

Abstract
High-throughput methods for screening protein-protein interactions (PPIs) enable the rapid characterization of engineered binding proteins and interaction networks. While existing methods are powerful, none allow quantitative library-on-library characterization of PPIs in a modifiable extracellular environment. Here, we show that sexual agglutination of S. cerevisiae can be reprogrammed to link PPI strength with mating efficiency using yeast synthetic agglutination (YSA). Validation of YSA with 96 previously characterized interactions shows a strong log-linear relationship between mating efficiency and PPI strength for interactions with KD’s ranging from 500 pM to 25 μM. Using induced chromosomal translocation to pair barcodes representing interacting proteins, thousands of distinct interactions can be screened in a single pot. YSA binding interactions occur in a controllable extracellular environment, and thus studying the effects of environmental factors on PPI networks is possible. YSA enables the high-throughput, quantitative characterization of PPI networks in a fully defined extracellular environment at a library-on-library scale.
Introduction
Powerful methods have been developed for the high-throughput screening of protein-protein interactions (PPIs). Yeast two-hybrid1 can be used to intracellularly screen pairwise PPIs and has been extended to the screening of large PPI networks using next generation sequencing (NGS)2. However, intracellular assays are limited by an inability to control the binding environment and suffer from frequent false-positives and false-negatives3,4. Phage5 and yeast6 display have enabled the high-throughput binding characterization of large protein libraries, but can only screen binding against a limited number of targets due to the spectral resolution of existing fluorescent reporters7. SMI-seq can be used to characterize PPI networks in a cell-free environment, but requires the use of purified proteins and a dedicated flow cell for the analysis of each network and condition8. While each approach expands PPI screening capabilities, none allows for cell-based, quantitative, library-on-library PPI characterization in an extracellular environment that can be modified as desired.
Yeast mating in an aerated liquid culture9 depends critically on an intercellular PPI that drives agglutination between MATa and MATα haploid cells10. The MATa sexual agglutinin subunit, Aga2, and the MATα cognate, Sag1, interact with a KD of 2 to 5 nM and initiate the irreversible binding of two haploid cells and subsequent cellular fusion to create a single diploid cell11. Cellular agglutination and mating is highly efficient, occurs in a matter of hours, and each mating event forms a stable and propagating diploid strain12.
Here, we reprogram yeast mating by replacing the native sexual agglutination interaction proteins, Aga2 and Sag1, with arbitrary engineered or natural binding pairs that act as synthetic adhesion proteins (SAPs). We first show that interaction strength between one MATa SAP and one MATα SAP can be quantitatively assessed by co-culturing the two haploid strains and measuring their mating efficiency with a flow cytometry assay. We then extend the assay for one-pot, library-on-library characterization by barcoding SAP cassettes, co-culturing many MATa and MATα strains, and using NGS to count interaction frequencies for all possible a-α SAP interactions. Finally, we demonstrate that the binding environment can be systematically manipulated to probe PPI networks.
Results
Reprogramming Sexual Agglutination
In order to co-opt yeast mating to probe protein-protein interactions, we first genetically replaced native sexual agglutination with SAP interactions. To validate our approach, we used PPIs involving BCL2 homologues that were previously characterized with biolayer interferometry (BLI) 13,14. Six BCL2 homologues (Bcl-2, Bfl-1, Bcl-B, Bcl-w, Bcl-xL, and Mcl-1) were expressed on MATa cells. Seven natural and nine engineered binding proteins, representing a broad range of affinities for the BCL2 homologues, were expressed on MATa cells. Isogenic yeast strains were generated for each SAP by transformation with a fragment containing a SAP cassette and a mating type specific fluorescent reporter. Pairs of SAP-expressing haploid cells were co-cultured in non-selective liquid media for 17 hours to allow agglutination-dependent mating. Flow cytometry was performed to differentiate between mCherry expressing MATa haploids, mTurquoise expressing MATα haploids, and mated diploids that expressed both fluorescent markers (Fig. 1a). Diploid percent was used as a metric for mating efficiency to quantitatively characterize the interaction strength between a MATa SAP and a MATα SAP. A detection limit of 0.4% for the pairwise mating assay was determined by combining saturated MATa and MATα cultures and immediately performing flow cytometry on the mixed population without providing an opportunity for mating.

