

ABSTRACT
When marked for degradation, surface receptor and transporter proteins are internalized and delivered to endosomes where they are packaged into intralumenal vesicles (ILVs). Many rounds of ILV formation create multivesicular bodies (MVBs) that fuse with lysosomes exposing ILVs to hydrolases for catabolism. Despite being critical for protein degradation, the molecular underpinnings of MVB-lysosome fusion remain unclear, although machinery underlying other lysosome fusion events is implicated. But how then is specificity conferred? And how is MVB maturation and fusion coordinated for efficient protein degradation? To address these questions, we developed a cell-free MVB-lysosome fusion assay using S. cerevisiae as a model. After confirming that the Rab7 ortholog Ypt7 and the multisubunit tethering complex HOPS are required, we found that the Qa-SNARE Pep12 distinguishes this event from homotypic lysosome fusion. Mutations that impair MVB maturation block fusion by preventing Ypt7 activation, confirming that a Rab-cascade mechanism harmonizes MVB maturation with lysosome fusion.
IMPACT STATEMENT Endocytosis culminates with multivesicular bodies (MVBs) fusing with lysosomes. But the molecular underpinnings of this event remain unclear. Here, using S. cerevisiae as a model, Karim et al. employ a new in vitro assay to show that MVB-lysosome fusion is driven by ESCRT-dependent Rab-GTPase activation and the syntaxin ortholog Pep12, distinguishing it from other lysosome membrane fusion events.
INTRODUCTION
Endocytosis is critical for regulating surface expression levels of polytopic proteins such as transporters and receptors for cellular signaling and survival in all eukaryotic organisms. Proteins destined for degradation are labeled with ubiquitin and cleared from the surface by invagination of the plasma membrane to form early endosomes. Through membrane fusion events, early endosomes deliver cargo proteins to an endosomal compartment where they encounter the ESCRT (Endosomal Sorting Complexes Required for Transport) machinery that sorts them into intraluminal vesicles (ILVs). Many rounds of ILV formation produce a multivesiclular body (MVB; Huotari and Helenius, 2011; Schmidt and Teis, 2012), that when mature, fuses with the lysosome to expose the ILVs to lumenal hydrolases for catabolism (Piper and Katzmann, 2007; Luzio et al., 2010). Although critical for surface protein degradation, we still do not understand many aspects of this terminal step of the endocytic pathway in molecular detail.
However, based on results primarily acquired using genetic approaches and microscopy, a model has emerged describing the mechanisms underlying MVB-lysosome fusion that are thought to be conserved in all eukaryotic species (reviewed by Luzio et al., 2010; Balderhaar and Ungermann, 2013). MVBs contain two paralogs of almost every component of the fusion machinery (Kümmel and Ungermann, 2014). One set (the Rab5 module) is required for endosome membrane fusion events responsible for anterograde membrane trafficking to the MVB, for surface protein delivery and organelle biogenesis. The second set (Rab7 module) is proposed to mediate MVB-lysosome fusion. However, most components of the latter set also drive homotypic lysosome fusion (Wickner, 2010; Wickner and Rizo, 2017), provoking the question: What distinguishes these two fusion events?
Because the underlying machinery is similar, inferences into the molecular basis of MVB-lysosome fusion have been drawn from detailed knowledge of the homotypic lysosome membrane fusion reaction, largely elucidated by studying Saccharomyces and its vacuolar lysosome (or vacuole), as models. Advances in this area were primarily driven by the use of reliable, quantitative cell-free membrane fusion assays to study homotypic vacuole fusion (Haas, 1995; Jun and Wickner, 2007). This powerful biochemical approach has revealed four distinct subreactions required for organelle membrane fusion and the critical players for each that presumably mediate MVB-lysosome fusion as well:
Priming, the first subreaction, requires Sec18, an NSF (N-ethylmalemide-sensitive factor) ortholog and homohexameric ATPase, and Sec17, an α-SNAP (soluble NSF attachment protein) ortholog and protein chaperone, to unravel cis-SNARE (SNAP receptor) complexes from previous fusion events to free up individual SNARE proteins for future rounds of membrane fusion (Mayer et al., 1996; Ungermann et al., 1998). As the only α-SNAP and NSF orthologs in S. cerevisiae Sec17 and Sec18 are thought to mediate all SNARE-mediated membrane fusion events in the cell, but their roles in MVB-lysosome fusion have not been investigated. Afterwards (or possibility concomitantly), organellar membranes undergo tethering, the second subreaction, whereby apposing membranes make first contact. This requires activation of the Rab-GTPase Ypt7 (a Rab7 ortholog) and interaction with its cognate multisubunit tethering complex (MTC) called HOPS (homotypic fusion and vacuole protein sorting complex; Eitzen et al., 2000; Seals et al., 2000).
Next, fusogenic proteins (e.g. Sec17, Ypt7, HOPS and SNAREs) and lipids are recruited to the initial contact site where they assemble into an expanding ring at the vertex between adjacent organelles (Wang et al., 2002; Wang et al., 2003; Fratti et al., 2004). Called docking, this subreaction also includes the formation of trans-SNARE protein complexes mediated by HOPS (Collins and Wickner, 2007; Starai et al., 2008). Like homotypic lysosome fusion, Ypt7 and HOPS accumulate at contact sites between MVB and lysosome membranes in S. cerevisiae (Cabrera et al., 2009). Furthermore, depleting components of HOPS impairs delivery of endocytic cargoes to lysosomes in yeast and human cells (Peterson and Emr, 2001; Wartosh et al., 2015). Thus both Ypt7 and HOPS are implicated in MVB-lysosome fusion where they presumably contribute to tethering and docking (Bugnicourt et al., 2004; Balderhaar et al., 2013; Pols et al., 2013; Numrich and Ungermann, 2014). During the last fusion subreaction, trans-SNARE complexes containing 3 Q-SNAREs (Qa, Qb and Qc donated from one membrane) and one R-SNARE (from the apposing membrane) fully zipper driving lipid bilayer merger (Schwartz and Merz, 2009). Two of four SNAREs within the complex that mediates homotypic lysosome fusion are also found on MVB membranes: the Qb-SNARE Vti1 and soluble Qc-SNARE Vam7 (von Mollard et al., 1997; Gossing et al., 2013). Otherwise, the MVB expresses its own paralogs of the Qa-SNARE, Pep12 in place of Vam3 on the vacuolar lysosome, and the R-SNARE Snc2 in place of Nyv1 (Gerrard et al., 2000; Gurunathan et al., 2000). The SNAREs donated by each organelle to the complex that drives MVB-lysosome fusion are unknown. Thus, it is possible that the MVB donates either Q-SNAREs including Pep12 or the R-SNARE Snc2 to SNARE complexes distinguishing MVB-lysosome from homotypic lysosome fusion.
Recognizing the impact of these cell-free assays have had on our understanding of homotypic lysosome fusion, we developed a similar method to reliably measure MVB-lysosome membrane fusion in S. cerevisiae. Here, we use it to test prevailing model describing the mechanisms underlying this heterotypic fusion event and, more importantly, to uncover mechanisms that differ from homotypic lysosome fusion. One intriguing and unique feature of MVB-lysosome fusion is the need for MVB maturation prior to fusion for efficient surface protein degradation. But how does the mature MVB know when to fuse? Deleting components of ESCRTs block ILV formation, preventing delivery of internalized proteins to lysosomes and causing endomembrane accumulation. Although implied, there is no empirical evidence supporting the hypothesis that knocking out ESCRTs block this fusion event. Furthermore, how ESCRTs may target the underlying machinery is not entirely understood. Thus, here we use our new fusion assay to better understand how MVB maturation is coupled to lysosome fusion to ensure efficient surface protein degradation.
