arrows indicate primers used for the fluorescent in situ hybridization (FISH) experiments. (Lower panel) FISH on polytene chromosomes of D. buzzatii (Dbuz) and D. seriema (Dser) using Cid6 probes, and of the closely related D. mojavensis (Dmoj) and the outgroup D. virilis (Dvir) using Cid1 probes. The chromosome arm in which the Cid probe hybridized (red signal) is indicated by a letter representing the corresponding Muller element. The chromocenter, a region in which all centromeres bundle together, is indicated by a white asterisk. (Note: the chromocenter of D. buzzatii and D. mojavensis ruptured during the fixation step).
Why Cid6 remained while Cid1 degenerated? The Cid1 locus of D. buzzatii is located in the most proximal region of the Muller element C (scaffold 115; Guillén et al. 2014), which is very close to the pericentromeric heterochromatin where TEs are highly abundant (Pimpinelli et al. 1995; Casals et al. 2005; Rius et al. 2016). Natural selection is known to be less effective in pericentromeric and adjacent regions due to low rates of crossing-over (Zhang and Kishino 2004; Clément et al. 2006; Comeron et al. 2012; Nambiar and Smith 2016). Thus, it is reasonable to suggest that the presence of an extra copy of Cid1 (i.e., Cid6) in Muller element B alleviated the selective pressure on Cid1 in Muller element C, whose proximity to the pericentromeric heterochromatin fostered its degradation by several posterior TE insertions.
Cenp-C is duplicated in the Drosophila subgenus
It has been recently shown that the Drosophila CenH3 homolog Cid underwent duplication events during evolution (Kursel and Malik, 2017). Given that CenH3 and Cenp-C are interdependent and coevolve as a functional unit, we investigated if Cenp-C was also duplicated in Drosophila species where Cid was duplicated.
In D. eugracilis, in species from the montium subgroup, and in all the other species of the Sophophora subgenus we found only one copy of Cenp-C, which is always flanked by the 5-HT2B gene. On the other hand, in the species of the Drosophila subgenus we found two copies of Cenp-C with ∼52% nucleotide identity, which we named Cenp-C1 and Cenp-C2: the former is flanked by the 5-HT2B and CG1427 genes, and the latter is flanked by the CLS and RpL27 genes. A maximum likelihood tree showed that Cenp-C was likely duplicated after the split between the Sophophora and Drosophila subgenera but before the split between D. busckii and the other species of the Drosophila subgenus (fig. 2). Thus, we concluded that Cenp-C2 originated from a duplication of Cenp-C1 in the lineage that gave rise to species of the Drosophila subgenus, at least 50 mya (Russo et al. 2013).

Maximum likelihood tree of the Cenp-C1 and Cenp-C2 paralogs. Red and green branches respectively correspond to Cenp-C1 and Cenp-C2 sequences from species of the Drosophila subgenus. Bootstrap values are shown in each node. Scale bar represents number of substitutions per site.
Why Cenp-C is duplicated only in the Drosophila subgenus if Cid is also duplicated in D. eugracilis and in the montium subgroup? The fact that both Cid and Cenp-C duplicated in the Drosophila subgenus does not mean that there is a cause-and-effect relationship between the duplications. However, it probably means that the new paralogs influenced each other’s evolution. As a histone H3 variant, CenH3 has the C-terminal histone fold domain, which is reasonably conserved among species, and the N-terminal tail (NTT), which is highly variable among species (Henikoff et al. 2000). The NTT evolves in a modular manner, with four core motifs always conserved when there is only one Cid protein encoded in the genome (Kursel and Malik 2017). In D. eugracilis, the Cid2 paralog functionally replaced the pseudogenized ancestral Cid1 paralog. In species of the montium subgroup, these four motifs are alternated between the paralogs, which share ∼25% amino acid identity. In contrast, in species of the Drosophila subgenus, all four motifs are conserved in Cid1 but only 1-2 are conserved in Cid5, with the paralogs sharing only ∼15% amino acid identity at their NTT. Therefore, we propose that if the NTT of Cid interacts with Cenp-C, a new Cenp-C copy would allow a higher divergence of the Cid paralogs by alleviating the selective pressure over the Cid/Cenp-C interaction, thus explaining the higher divergence of the Cid1 and Cid5 paralogs. However, future studies focusing on the specific interactions between Cid and Cenp-C shall shed light on the exact basis behind the flexibility of these two proteins during evolution.