(a) Pairwise PPI characterization involves isogenic yeast transformations followed by mating and flow cytometry. (b) Library-on-library PPI network characterization involves strain library construction followed by mating, diploid DNA isolation, and NGS. Manipulation of the mating environment prior to mating is optional. (c) YSA outperforms existing technologies for high-throughput PPI characterization for protein engineering applications.
We found that complementary binding proteins expressed on the surface of yeast are necessary and sufficient to replace the function of the native sexual agglutinin proteins, Aga2 and Sag1. Wild-type W303 S. cerevisiae haploid cells mated with an efficiency of 63.6% ± 3.1% in standard laboratory conditions and a knockout of Sag1 in the MATα haploid eliminated mating with wild-type MATa (Fig. 2a). Maintaining the Sag1 knockout, expression of interacting SAP pairs recovered mating efficiency up to 51.6% ± 7.9%, while expression of a non-interacting SAP pair showed no observable recovery (Fig. 2b). SAP-dependent recovery of mating occurred with a variety of natural and engineered proteins ranging from 26 to 206 amino acids, indicating a lack of structural restrictions for synthetic agglutination beyond interaction strength (Supplementary Fig. 1, 2).
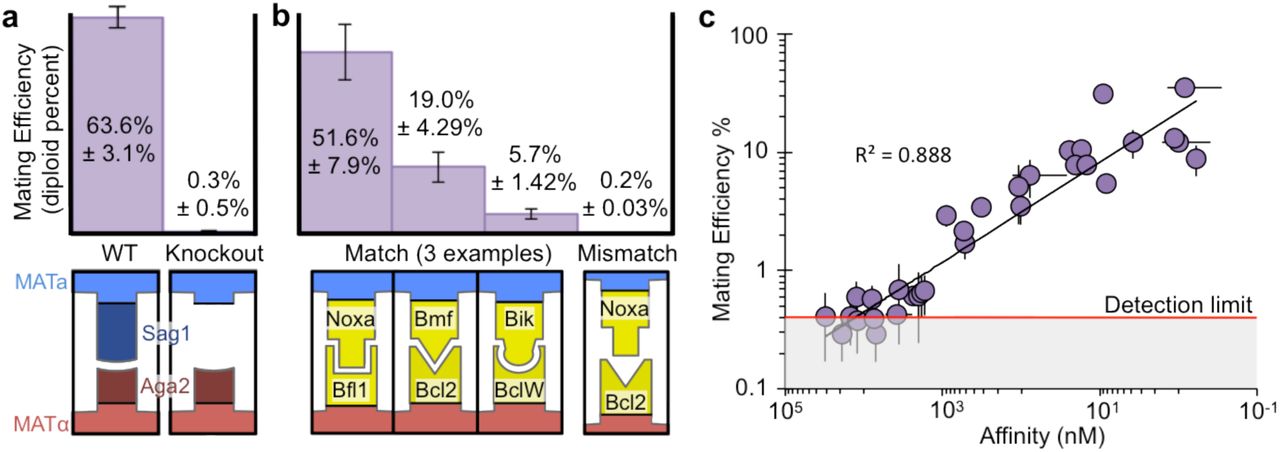
(a,b) Mating efficiency, given as the percent of diploid cells, for representative haploid pairs expressing (a) wild type sexual agglutinin proteins and (b) synthetic adhesion proteins (n=3). (c) Mating efficiencies for SAP pairs with a KD between 500 pM and 25 μM show a log-linear relationship with affinity (n=3). The limit of detection for pairwise mating efficiency is 0.4%.