RESULTS
A new cell-free assay to measure MVB-vacuole membrane fusion
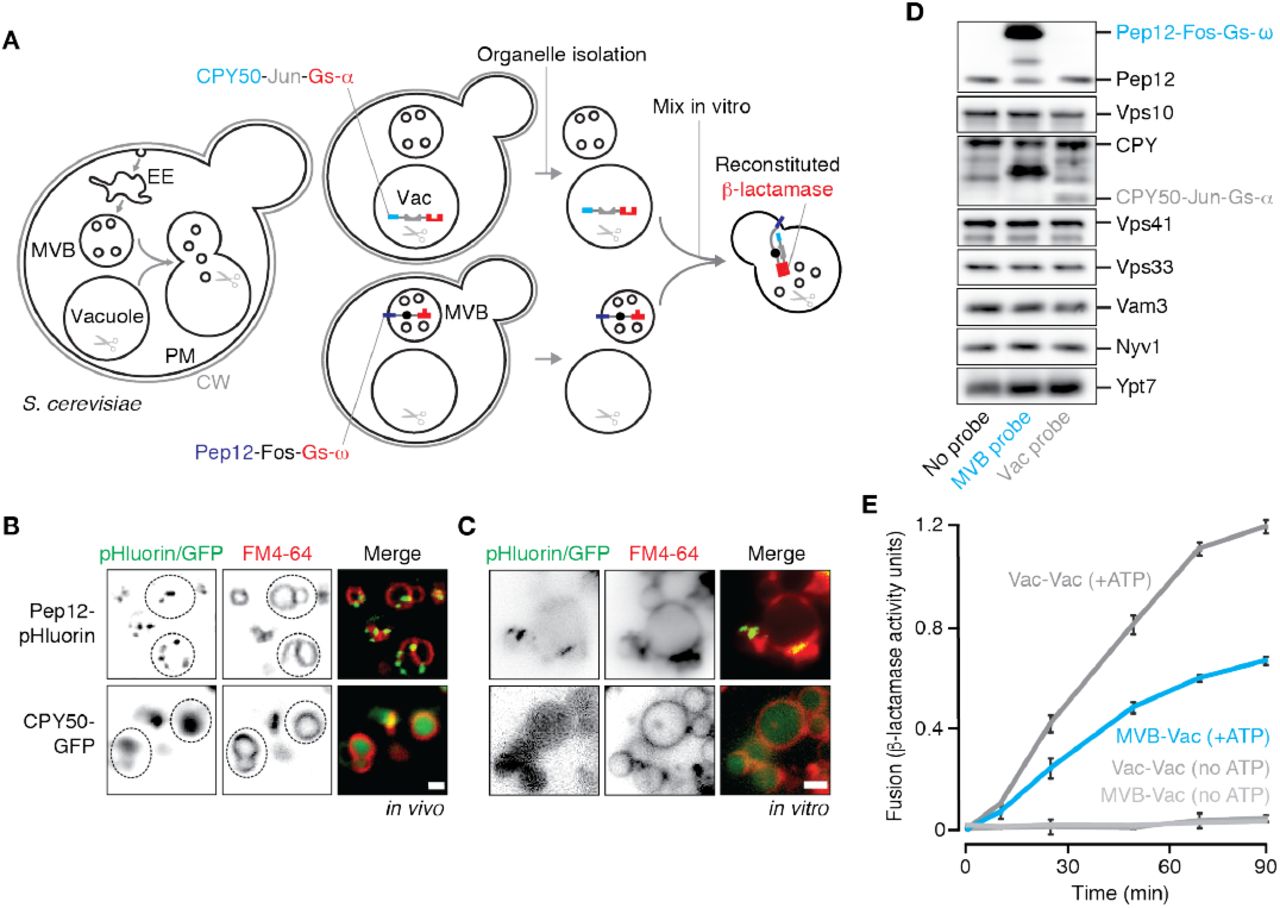
We designed our new cell-free MVB-vacuole membrane fusion assay based on a strategy originally devised by Jun and Wickner (2007) to study homotypic vacuole fusion that relies on the reconstitution of β-lactamase upon luminal content mixing (Figure 1A): To target the first fusion probe to the MVB lumen, we fused the C-terminus of the endosomal Qa-SNARE Pep12 to the proto-oncogene product c-Fos followed by the ω-subunit of β-lactamase (Pep12-Fos-Gs-ω). To target our second fusion probe the vacuole lumen, we fused the targeting sequence of lysosomal protease carboxypeptidase Y (CPY; first 50 amino acids) to Jun, a cognate binding partner of c-Fos, followed by the α-subunit of β-lactamase (CPY50-Jun-Gs-α). We then expressed each probe in separate yeast strains (deficient of vacuolar proteases to prevent probe degradation) to prevent probe interaction in vivo. After isolated organelles from each strain are combined and undergo lipid bilayer merger in vitro, lumenal contents mix allowing Jun and c-Fos to interact driving the complementary halves of β-lactamase together to reconstitute enzyme activity. Reconstituted β-lactamase activity is then measured by monitoring nitrocefin hydrolysis (by recording absorbance at 492 nm over time) to quantify MVB-lysosome fusion.
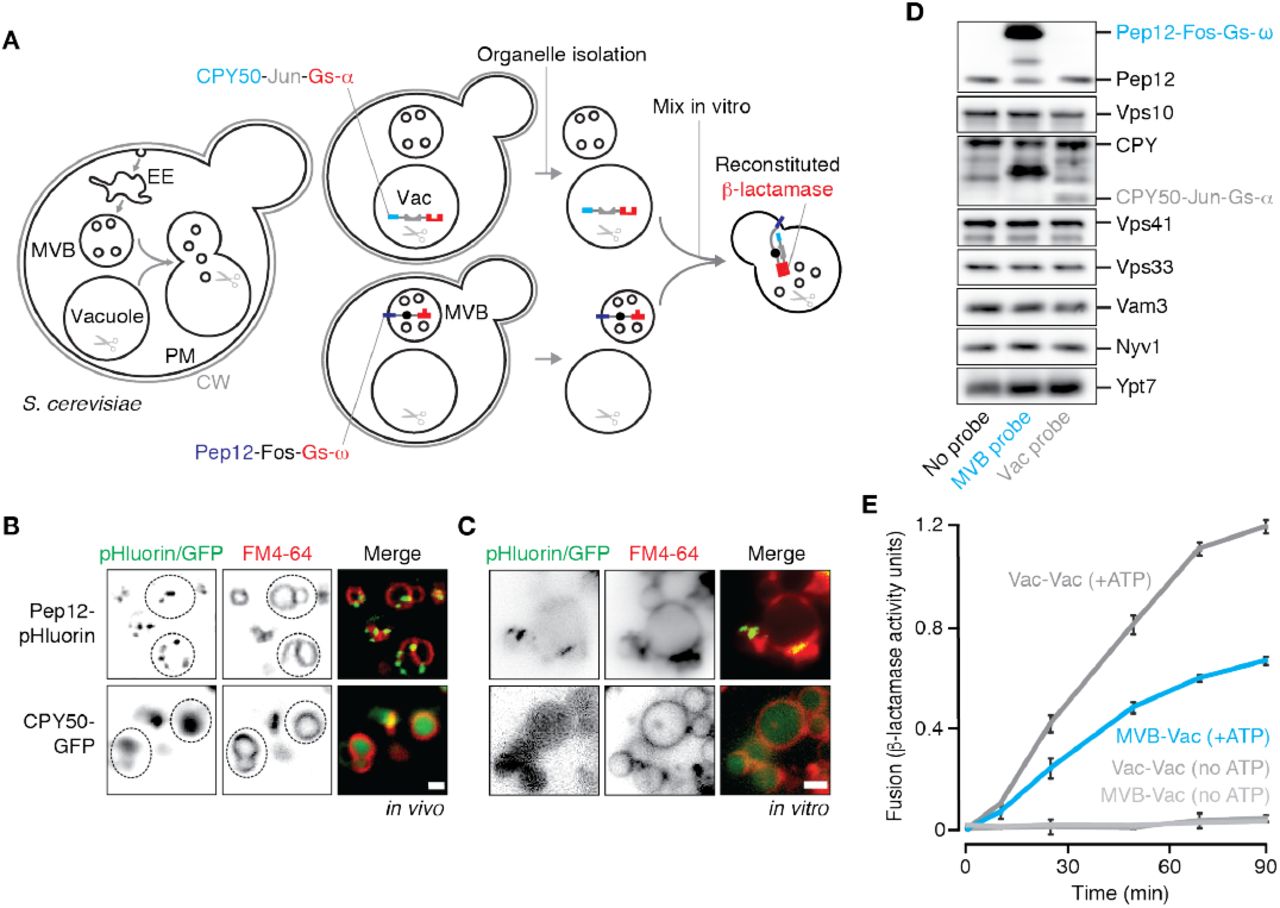
(A) Cartoon illustrating new cell-free assay to quantify MVB-vacuole fusion. CW, cell wall; PM, plasma membrane; EE, early endosome; Vac, vacuole. Fluorescence micrographs of (B) Live cells or (C) isolated organelles expressing Pep12-pHluorin or CPY50-GFP. Vacuole membranes are stained with FM4-64. Dotted lines outline yeast cells as observed by DIC. Scale bars, 2 μm. (D) Western blots to confirm presence of fusion probes and fusogenic proteins in organelles isolated by ficoll gradient. (E) Content mixing values obtained over time by mixing organelles isolated from separate strains expressing complimentary fusion probes targeted to MVBs or vacuoles, or probes targeted to vacuoles. Means ± S.E.M shown (n ≥ 3).