Some Cenp-C motifs are alternatively conserved between Cenp-C1 and Cenp-C2
Cenp-C was previously thought to be absent in Drosophila (Talbert et al. 2004), but it turned out that a protein that interacts with the regulatory subunits of separase is a highly divergent Cenp-C homolog (Heeger et al. 2005). The D. melanogaster Cenp-C1, as characterized by Heeger et al. (2005), has seven independent functional motifs, from N- to C-terminal: arginine-rich (R-rich), drosophilids Cenp-C homology (DH), AT hook 1 (AT1), nuclear localization signal (NLS), CenH3 binding (also known as the Cenp-C motif), AT hook 2 (AT2), and C-terminal dimerization (Cupin). The R-rich and DH motifs, as well as both AT1 and AT2 motifs (which may mediate binding to the minor grove of DNA), are functionally poorly characterized. However, all except AT1 appear to hold essential functions, as Cenp-C1 variants lacking these regions are unable to prevent phenotypic abnormalities in Cenp-C1 mutant embryos (Heeger et al. 2005). In fact, it is known that the DH motif must be involved in the recruitment of kinetochore proteins (Przewloka et al. 2011; Liu et al. 2016). Furthermore, arginine 1101 (R1101), present in the CenH3 binding motif, is crucial for centromere localization (Heeger et al. 2005). Given the functional relevance of these motifs, we searched for them in both Cenp-C1 and Cenp-C2.
With the exception of D. kikkawai (from the montium subgroup), in which the AT2 motif is absent, all seven motifs are conserved in Cenp-C1 from all other species of the Sophophora subgenus. In contrast, the motifs are alternatively conserved between Cenp-C1 and Cenp-C2 in species from the Drosophila subgenus (fig. 3). Both Cenp-C1 and Cenp-C2 of all species have the DH, NLS, and CenH3 binding motifs (with the corresponding R1101 of D. melanogaster), but lack the AT1 motif. Furthermore, only Cenp-C2 has the R-rich and AT2 motifs conserved. Both Cenp-C1 and Cenp-C2 of most species have the Cupin motif, the exceptions being Cenp-C1 of D. busckii, which lacks the final half of it, and Cenp-C2 of D. grimshawi, which entirely lacks it. Interestingly, the DH and NLS motifs of Cenp-C2 are more similar to those of Sophophora Cenp-C1 than to those of Drosophila Cenp-C1 (table 1). For the logo representation of the motifs, see Supplementary Figure S1.