To confirm that the fusion probes were properly localized within cells, we generated yeast strains expressing versions of the probes tagged with GFP at their C-termini, stained them with FM4-64 to label vacuole membranes and imaged them using fluorescence microscopy (Figure 1B). The Pep12-GFP fusion protein appeared on puncta consistent with MVB localization as reported previously (Becherer et al., 1996), whereas the CPY50-GFP fusion protein was found within the vacuole lumen. Next, we fractionated cellular membranes by sucrose gradient (Figure S1A) and used western blot analysis to confirm that Pep12-Fos-Gs-ω is found in similar fractions (9-12) as Vps10, a resident protein on MVBs (Marcusson et al., 1994), as well as endogenous Pep12. CPY50-Jun-Gs-α is not found in fractions containing the MVB fusion probe, but rather in fractions (3-4) containing Nyv1, a R-SNARE that is exclusively found on vacuole membranes (Wen et al., 2006), and endogenous CPY a vacuole resident protein. We next collected fractions containing MVB membranes (9-12) from Pep12-Fos-Gs-ω expressing cells or vacuole membranes (3-4) from CPY50-Jun-Gs-α expressing cells and mixed them together under conditions that promote the homotypic vacuole membrane fusion reaction in vitro (see Jun and Wickner, 2007). Unfortunately these preparations were fusion incompetent (Figure S1B). Thus, we sought an alternative method to isolate the organelles to optimize our cell-free fusion assay.
Based on a method to isolate intact, fusogenic vacuoles from yeast cells (see Conradt et al., 1992), we decided to use a 4-step ficoll gradient to isolate both MVBs and vacuoles in the same preparations. Although vacuoles are known to accumulate at the 0-4 % ficoll interface, it is not clear whether MVBs also migrate to this layer during centrifugation. Thus, we performed ficoll-based fractionation experiments using cells expressing GFP-tagged versions of the fusion probes, collected organelles that accumulated at the 0-4% ficoll interface, and imaged them (Figure 1C). Pep12-GFP is present in the preparation and localizes to small puncta adjacent to vacuole membranes reminiscent of MVB structures, whereas CPY50-GFP is found within the lumen of isolated vacuoles, consistent with their distributions in living cells. We also observed the presence of our fusion probes (Pep12-Fos-Gs-ω or CPY50-Jun-Gs-α) in this fraction by western blot analysis (Figure 1D). Markers of both MVBs (Vps10, endogenous Pep12) and vacuoles (Nyv1, endogenous CPY), and fusogenic proteins (e.g. Ypt7, Vps41, Vps33 and Nyv1) were also present confirming that both organelles are found in this preparation and are likely fusogenic. We next mixed equal proportions of organelles isolated from cells expressing only the MVB fusion probe or only the vacuole fusion probe in fusion buffer that mimics cytosolic conditions (125 mM KCl, 5 mM MgCl2, 200 mM sorbitol, 20 mM PIPES, pH 6.80). We added 1 mM ATP to trigger membrane fusion and incubated reactions at 27°C for up to 90 minutes. Upon measuring reconstituted β-lactamase activity (Figure 1E), we observed robust nitrocefin hydrolysis indicative of heterotypic fusion, showing a signal up to 10 fold greater than background (i.e. reactions run without ATP or left on ice). Rates of heterotypic fusion were comparable to those observed for homotypic vacuole fusion, an additional indication that this new assay may be reliably used to study the heterotypic fusion reaction in detail.
Sec17, Ypt7 and HOPS drive MVB-vacuole fusion in vitro
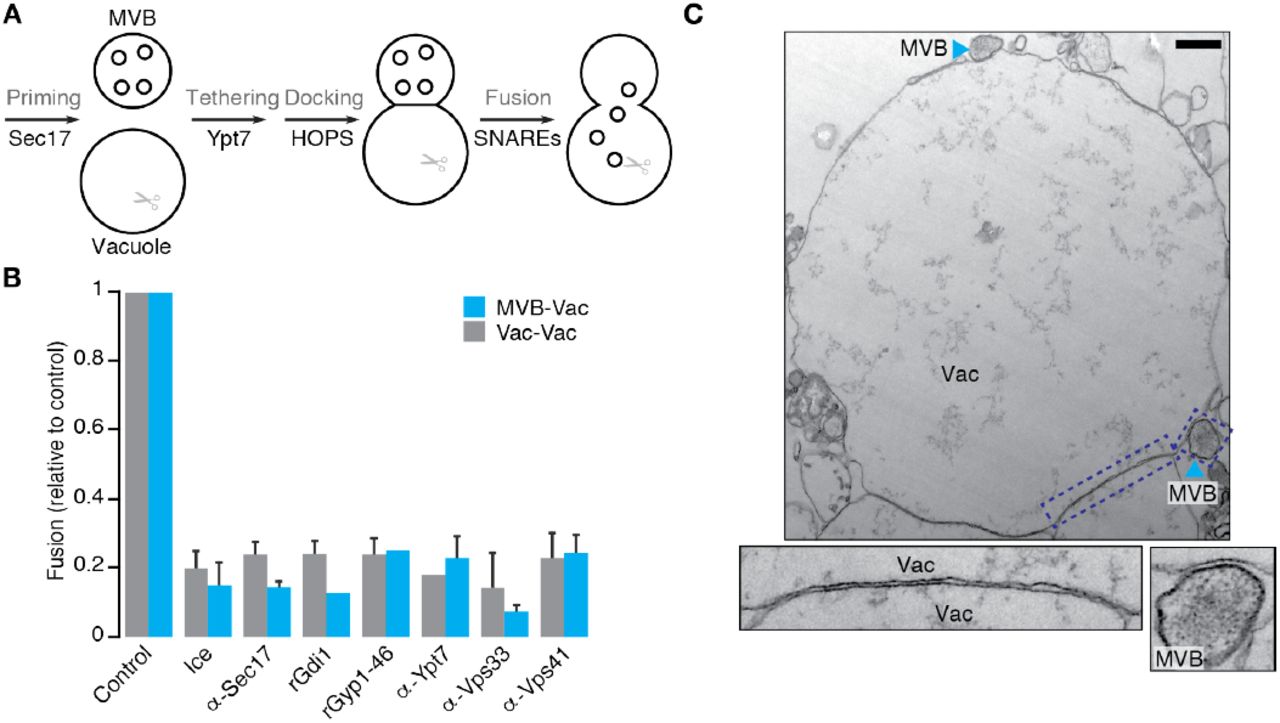
With this new assay in hand, we next tested the prevailing model of MVB-vacuole fusion (Figure 2A), which predicts that Sec17, Ypt7 and HOPS orchestrate this event (Balderhaar et al., 2010, 2013). The α-SNAP ortholog Sec17 both unravels cis-SNARE complexes and, through HOPS binding, promotes SNARE-zippering during homotypic fusion (Schwartz and Merz, 2009; Wickner and Rizo, 2017). As the only α-SNAP ortholog in S. cerevisiae, it is hypothesized to also MVB-vacuole fusion. Indeed, adding purified anti-Sec17 antibody, at concentrations that block homotypic fusion, also prevents heterotypic fusion (Figure2B), suggesting a role in both events. We next confirmed that Ypt7 and HOPS (the Rab7 module in yeast) also underlie both fusion events (Figure 2B): Addition of the Rab-GTPase inhibitors rGdi1 (a Rab-GTPase chaperone that extracts Rab proteins from membranes) or rGyp1-46 (the active domain of the Rab-GAP protein Gyp1; see Brett and Merz, 2008) blocks both fusion events, confirming that Rab-GTPase activation is required. Addition of purified anti-Ypt7 antibody, at levels that block homotypic fusion, inhibited heterotypic fusion, confirming that activation of this specific Rab is needed for both fusion events. The HOPS holocomplex contains six protein subunits, two of which are unique (Vps41 and Vps39) and four (Vps33, Vps11, Vps18 and Vps16) that are shared with CORVET (class C core vacuole/endosome tethering complex), a similar multisubunit tethering complex within the Rab5 module also found on endosomes (Peplowska et al., 2007; Nickerson et al., 2009; Plemel et al., 2012). Addition of purified anti-Vps33 antibody blocks both fusion events (Figure 2B), suggesting involvement of either complex. Purified anti-Vps41 antibody also blocked heterotypic and homotypic fusion (Figure 2B), confirming that HOPS, specifically, contributes MVB-vacuole fusion.

(A) Cartoon illustrating the predominant model of MVB-vacuole fusion. Proteins or protein complexes implicated in subreactions are indicated. (B) MVB-vacuole (MVB-Vac) or homotypic vacuole (Vac-Vac) fusion measured in the presence or absence of affinity-purified antibodies against Sec17 (1.8 μM), Ypt7 (1.8 μM), Vps33 (1.8 μM) or Vps41 (1.8 μM), or purified recombinant Gdi1 (4 μM) or Gyp1-46 (5 μM) proteins. Means ± S.E.M. shown (n ≥ 3). (C) Transmission electron micrograph of a fusion reaction containing MVBs and vacuolar lysosomes imaged at 30 minutes in the presence of ATP. Panels below show high magnification images of boxed regions illustrating docking sites between apposing organelles. Vac, vacuole. Scale bar, 500 nm.
Ypt7 and HOPS drive the docking subreaction of homotypic vacuole fusion whereby fusogenic proteins and lipids assemble into a ring at the vertex between closely apposed membranes (Wang et al., 2002; Wang et al., 2003; Fratti et al., 2004). This unique arrangement causes the portion of membrane within the vertex ring, called the boundary, to form an extended, flat interface between apposing membranes that can be visualized by transmission electron microscopy (Mattie et al., 2017). Given that same Rab7 module mediates heterotypic fusion, we hypothesized that similar boundaries may form during the docking stage of MVB-vacuole fusion. To test this hypothesis, we visualized in vitro heterotypic fusion reactions by transmission electron microscopy (Figure 2C). We observed structures reminiscent of MVBs based on size and morphology (e.g. round organelles, 500 nm diameter, containing numerous circular ILVs of 20-80 nm diameter; Friend, 1969; Russell et al., 2012), confirming their presence in our organelle preparation. Many MVBs were located immediately adjacent to large, single-membrane-encased organelles containing lipophilic materials characteristic of vacuoles (Mattie et al., 2017). At these sites, adjacent membranes form extended, flat interfaces separated by approximately 8 nm, similar to those formed between docked vacuoles. Thus, Ypt7 and HOPS may also drive arrangement of the fusion machinery into a ring during MVB-vacuole docking, consistent with the prevailing model describing this fusion event.
Trans-SNARE complexes distinguish MVB-vacuole from homotypic vacuole fusion events
The final stage of the organelle fusion reaction is lipid bilayer merger driven by complete trans-SNARE-complex zippering (Nichols et al., 1997). For homotypic vacuole fusion, trans-SNARE complexes are minimally composed of the three Q-SNAREs – the syntaxin ortholog Vam3 (Qa), Vti1 (Qb) and the SNAP25 ortholog Vam7 (Qc) – and the R-SNARE Nyv1, a synaptobrevin ortholog (Wickner, 2010; Wickner and Rizo, 2017). Vam7, a soluble protein, complexes with Vam3 and Vti1 on one membrane forming a Q-SNARE bundle that binds to Nyv1 on the apposing membrane (Schwartz and Merz, 2009). Of these 4 SNAREs, only Vti1 and Vam7 are expressed on MVBs. Otherwise MVBs express the Qa-SNARE paralog Pep12 and R-SNARE Snc2 (Becherer et al., 1996; von Mollard et al., 1997; Gerrard et al., 2000; Gurunathan et al., 2000) suggesting that trans-SNARE complexes mediating heterotypic and homotypic fusion are different, whereby the MVB donates either Pep12 or Snc2 (but not both). Elegant work by Joji Mima’s group shows that synthetic proteoliposomes containing Pep12, in place of the vacuolar Qa-SNARE Vam3, are capable of forming stable, fusogenic complexes with the other SNAREs responsible for homotypic vacuole fusion (Furukawa and Mima, 2014). Thus, we hypothesized that a trans-SNARE complex composed of Pep12, Vti1 and Vam7 donated from MVBs with Nyv1 donated by vacuoles may drive heterotypic fusion events.
To test this hypothesis, we isolated SNARE complexes using an approach that ensures they form in trans during the membrane fusion reaction in vitro (see Collins and Wickner, 2007; Schwartz and Merz, 2009): We mixed organelles missing NVY1 and expressing Pep12 fused to calmodulin-binding protein (CBP::Pep12) with organelles missing PEP12 under fusogenic conditions, and then conducted pull-down assays and western blot analysis to identify SNARE proteins in complex with CBP::Pep12 (Figure 3A). We also repeated the experiment with CBP::Vam3 for comparison and as a positive control (Schwartz and Merz, 2009). As expected, both trans-SNARE complexes contained Vti1 and Vam7, but only under fusogenic conditions (with ATP; Figure 3B). Very little Vma3 co-purified with CBP::Pep12, and no Pep12 was found in CBP::Vam3 complexes, confirming the presence of distinct Q-SNARE bundles. Importantly, the vacuolar R-SNARE Nyv1 was found in both complexes, but not the endosomal R-SNARE Snc2, confirming that Pep12-Vti1-Vam7-Nyv1 complexes form in trans during organelle fusion in vitro.

(A) Cartoon illustrating method used to isolate trans-SNARE complexes from organelle fusion reactions in vitro. CBP-Vam3 was used in place of CBP-Pep12 to isolate SNARE complexes known to mediate homotypic vacuole fusion. (B) Western blots used to identify fusogenic proteins bound to CBP-Pep12 (left) or CBP-Vam3 (right) based on pull-down experiments illustrated in A. (C) MVB-vacuole (MVB-Vac) or homotypic vacuole (Vac-Vac) fusion values measured in the presence of increasing concentrations of purified anti-Pep12 (left) or anti-Vam3 (right) antibody. (D) Heterotypic (MVB-Vac) or homotypic (Vac-Vac) fusion values measured using organelles isolated from nyv1Δ cells or from wild type cells in the presence of 2.2 μM purified anti-Nyv1 antibody. Fusion of organelles from wild type cells was also measured in the presence of 100 nM rVam7 to stimulate fusion in place of ATP, with or without pretreatment with 1.8 μM anti-Sec17 to encourage SNARE complex formation (Thorngren et al., 2004). Reactions were kept on ice to prevent fusion as negative control. Means ± S.E.M. shown (n ≥ 2).
Addition of purified, recombinant Vam7 protein (rVam7, the soluble Qc-SNARE) to isolated vacuoles drives formation vacuolar trans-SNARE complexes promoting homotypic fusion without ATP (Thorngren et al., 2004). Given that Vam7 is found in both trans-SNARE complexes, we next tested if rVam7 could also stimulate assembly of Pep12-containg complexes. As expected, rVam7 drives formation of both trans-SNARE complexes (Figure 3B), suggesting that it contributes to heterotypic as well as homotypic fusion. Vps33, a component of HOPS and SM-protein ortholog, binds SNAREs and initiates trans-SNARE complex formation during homotypic vacuole fusion by threading the Qa-SNARE Vam3 with the R-SNARE Nyv1 (Baker et al., 2015). Recombinant Vps33 protein was also shown to thread Pep12 and Nyv1 protein in vitro (Lobingier et al., 2012). Thus, we also tested whether Vps33 binds to trans-SNARE complexes thought to mediate MVB-vacuole fusion. Indeed, Vps33 bound to Pep12-Nyv1-containing as well as Vam3-Nyv1-contianing complexes (Figure 3B), suggesting that HOPS initiates formation of Pep12-Vti1-Vam7-Nyv1 complexes in trans to drive MVB-vacuole fusion.