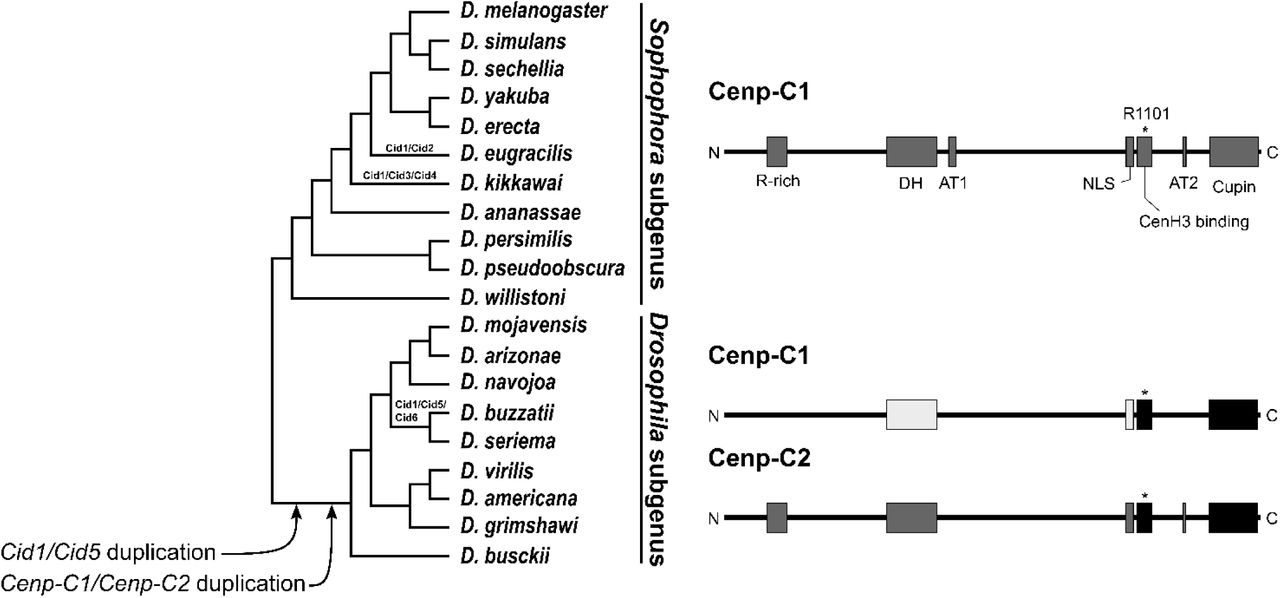
Both Cid and Cenp-C genes were duplicated in the lineage that gave rise to species of the Drosophila subgenus, as indicated in the species tree. Moreover, Cid1 was also duplicated in D. eugracilis, the montium subgroup (which includes D. kikkawai), and the buzzatii species cluster, the new paralogs of which are indicated at their respective branches. After the Cenp-C duplication, some functional motifs were alternatively conserved between the paralogs, as indicated at the right half of the image. High amino acids identity is indicated by the same color shade. Motifs are as follow: R-rich, arginine-rich; DH, drosophilid Cenp-C homology; AT1, AT hook 1; NLS, nuclear localization signal; CenH3 binding, also known as Cenp-C motif; AT2, AT hook 2; Cupin, a dimerization domain near the C-terminal region. The asterisk in the CenH3 binding motif indicates the corresponding R1101 of D. melanogaster, which is crucial for the centromere localization of Cenp-C1.
The conservation of the DH motif (involved in the recruitment of kinetochore proteins) and the NLS and CenH3 binding motifs (involved in centromere localization) in both Cenp-C1 and Cenp-C2 (fig. 3) indicates that it is unlikely that any of the paralogs underwent neofunctionalization. The (partial) loss of the Cupin motif in D. busckii and D. grimshawi points towards subfunctionalization. It is currently difficult to evaluate the loss of the AT1 motif in both Cenp-C1 and Cenp-C2, given that its function is unknown. However, the higher similarity of the DH and NLS motifs of Cenp-C2 to those of Sophophora Cenp-C1, the loss of the R-rich and AT2 motifs in Cenp-C1, and their retention in Cenp-C2 are highly indicative of subfunctionalization.
The Cenp-C paralogs are differentially expressed
Given that Cenp-C is incorporated onto centromeres concomitantly with Cid (Schuh et al. 2007) and that the excess of both proteins can cause centromere expansion and kinetochore failure (Schittenhelm et al. 2010), the expression of both proteins needs to be tightly regulated. Kursel and Malik (2017) showed that Cid5 expression is male germline-biased and proposed that Cid1 and Cid5 subfuncionalized and now performed nonredundant centromeric roles. In order to investigate if Cenp-C1 and Cenp-C2 are differentially expressed and correlated in some way with the expression of the Cid paralogs, we analyzed the available transcriptomes from embryos, larvae, pupae and adult females and males of D. buzzatii (Guillén et al. 2014), and from testes of D. virilis and D. americana (BioProject Accession PRJNA376405).