To demonstrate that this distinct trans-SNARE complex drives MVB-vacuole fusion, we next tested the role of these SNAREs in this process using our new in vitro assay. First, we added different purified antibodies raised against the Qa-SNAREs Pep12 or Vam3 to organelle fusion reactions (Figure 3C). As predicted, increasing concentrations of anti-Pep12 antibody preferentially blocked heterotypic fusion, whereas anti-Vam3 antibody preferentially inhibited homotypic fusion. Addition of purified anti-Nyv1 antibody or knocking out NYV1 blocked both fusion events, consistent with the presence of this R-SNARE in both complexes (Figures 3D and S2). To confirm that trans-SNARE complexes formed by the Qc-SNARE Vam7 mediate both MVB-vacuole and homotypic vacuole fusion, we next added rVam7 to isolated organelles in the absence of ATP (Figure 3D). As for homotypic fusion (Thorngren et al., 2004), we found that addition of rVam7 is sufficient to stimulate MVB-lysosome fusion in vitro. Furthermore, addition of anti-Sec17 antibody to prevent unraveling of newly formed SNARE complexes (Schwatrz and Merz, 2009) enhanced this effect, confirming that it contributes to heterotypic as well as homotypic fusion. Together, these results demonstrate that the Pep12-Vti1-Vam7-Nyv1 trans-SNARE complex drives MVB-vacuole fusion, revealing that the composition of SNARE complexes distinguishes heterotypic from homotypic fusion events.
ESCRTs activate Ypt7 for MVB-lysosome fusion
The ESCRT machinery mediates sorting and packaging of internalized surface proteins into intralumenal vesicles creating the MVB (Henne et al., 2011). Deleting components of ESCRTs prevents delivery of internalized surface proteins to vacuoles and causes them to accumulate on aberrant, enlarged endocytic compartments devoid of intralumenal vesicles (Raymond et al., 1992; Robinson et al., 1998; Russel et al., 2012). These observations led to the hypothesis that ILV formation by ESCRTs is necessary to trigger MVB-lysosome fusion (Metcalf and Issacs, 2010). To directly test this hypothesis, we individually knocked out single components of three ESCRTs (VPS23 in ESCRT-I, VPS36 in ESCRT-II and SNF7 in ESCRT-III; Henne et al., 2011) in strains harboring the fusion probe expressed in the MVB. After confirming the probes localized to aberrant endocytic compartments within these mutant cells (Figure 4A; see Coonrod and Stevens, 2010; Russell et al., 2012), we mixed organelles isolated from these strains with organelles from wild type cells expressing the vacuolar fusion probe and conducted in vitro fusion assays (Figure 4B). This approach ensured that vacuoles were fusogenic, as deleting components of ESCRTs blocks the CPY and CPS (carboxypeptidase-S) biosynthetic pathways that supply vacuoles with fusion proteins (Katzmann et al., 2001) – noting that this also prevented us from assessing effects of these mutants on homotypic vacuole fusion in vitro. As predicted, deleting any of the three ESCRT components inhibited MVB-vacuole fusion, indicating that blocking any stage of ILV formation impairs heterotypic fusion.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Fluorescence micrographs of live wild type (WT), vps36Δ, snf7Δ, vps23Δ or vps23Δ Ypt7-Q68L cells expressing Pep12-pHluorin. Vacuoles membranes were stained with FM4-64. Scale bar, 2 μm. (B) MVB-vacuole fusion values from reactions containing organelles isolated from WT, vps23Δ, vps36Δ or snf7Δ cells conducted in the absence or presence of ATP, 0.2 mM GTPγS, or 100 nM rVam7. (C) MVB-vacuole fusion values from reactions containing organelles isolated from WT, vps23Δ or vps23Δ Ypt7-Q68L cells in the absence or presence of ATP or 0.2 mM GTPγS. (D) Cartoon illustrating new model of MVB-vacuole fusion. Means ± S.E.M. shown (n ≥ 3).
How do ESCRTs stimulate MVB-vacuole fusion? Rab-GTPase conversion coordinates successive fusion events in the secretory and endocytic pathways for efficient delivery of cargo proteins (Rivera-Molina and Novick, 2009). At the MVB, it has been proposed that inactivation of the Rab5 ortholog Vps21 (responsible for endosomal fusion events that deliver proteins to the MVB) is necessary for activation of Ypt7 to initiate the next trafficking step: MVB-lysosome fusion. Impairing ILV formation causes active Vps21 to accumulate on aberrant enlarged endosomes (Russell et al., 2012). Thus, it was hypothesized that deleting ESCRTs impairs Rab-conversion, whereby Ypt7 cannot be activated to trigger MVB-vacuole fusion. To test this hypothesis, we took three approaches: First, we added GTPγS, a non-hydrolyzible GTP analog, to activate all GTPases (including Ypt7) and found that it rescued heterotypic fusion defects caused by deleting components of ESCRTs (Figure 4B). Next, we added rVam7 in place of ATP to stimulate fusion, as it bypasses the requirement for Ypt7 (Thorngren et al., 2004), and found that it also rescued fusion defects (Figure 4B). These results also confirm that Ypt7 and SNAREs are functional, but not engaged, on aberrant endosomal compartments isolated from ESCRT mutants. Finally, to demonstrate that Ypt7 is targeted, we introduced a Q68L mutation in Ypt7 to render it constitutively active (see Brett et al., 2008) in cells lacking VPS23. After confirming the fusion probe was properly localized, we found that it too rescued MVB-lysosome fusion defects caused by this mutation (Figures 4A and 4C). Furthermore, addition of GTPγS did not further enhance heterotypic fusion in the presence of Ypt7 Q68L, suggesting that Ypt7 was the likely the target of GTPγS that rescues fusion when ESCRT function is impaired. In all, these findings demonstrate that the ESCRT machinery activates Ytp7 to trigger MVB-vacuole fusion.
DISCUSSION
A reliable cell-free assay to study MVB-lysosome fusion
Here we describe a new cell-free assay to measure MVB-vacuole membrane fusion based on reconstitution of lumenal β-lactamase (Figure 1). The method of organelle isolation was crucial for success, and although MVBs are not separated from vacuoles in these preparations, targeting the fusion probes to each organelle in separate yeast strains and mixing them later ensures that reconstituted β-lactamase activity represents only fusion that occurs in vitro under controlled conditions. Importantly, this new assay offers a reliable method to study the MVB-vacuole membrane fusion reaction in detail, by providing many experimental advantages:
First, it is quantitative, robust (with a signal-to-noise ratio up to 10:1) and allows kinetic analysis. Second, it is a simple, colorimetric alternative to fluorescence-based assays limited by resolution and accuracy of colocalization methods offered by light microscopy (offering only 3:1 signal-to-noise; Cao et al., 2015) or assays that require use of radioactive reagents, e.g. 125I-labeled biotin (offering only 4:1 signal-to-noise; Pryor et al., 2004). Third, it overcomes limitations associated with using genetic approaches to study this event, whereby mutations hypothesized to block MVB-vacuole fusion also impair carboxypeptidase-Y and -S biosynthetic pathways that feeds vacuoles resident proteins, possibly introducing defects that indirectly affect vacuole fusogenicity in vivo (see Robinson et al., 1988; Raymond et al., 1992). This is achieved by mixing organelles isolated from mutants with those from wild type cells; an approach that may also be used to define contributing mechanisms from each organelle. (4) It overcomes the need to purify organelles to homogenity, as required to make inferences from methods that do not selectively label a single organelle population, e.g. lipid dyes (R-18) for fluorescence dequenching-based lipid-mixing fusion assays (e.g. Jun and Wickner, 2007), or internalized protein reagents fluorescence dyes that label multiple endocytic compartments (e.g. Morvan et al., 2009). Fifth, unlike most cell-free endosome or MVB fusion assays (e.g. Vida and Gerhardt, 1999), it does not require addition of cytosol indicating that all machinery necessary for fusion co-purifies with organelles facilitating their study. Finally, it offers the advantages of changing the reaction buffer conditions to simulate changes in the “cytoplasm” to better understand how heterotypic fusion is regulated or using protein reagents to overcome issues associated with plieotropy, allowing study of contributions from gene products predicted to have multiple functions at multiple site in cells, e.g. Sec17.
As an alternative to MVB-vacuole fusion, MVBs may be engulfed by the vacuole membrane and internalized by a closure event mediated by the shared fusion machinery, akin to autophagy. Within the lumen, the MVB perimeter membrane would be catabolized by acid hydrolases facilitating content mixing and interaction of the fusion probes. However, three observations exclude the possibility that MVBs are internalized by this autophagic mechanism: (1) No intermediates or products of autophagy were observed in the fusion reactions by TEM or fluorescence microscopy (e.g. an invagination of the vacuole membrane containing an MVB, an intact MVB encased by a double lipid bilayer within the vacuole lumen; also see Mattie et al., 2017). Nor did we observe MVBs trapped between apposing membranes at large contact sites between adjacent vacuolar lysosomes, an area that internalized into the lumen upon fusion at the vertex ring (Wang et al., 2002; McNally et al., 2017). (2) Polytopic proteins that exclusively reside within MVB perimeter membranes appear on membranes of large vacuoles over time during the fusion reaction in vitro. Furthermore, degradation of these proteins is not observed, excluding the possibility that the MVB perimeter membrane is exposed to lumenal hydrolases (e.g. GFP-tagged Nhx1; M.A. Karim and C.L. Brett, unpublished results). (3) We find that the MVB contributes at least one SNARE, Pep12, to trans-SNARE complexes that drive membrane fusion, suggesting that the underlying machinery is distinct from homotypic vacuole fusion. Therefore, it is unlikely to mediate closure of the vacuole membrane, which would be analogous to a homotypic vacuole fusion event. Thus, consistent with previous reports (e.g. Futter et al., 1996; Bright et al., 2005), we are confident that observed reconstituted β-lactamase activity reflects genuine merger between MVB perimeter and vacuole membranes.
The Qa-SNARE Pep12 is implicated in earlier endosome fusion events required for MVB maturation (Kümmel and Ungermann, 2014). Thus, to better understand this process, we also attempted to measure homotypic endosome fusion in vitro by swapping the targeting sequence in the vacuolar fusion probe re-routing it to the endosome (i.e. CPY50 was exchanged with Pep12 rendering Pep12-Jun-Gs-α). However, we did not detect reconstituted β-lactamase activity over background levels (data not shown). We suspect that this is due to our method of organelle isolation: From electron and fluorescence micrographs, endosomes and MVBs seem less abundant than vacuoles. When also considering that vacuoles are much larger than MVBs, we suspect that endosomes within the in vitro fusion reaction were more likely to encounter a vacuole than another endosome, decreasing possible endosome-endosome fusion events to a level below detectability by this assay. Future experiments will involve further purification of endosomes to overcome this technical issue. With this in mind, the same approach could be used to study other organelle membrane fusion events by swapping the targeting sequence in the fusion probe(s) and purifying organelles using a method that does not damage membranes (e.g. isolation by floatation, not sedimentation) and ensures fusogenicity (e.g. low osmolarity, not by sucrose gradient; see Figure S1).
A new model describing MVB-lysosome fusion
Here we definitively show that Sec17, Ypt7 and the HOPS complex are necessary for membrane fusion between wild type, intact MVBs and vacuoles (Figure 2B). These proteins also contribute to other fusion events in cells (Balderhaar et al., 2013; Kümmel and Ungermann, 2014). Thus, deleting the genes encoding them induces plietropic phenotypes, including blockade of biosynthetic pathways to the vacuole (Robinson et al., 1988; Peterson and Emr, 2001). This work therefore lends important biochemical-based evidence to support the prevailing model describing the molecular underpinnings of MVB-vacuole fusion (Figures 2A and 4D), which was almost exclusively backed by evidence generated using genetic approaches (Kümmel and Ungermann, 2014). Moreover, we make two advances in our understanding of this process:
A unique trans-SNARE complex distinguishes MVB-vacuole fusion from other vacuole fusion events. It differs from the complex that drives homotypic vacuole fusion whereby the Qa-SNARE Pep12 is donated from the MVB, in place of the vacuolar paralog Vam3, to form a Pep12-Vti1-Vam7-Nyv1 complex in trans (Figure 3B). As preassembled Q-SNARE bundles are donated from one membrane and R-SNAREs from the other (Schwartz and Merz, 2009), we propose that the MVB donates the Q-SNARE bundle Pep12-Vti1-Vam7 and the vacuole donates the R-SNARE Nyv1. We also demonstrate that these SNAREs are necessary or sufficient for MVB-vacuole fusion using three approaches (Figure 3C and D): knocking out NYV1 blocks fusion, adding purified antibodies raised against Nyv1 or Pep12 blocks fusion, or adding purified, recombinant Vam7 protein drives fusion. Importantly, we also measure homotypic vacuole fusion as a control (Figures 1 – 3) to demonstrate that reagents targeting shared machinery are effective at concentrations used, and we also exclude the possibility that the inverse trans-SNARE complex arrangement contributes to this process, i.e. a complex containing the Qa-SNARE Vam3 (in complex with Vti1 and Vam7) from the vacuole and R-SNARE Snc2 donated by the MVB (Figure 3). These findings are consistent with numerous previous reports:
First, when Pep12-Vti1-Vam7-containing synthetic proteoliposomes are mixed with proteoliposomes expressing Nyv1, these SNAREs form stable protein complexes in trans that support lipid bilayer fusion (Furukawa and Mima, 2014). Second, Vam3 is thought to contribute to the ALP (alkaline phosphatase) biosynthetic pathway, which sends proteins directly from the Golgi to the vacuole bypassing the endosome or MVB (Cowles et al., 1997). Pep12, on the other hand, is implicated in the CPY biosynthetic pathway, which sends proteins from the Golgi to endosomes or MVBs en route to the vacuole (Becherer et al., 1996; Gerrard et al., 2000), consistent with this Qa-SNARE, and not Vam3, mediating the terminal step of this pathway: MVB-vacuole fusion. Third, overexpression of VAM3 suppresses membrane trafficking defects in pep12Δ yeast cells (Götte and Gallwitz, 1997), suggesting that Pep12 and Vam3 can be exchanged within SNARE complexes as we show in Figure 3B. Forth, consistent with our findings, endosomal Q-SNAREs and a lysosomal R-SNARE are proposed to mediate endosome-lysosome fusion in mammalian cells (Pryor et al., 2004), suggesting this mechanism is evolutionarily conserved. Finally, Vps33 is known to function at the endosome and vacuole (Subramanian, 2004) and studies conducted with purified proteins demonstrate that the SM-protein Vps33 binds to Vam3 or Pep12 with equal affinity to promote templating with Nyv1 (Lobingier and Merz, 2012; Baker et al., 2015). Here we show that Vps33 binds SNARE complexes containing Pep12-Nyv1 or Vam3-Nyv1 (Figure 3B) and MVB-vacuole fusion requires Vps33 and Pep12 but not Vam3 (Figures 2B and 3C). Thus, we conclude that Vps33 within the HOPS complex promotes Pep12-Vti1-Vam7-Nyv1 complex formation in trans to drive MVB-lysosome fusion (Figure 4D).
The second advance is our discovery that fusogenic proteins and lipids may assemble into a ring at the interface between docked MVB and vacuole membranes. During homotypic vacuole fusion, Ypt7, HOPS and SNAREs assemble into a ring at the vertex between apposed membranes, creating a flat circular boundary area at the site of contact between organelle membranes (Wang et al., 2003; Mattie et al., 2017). We observed similar flattened interfaces between MVBs and vacuoles by TEM (Figure 2C). Given that most components of the underlying fusion machinery are shared (Sec17, Ypt7, HOPS and 3 of 4 SNAREs), we reason that they also drive formation of this same topological arrangement to mediate MVB-vacuole fusion. Importantly, during homotypic vacuole fusion, lipid bilayer merger occurs at the vertex ring causing the entrapped boundary membrane to be internalized as an intralumenal fragment (Wang et al., 2002; Mattie et al., 2017; Karim et al., submitted). Vacuolar polytopic proteins are selectively sorted into the boundary and, upon membrane fusion, are internalized and degraded by lumenal hydrolases (McNally et al., 2017). This process, called the ILF (IntraLumenal Fragment) pathway requires Ypt7 and HOPS for protein sorting. Thus, it is tempting to speculate an ILF is also formed during MVB-vacuole fusion. If so, this could explain the presence of large ILVs within MVBs in cells lacking the ESCRT machinery (Stuffers et al., 2009). It would also allow organelles to selectively remove damaged or superfluous polytopic proteins for quality control or to remodel the protein landscape of the fusion product membrane for organelle identity and function.