While Cid6 is transcribed in all stages of development in D. buzzatii, confirming that Cid6 functionally replaced Cid1, Cid5 transcription is limited to pupae and adult males, with a higher transcription than Cid6 in the latter (fig. 4A). Additionally, Cid5 transcription is elevated in testes of D. virilis and D. americana, whereas Cid1 is virtually silent (fig. 4C). Our results further support the finding of Kursel and Malik (2017) that Cid5 displays a male germline-biased expression. In this context, our finding that Cid5 is also transcribed in pupae of D. buzzatii may be related to the ongoing development of the male gonads.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cid and Cenp-C expression patterns in D. buzzatii (A and B) and D. virilis and D. americana (C and D). Data are presented as mean ± 95% confidence interval and analyzed by one-way ANOVA (A and B) and Student’s t-test (C and D): n.s., not significant; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. TPM, transcripts per million.
In contrast to the Cid paralogs, we found that both Cenp-C1 and Cenp-C2 are transcribed in almost all stages of D. buzzatii development, with the exception of larvae (fig. 4B). Cenp-C1 transcription is higher than that of Cenp-C2 in D. buzzatii embryos and adult females. On the other hand, transcription of Cenp-C2 is higher than that of Cenp-C1 in D. buzzatii pupae and adult males. Cenp-C2 transcription is also higher than that of Cenp-C1 in D. virilis testes, but there is no significant difference between their expression in D. americana testes (fig. 4D). Therefore, similarly to the findings for the Cid paralogs, the differential expression between the Cenp-C paralogs in testis supports the subfunctionalization hypothesis. The male germline-biased expression of both Cid5 and Cenp-C2 points towards their interaction in spermatogenesis, but biochemical assays need to be performed to confirm this possible interdependence.
The Cid and Cenp-C paralogs show signs of positive selection in species of the repleta group
The centromere drive hypothesis states that CenH3 and Cenp-C constantly evolve in an effort to suppress and diminish the associated deleterious effects of cenDNA selfish spread throughout the population by female meiotic drive (Henikoff et al. 2001; Dawe and Henikoff 2006). However, it has been proposed that the rapid evolution of CenH3 required for the “drive suppressor” function may be disadvantageous for canonical functions (e.g., mitosis; Finseth et al. 2015; Kursel and Malik 2017). The possibility that the paralogs achieved fitness optima for divergent functions predicts that selection may act differently in each of the Cid and Cenp-C paralogs. To test this hypothesis, we looked in our full-length alignments of the Cid and Cenp-C paralogs for signatures of positive selection using maximum likelihood methods. Given that CenH3 and Cenp-C are highly divergent, we focused our analyses on five closely related cactophilic Drosophila species from the repleta group (D. mojavensis, D. arizonae, D. navojoa, D. buzzatii and D. seriema).
We first used random-site and branch-site models to test for positive selection on particular sites during the evolution of the paralogs. The random-site models, which allow ω to vary among sites but not across lineages, revealed that both Cid5 and Cenp-C2 show extensive signs of positive selection (table 2). Particularly, Bayes Empirical Bayes analyses identified with a posterior probability > 95% four amino acids in the NTT of Cid5 and six amino acids across Cenp-C2 as having evolved under positive selection. Of the six Cenp-C2 amino acids, one is in the DH motif, one is in the Cupin motif, and the remaining four are in inter-motif sequences.
The branch-site models allow ω to vary both among sites and across branches on the tree and aim to detect positive selection affecting a few sites along particular lineages. The tests revealed that the paralogs show signs of positive selection in the branches of D. navojoa Cid1 and Cenp-C2, D. buzzatii Cenp-C1 and Cenp-C2, and D. seriema Cenp-C1 and Cenp-C2 (table 3). Particularly, Bayes Empirical Bayes analyses identified with posterior probability > 60% four amino acids in the NTT of D. navojoa Cid1, seven in inter-motif sequences of D. navojoa Cenp-C2, four in D. buzzatii Cenp-C1 (one in the DH motif and three in inter-motif sequences), six in inter-motif sequences of D. buzzatii Cenp-C2, four in D. seriema Cenp-C1 (two in the Cupin motif and two in inter-motif sequences), and six in inter-motif sequences of D. seriema Cenp-C2.