ESCRTs trigger MVB-lysosome fusion by promoting Rab conversion
Here, we show that individually deleting VPS23, VPS326 or SNF7––components of ESCRT-I, -II and –III, respectively––prevents MVB-vacuole fusion in vitro (Figure 4B). Activating Ypt7 by introducing a Q68L mutation or adding GTPγS to reactions rescued fusion defects caused by these mutations (Figure 4B and C). Although they perform unique roles in the pathway, deleting any of these complexes prevents ILV formation. We therefore reason that ILV formation itself triggers fusion with lysosomes (Russell et al., 2012). But how does this occur?
Although the precise molecular mechanism remains unknown, previous work revealed that active Vps21 and its GEF (Guanine Exchange Factor) Vps9 accumulate on aberrant endosome membranes in cells lacking ESCRT function (Russell et al., 2012). Although Ypt7 is present, its effector Vps41 (and presumably the HOPS complex) is absent from these compartments. These findings led to the hypothesis that a Rab conversion mechanism may couple MVB maturation with vacuole fusion, whereby Vps21 inactivation triggers activation of Ypt7 to promote anterograde trafficking. Vps9 is recruited to endosomal membranes by binding ubiquitin linked to internalized surface proteins (Shideler et al., 2015). Normally these proteins are cleared from the perimeter membrane by ESCRTs, causing dissociation of Vps9 from the perimeter membrane and, in turn, Vps21 is inactivated linking MVB maturation to Rab conversion. However, ESCRT impairment causes ubiquitylated proteins to accumulate, Vps9 then remains and chronic activation of Vps21 prevent Rab conversion thus blocking MVB-vacuole fusion (Russell et al., 2012; Shideler et al., 2015). Although the mechanism that couples Vps21 and Ypt7 activities remains elusive, it likely involves stimulation of Msb3, a GAP that inactivates Vps21 (Lachmann et al., 2012), Mon1-Ccz1, the GEF that activates Ypt7 (Nordmann et al., 2010), and possibly BLOC-1 (biogenesis of lysosome-like organelles complex-1) to coordinate GAP and GEF function on mature MVB membranes (Rana et al., 2015). We will test this hypothesis in the near future using this new cell-free MVB-vacuole fusion assay to provide a comprehensive understanding of this process and to improve our knowledge of Rab-conversion, which is thought to coordinate progressive membrane trafficking at most sites within eukaryotic cells.
As with the fusion machinery, ESCRTs and potential mediators of Vps21-Ypt7 conversion are evolutionarily conserved, suggesting the same mechanism likely underlies MVB-lysosome fusion in metazoan cells (Saksena and Emr, 2009; Dennis et al., 2016). Loss-of-function mutations in ESCRT components are linked to many human disorders, including cancers and neurodegenerative diseases (Saksena and Emr, 2009). Similar to phenotypes observed in S. cerevisiae, enlarged, abnormal endosome-like structures accumulate and surface protein degradation is impaired within human cells harboring ESCRT mutations, suggesting MVB-lysosome fusion is defective. Here we show that stimulating Ypt7, the yeast ortholog of human Rab7, overcomes this fusion defect. Although these compartments lack ILVs, this fusion event delivers internalized surface proteins to the vacuolar lysosome membrane where they are selectively degraded by the ILF pathway (McNally and Brett, submitted). As it is proposed that inefficient surface receptor degradation underlies the etiology of these diseases, we hypothesize that developing therapeutics designed to activate Rab7 may represent a valid strategy for treatment.
MATERIALS AND METHODS
Yeast strains and reagents
We expressed all fusion probes in the Saccharomyces cerevisiae strain BJ3505 [MATα pep4::HIS3 prbΔ1-1.6R his3–Δ200 lys2–801trp1-Δ101 (gal3) ura3–52 gal2 can1] because it is devoid of vacuolar lumenal proteases that would otherwise degrade these proteins. To ensure high levels of protein expression, BJ3505 cells were transformed with two plasmids each containing a copy of a single fusion probe: For MVB-vacuole fusion, pRS406-Pep12-Fos-Gs-ω (pCB002) and pRS404-Pep12-Fos-Gs-ω (pCB003) to target the MVB, or pRS406-CPY50-Jun-Gs-α (pYJ406-Jun) and pRS404-CPY50-Jun-Gs-α (pCB011) to target the vacuole. For homotypic vacuolar lysosome fusion, the yeast strain expressing the MVB probe was replaced with one transformed with pRS406-CPY50-Fos-Gs-ω (pYJ406-Fos) and pRS404-CPY50-Fos-Gs-ω (pCB008) to target the vacuole instead. NYV1, VPS23, VPS36, and SNF7 were knocked out of BJ3505 cells using the Longtine method (Longtine et al., 1998). To assess localization of fusion probes, we transformed wild type or mutant BJ3505 cells with pRS406-Pep12-pHluorin (pCB046) to target the MVB lumen or pRS406-CPY50-GFP (pCB044) to target the vacuole lumen and imaged live cells by fluorescence microscopy. To evaluate trans-SNARE complexes, BJ3505 cells lacking NYV1 were transformed with pRS406-CBP-Pep12 (pCB045) to generate BJ3505-CBP-Pep12 nyv1Δ. BJ3505-CBP-Vam3 nyv1Δ was a gift from A.J. Merz (see Collins and Wickner, 2007).
All biochemical and yeast growth reagents were purchased from Sigma-Aldrich, Invitrogen or BioShop Canada Inc. All restriction enzymes, Ni-sepharose 6FF, and glutathione sepharose 4B, polymerases, and ligases were purchased from New England Biolabs (Ipswich, MA, USA). Purified rabbit polyclonal antibody against Sec17 was a gift from William Wickner (Dartmouth College, Hanover, NH, USA), whereas those raised against Vam3, Vam7, Ypt7, Vps21, Vps33, Vps41, Vps10 and CPY were gifts from Alexey Merz (University of Washington, Seattle, WA, USA) and another raised against Snc2 was a gift from Jeffrey Gerst (Weizmann Institute of Science, Rehovot, Israel). Recombinant mouse antibody against Pep12 was purchased from Abcam (Toronto, ON, Canada). Proteins used include recombinant Gdi1 purified from bacterial cells using a calmodulin-binding peptide intein fusion system (Brett et al., 2008), recombinant Gyp1-46 (the catalytic domain of the Rab-GTPase activating protein Gyp1) purified as previously described (Eitzen et al., 2000), and recombinant soluble Qc-SNARE Vam7 purified as previously described (Schwartz and Merz, 2009). Reagents used in fusion reactions were prepared in 10 mM Pipes-KOH, pH 6.8, and 200 mM sorbitol (Pipes-sorbitol buffer, PS).
Membrane fractionation by sucrose gradient
Yeast cells were grown in YPD overnight to OD600nm= 1.6/ml, harvested, and spheroplasted with lyticase for 30 minutes at 30°C. Spheroplasts were then sedimented and resuspended in 10 ml of TEA buffer (10 mM triethanolamine, pH 7.5, 100 mg/ml phenylmethylsulfonyl fluoride, 10 mM NaF, 10 mM NaN3, 1 mM EDTA, and 0.8 M sorbitol) and homogenized on ice by Dounce homogenization (20 strokes). Cell lysates were centrifuged at 15,000 g for 20 minutes, and the resulting supernatant was then centrifuged at 100,000 g for 2 hours to sediment cellular membranes. Pellets were resuspended in 1 ml of TEA buffer and loaded onto a stepwise (20–70%) sucrose density gradient, and then centrifuged at 100,000 g for 16 hours at 4°C to separate different cellular membranes by density. Samples were collected from the top, and each fraction was precipitated using 10% trichloroacetic acid, washed and resuspended in 100 μl of SDS-PAGE buffer. Fractions were then loaded into SDS-polyacrylamide gels and subjected to electrophoresis to separate proteins by size. Western blot analysis was performed to determine fractions that contain proteins of interest.