Finally, we used clade model C to test for divergent selection among a priori designated lineages. The test reveal evidence of divergent selection acting on Cid1, Cenp-C1 and Cenp-C2 across almost all the foreground branches, the exception being D. buzzatii (Table 4). It is clear that the majority of sites are under negative selection across all lineages, and a small proportion do show signatures of positive selection (data not show); however, there is no obvious pattern of divergent selection across the phylogeny. Unlike the sites-models, clade models freely estimate ω‘s for each a priori designated clade and permit sites under positive selection in null models, which could explain the discrepancy among the sites-models and the clade model. Overall, we interpret our data as providing strong support for adaptive evolution at several sites in both the Cid and Cenp-C paralogs.
Our tests revealed that both the Cid and Cenp-C paralogs show signs of positive selection to some extent. Random-site models revealed that, on average, Cid5 and Cenp-C2 show extensive signs of positive selection, which may indicate that these male germline-biased genes possess drive-suppression function. Kursel and Malik (2017) found signs of positive selection in the Cid3 paralog of the montium subgroup and proposed that Cid3 and Cid5 could be attenuating deleterious effects of centromere drive due to their male germline-biased expression. Our results of extensive positive selection on both Cid5 and Cenp-C2 do support this hypothesis. However, male germline-biased genes are widely known to evolve adaptively as the result of male-male or male-female competition (Ellegren and Parsch 2007; Meisel 2011). On the other hand, branch-site models revealed that different sites of both Cenp-C1 and Cenp-C2 show signs of positive selection in D. buzzatii and D.seriema, which may indicate that drive-suppression functions are not restricted to male-biased genes. Either way, molecular genetic data alone cannot reveal the underlying cause of adaptive evolution. What our findings do suggest is that species of the Drosophila subgenus likely have a specific inner kinetochore composition that mainly functions in spermatogenesis.
Concluding remarks
The extensive diversity of kinetochore compositions in eukaryotes poses numerous questions regarding the flexibility of essential cellular functions (van Hooff et al. 2017). Is the kinetochore less conserved than other core eukaryotic cellular systems? And if so, why so many core kinetochore proteins are so diverse? Are the variants adaptive to the species? To answer such questions, it is necessary to investigate how a specific kinetochore composition affects specific cellular features and lifestyles. Herein, we showed that Cid5 and Cenp-C2 offer such a possibility, as both are inner kinetochore protein variants likely specialized to function mainly in spermatogenesis. Thus, finding out if and how Cid5 and Cenp-C2 play a role either in centromere drive suppression or reproductive competition can shed a new light into our understanding of centromere evolution.
Materials and methods
Identification of Cid and Cenp-C orthologs and paralogs in sequenced genomes
For most Drosophila species, Cid and Cenp-C coding sequences were obtained from EST data. For Cenp-C1 of D. navojoa, D. mojavensis, D. buzzatii, D. seriema and D. americana, Cenp-C2 of D. buzzatii, D. seriema, D. americana and D. grimshawi, Cid5 of D. virilis, and both Cid5 and Cid6 of D. buzzatii and D. seriema, coding sequences were identified by tBLASTx in sequenced genomes. Since Cid is encoded by a single exon in Drosophila, we selected the entire open reading frame for each Cid gene hit, and since Cenp-C has multiple introns, we used the Augustus gene prediction algorithm (Stanke and Morgenstern 2005) to identify the coding DNA sequences. For annotated genomes, we recorded the 5’ and 3’ flanking genes for the Cid and Cenp-C genes of each species. For genomes that are not annotated, we used the 5’ and 3’ nucleotide sequences flanking the Cid and Cenp-C genes as queries to the D. melanogaster genome using BLASTn and verified the synteny in accordance to the hits. For the D. seriema genome assembly, see Supplementary File S1. All Cid and Cenp-C coding sequences and their database IDs can be found in Supplementary Files S2 and S3, respectively.