Organelle isolation and cell-free fusion assay
Organelles were isolated from yeast cells by a ficoll floatation method as previously described (Haas, 1995). Organelle membrane fusion was assessed using a modified version of a published assay that relies on split β-lactamase complementation upon lumenal content mixing (Jun and Wickner, 2007). In brief, organelles were isolated from two separate strains that each express a single complementary fusion probe, and 6 μg of organelles from each strain were added to 60 μl fusion reactions in standard fusion buffer supplemented with 10 μM recombinant GST-Fos protein to reduce background caused by lysis. Reactions were incubated up to 90 minutes at 27°C and then stopped by placing them on ice. Content mixing was quantified by measuring the rate of nitrocefin hydrolysis by reconstituted β-lactamase. 58 μl of the fusion reactions were transferred into a clear-bottom 96-well plate and mixed with 142 μl of nitrocefin developing buffer (100 mM NaPi pH 7.0, 150 μM nitrocefin, 0.2 % Triton X-100). To measure nitrocefin hydrolysis, absorbance at 492 nm was monitored at 15 seconds intervals for 15 minutes at 30°C with a Synergy H1 multimode plate reading spectrophotometer (Biotek, Winooski, VT, USA). Slopes were calculated, and one fusion unit is defined as 1 nmol of hydrolyzed nitrocefin per minute from 12 μg of organelle proteins. Standard fusion buffer contains 125 mM KCl, 5 mM MgCl2, ATP regenerating system (1 mM ATP, 40 mM creatine phosphate, 0.5 mg/ml creatine kinase), and 10 μM CoA in PS buffer. To block fusion, either antibodies raised against Sec17, Ypt7, Vps33, Vps41, Vam3, Nyv1 or Pep12, or purified recombinant Gdi1 or Gyp1-46 proteins were added to fusion reactions. Where indicated, vacuoles were pretreated with 0.2 mM GTPγS for 10 minutes at 27°C prior to addition to fusion reactions, or reactions were supplemented with 100 nM rVam7 and 10 μg/ml bovine serum albumin.
Trans-SNARE paring assay
Analysis of trans-SNARE complex formation was assayed as described (Schwartz and Merz, 2007), whereby the levels of Nyv1 that co-immunoprecipitate with CBP-Pep12 or CBP-Vam3 was determined. Briefly, 45 μg of organelles each from CBY224 (BJ3505-CBP-Pep12 nyv1Δ) or BJ3505-CBP-Vam3 nyv1Δ and BJ3505 were incubated under fusion conditions for 60 minutes. Reactions were then placed on ice for 5 minutes, and samples were centrifuged (11,000 g, 15 minutes, 4°C). Supernatants were decanted, and membrane fractions were resuspended in 560 μl solubilization buffer (20 mM Tris-Cl pH 7.5, 150 mM NaCl, 1 mM MgCl2 0.5% Nonidet P-40 alternative, 10% glycerol) with protease inhibitors (0.46 μg/ml leupeptin, 3.5 μg/ml pepstatin, 2.4 μg/ml pefabloc, 1 mM PMSF), and nutated for 20 minutes at 4°C. Reactions were centrifuged again (16,000 g, 20 minutes, 4°C) and supernatants collected. 10 % of the extracts were collected as input samples, and the remaining extracts were brought to 2 mM CaCl2. The CBP-Pep12-Nyv1 or CBP-Vam3-Nyv1 complexes were recovered with calmodulin sepharose beads 4B (GE Healthcare) by nutating overnight at 4°C, collected by brief centrifugation (4,000 g, 2 minutes, 4°C), washed five times with sample buffer, and followed by bead sedimentation. Bound proteins were eluted by boiling beads (95°C, 10 minutes) in SDS sample buffer containing 5 mM EGTA for SDS-PAGE analysis and immunoblotting.
Western blot analysis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed using a Bio-Rad mini protein system (Bio-Rad Laboratories, Hercules, CA, USA). After separation, proteins were transferred onto a nitrocellulose membrane by wet transfer method at 12 V for 8 hours using a Royal Genie Blotter apparatus (Idea Scientific, Minneapolis, MN, USA). Membranes were blocked with 3% BSA in PBST buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, 0.1% Tween-20) and then washed twice with PBST and incubated with primary antibody diluted to 1:1,000 in PBST for 1 hour at room temperature. Membranes were washed with PBST five times, and then incubated with HRP or FITC labeled goat anti-rabbit or anti-mouse IgG diluted 1:10,000 in PBST for 45 minutes at room temperature. After an additional 5 washes with PBST, the membranes were probed to detect bound secondary antibody using GE Amersham Imager 600 for chemiluminescence or a Typhoon scanner for fluorescence (GE Healthcare, Piscataway, NJ, USA).
Fluorescence microscopy
Live yeast cells stained with FM4-64 to label vacuole membranes were prepared for imaging using a pulse-chase method as previously described (Brett et al., 2008). For in vitro imaging, organelles isolated from strains expressing Pep12-pHluorin or CPY50-GFP were stained with 3 μM FM4-64 for 10 minutes at 27°C to label vacuole membranes. We used a Nikon Eclipse TiE inverted microscope equipped with a motorized laser TIRF illumination unit, Photometrics Evolve 512 EM-CCD camera, an ApoTIRF 1.49 NA 100x objective lens, and bright (50 mW) blue and green solid-state lasers operated with Nikon Elements software (housed in the Centre for Microscopy and Cellular Imaging at Concordia University).
Transmission Electron Microscopy
Isolated organelles were processed for transmission electron microscopy (TEM) as previously described (Mattie et al., 2017). Briefly, fusion reactions were incubated at 27°C for 30 minutes, vacuoles were gently pelleted (5,000 g for 5 minutes) at 4°C and immediately fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) overnight at 4°C. Vacuole pellets were washed with 0.1 M sodium cacodylate (3 times, 10 minutes) and then fixed with 1% osmium tetroxide for two hours at 4°C. Pellets were washed with water (3 times, 5 minutes) followed by gradual dehydration in ethanol (30-100%) and 100% propylene oxide. Pellets were infiltrated with epon:propylene oxide for 1 hour and then embedded in pure epon by polymerization (48 hours at 57°C). Samples were cut into 100 nm thick sections using an ultra diamond knife and Reichert Ultracut II microtome, loaded onto 200-mesh copper grids, and stained with uranyl acetate (8 minutes) and Reynold’s lead (5 minutes). Sections were imaged at 120 kV using an FEI Tecnai 12 electron microscope outfitted with a Gatan Bioscan digital camera (1k × 1k pixels) housed in the Facility for Electron Microscopy Research at McGill University (Montreal, QC, Canada). Images were obtained from at least three separate organelle fusion reactions.
Data analysis and presentation
All quantitative data were processed using Microsoft Excel v.14.0.2 software (Microsoft Cooperation, Redmond, WA, USA), including calculation of mean and S.E.M.. Data were plotted using Kaleida Graph v.4.0 software (Synergy Software, Reading, PA, USA). Micrographs were processed using ImageJ software (National Institutes of Health, Bethesda, MD, USA) and Adobe Photoshop CC (Adobe Systems, San Jose, CA, USA). Images shown were adjusted for brightness and contrast, inverted and sharpened with an unsharp masking filter. All figures were prepared using Adobe Illustrator CC software (Adobe Systems, San Jose, CA, USA).
AUTHOR CONTRIBUTIONS
C.L.B and M.A.K. conceived the project. S.M. acquired the transmission electron micrograph shown in Figure 2C. M.A.K. performed all other experiments and prepared all data for publication. M.A.K. and C.L.B. wrote the paper.
ACKNOWLEDGEMENTS
We thank J.E. Gerst, A.J. Merz and W.T. Wickner for custom antibodies, yeast strains and plasmids, as well as C. van Oostende-Triplet for technical assistance with microscopy. The Canadian Foundation for Innovation and Natural Sciences and Engineering Research Council of Canada (NSERC) provided generous support for the Centre for Microscopy and Cell Imaging at Concordia University. This research was funded by grant #RGPIN/403537-2011 to C.L.B from the NSERC Discovery program.
Footnotes
↵3 Lead Contact
LIST OF ABBREVIATIONS
- ALP
- Alkaline Phosphatase
- BLOC
- Biogenesis of Lysosome-like Organelles Complex-1
- CPS
- CarboxyPeptidase-S
- CPY
- CarboxyPeptidase-Y
- ESCRT
- Endosomal Sorting Complexes Required for Transport
- GAP
- Gtpase Activating Protein
- GEF
- Guanine nucleotide Exchange Factor
- HOPS
- HOmotypic fusion and vacuole Protein Sorting
- ILF
- IntraLumenal Fragment
- ILV
- IntraLumenal Vesicle
- MTC
- Multisubunit Tethering Complex
- MVB
- MultiVesicular Body
- NSF
- N-ethylmalemide-Sensitive Factor
- SNAP
- Soluble Nsf Attachment Protein
- SNARE
- SNAp REceptor
- TEM
- Transmission Electron Microscopy
- VPS
- Vacuole Protein Sorting.
REFERENCES