Fluorescent in situ hybridizations (FISH) on polytene chromosomes
Probes for Cid1 /Cid6 were obtained by PCR (see fig. 1A for primer site) from genomic DNA of D. buzzatii (strain st-1), D. seriema (strain D73C3B), D. mojavensis (strain 14021-0248.25) and D. virilis (strain 15010-1551.51). We cloned the PCR products into the pGEM-T vector (Promega) and sequenced them to confirm identity. Recombinant plasmids were labeled with digoxigenin 11-dUTP by nick translation (Roche Applied Science). FISH on polytene chromosomes was performed as described in Dias et al. (2015). The slides were analyzed under an Axio Imager A2 epifluorescence microscope equipped with the AxioCam MRm camera (Zeiss). Images were captured with the AxioVision software (Zeiss) and edited in Adobe Photoshop. Chromosome arms were identified by their morphology (Kuhn et al. 1996; González et al. 2005; Schaeffer et al. 2008).
Phylogenetic analyses
Cid and Cenp-C sequences were aligned at the codon level using MUSCLE (Edgar 2004) and refined manually. Subsequently, we generated maximum likelihood phylogenetic trees in MEGA6 (Tamura et al. 2013) with the GTR substitution model and 1,000 bootstrap replicates for statistical support.
Expression analyses
RNA-seq data from D. buzzatii (Guillén et al. 2014), and from D. virilis and D. americana (BioProject Accession PRJNA376405) were analyzed for the Cid and Cenp-C expression patterns with Bowtie2 (Langmead and Salzberg 2012), as implemented to the Galaxy server (Afgan et al. 2016). Mapped reads were normalized by the transcripts per million (TPM) method (Wagner et al. 2012), and all normalized values < 1 were set to 1 so that log2 TPM ≥ 0.
Positive selection analyses
Cid and Cenp-C alignments and gene trees were used as input into the CodeML NSsites models of PAMLX version 1.3.1 (Xu and Yang 2013). Random-site and branch-site models were used to test for positive selection on particular sites during the evolution of the Cid and Cenp-C paralogs. Random-site models allow ω to vary among sites but not across lineages; for this analysis, we compared three models that do not allow ω to exceed 1 (M1a, M7 and M8a) to two models that allow ω > 1 (M2a and M8). Branch-site Model A was compared with Model Anull to examine whether particular sites evolved under positive selection along a priori specified branches (called foreground branches). Foreground branches were as follow: #1 (D. arizonae, D. mojavensis); #2 (D. navojoa); #3 ((D. arizonae, D. mojavensis), D. navojoa); #4 (D. buzzatii); #5 (D. seriema); #6 (D. buzzatii, D. seriema). Positively selected sites were classified as those with a Bayes Empirical Bayes posterior probability > 90%. Clade model C (CmC) tests for divergent selection on particular sites among a priori designated lineages. The modified null model of CmC (M2a_rel) assumes that sites fall into three classes: purifying selection (0 < ω < 1); neutral evolution (ω = 1); or positive selection (ω >1). In CmC, the third site class allows the estimated ω for a site to diverge across foreground branches. Foreground branches were as follow: #1 ((D. arizonae, D. mojavensis), D. navojoa); #2 (D. buzzatii, D. seriema).
Acknowledgments
We are grateful to Dr. Maura Helena Manfrin (Univesity of São Paulo) for providing us the D. seriema strain. This work was supported by a grant from “Fundação de Amparo à Pesquisa do Estado de Minas Gerais” (FAPEMIG) to G.K. (grant number APQ-01563-14).
References