Quantitative Pairwise PPI Characterization
Mating efficiency and affinity, measured with BLI, were found to be related log-linearly (R2 = 0.89) for PPIs across over four orders of magnitude of KD (Fig. 2c). We tested proteins with binding affinities ranging from 500 pM to above 100 μM, which gave mating efficiencies of up to 35.4% and down to below 0.4%, respectively. None of the 14 tested pairs with a KD above 25 μM resulted in a recovery of mating above 0.4%, the limit of detection for the pairwise mating assay, suggesting high assay specificity. The weakest interaction showing a detectable mating recovery was observed to have a KD of 12.5 μM and a mating efficiency of 0.6% (Supplementary Fig. 1).
The strong log-linear relationship between mating efficiency and affinity over multiple orders of magnitude contradicted our expectation of avidity as the main driving force for yeast agglutination11. We expected that upon the formation of a single interaction between cells, newly localized protein pairs would rapidly bind, making off-rate largely irrelevant. However, both on- and off-rate showed a correlation with mating efficiency, and neither provided as good a fit as KD (Supplementary Fig. 3a,b, Fig.2b). The contribution of both on- and off-rate implies that a single reversible PPI between cells initiates agglutination.
Detectable SAP surface expression is required for mating recovery. Prior to interaction screening, each SAP-expressing yeast strain was tested for surface expression by labeling with FITC-conjugated anti-myc6. One BCL2 homologue, Mcl-1, showed no surface expression and subsequently no recovery of mating efficiency regardless of its mating partner. A functional truncation of Mcl-1 (151-321) improved surface expression and enabled affinity-dependent mating 15 (Supplementary Fig. 3c).
Barcoding and recombination of interaction libraries
A barcoding and recombination scheme was developed for one-pot PPI network characterization. We began by constructing MATa and MATα parent strains, ySYNAGa and ySYNAGα respectively, into which pools of barcoded SAP cassettes were transformed (Fig. 3a and Supplementary Fig. 4). These strains include complementary lysine and leucine auxotrophic markers for diploid selection and express CRE recombinase16 after mating when induced with β-Estradiol (βE)17. For small libraries, SAP cassettes were assembled with isothermal assembly18 in one of two standardized vectors, pSYNAGa or pSYNAGα, for integration into the corresponding parent yeast strain (Supplementary Fig. 5). In addition to a barcoded surface expression cassette, each vector backbone contains a mating type specific lox recombination site and primer binding site. Sanger sequencing19 was used to match barcodes with their corresponding SAPs.

(a) Each SAP cassette is flanked by a unique barcode and mating type specific primer binding site and lox recombination site. CRE recombinase expression in diploid cells pairs MATa and MATα barcodes on the same chromosome for NGS. (b) A one-pot batched mating assay gives a strong log-linear relationship between the batched mating percent and affinity for PPIs with a KD between 500 pM and 25 μM (n=2). (c) All interactions between five BCL2 pro-survival homologues and seven peptide binders were characterized in a one-pot mating assay. The height of each purple bar represents the frequency of diploid formation from a given pair of SAP expressing haploid strains. (d,e,f) Interaction profiles for (d) Noxa, (e) Puma, and (f) Bad are shown in greater detail, with bolded targets indicating an affinity below 1 μM.
CRE induced chromosomal translocation 20 in diploid cells resulted in the juxtaposition of two barcodes, specific to the MATa and MATα SAP pair, on the same chromosome. Interacting SAPs are identified in a mixed culture using Illumina NGS 21 (Fig. 3a). qPCR with the mating type specific primers and subsequent Sanger sequencing was used to verify that recombination occurred only in diploid cells upon the addition of βE and that the recombination resulted in the expected chromosomal translocation.
One-pot protein library characterization
The frequency with which pairs of barcodes corresponding to interacting SAPs appear in diploid lysate following a batched mating was observed to be log-linear with BLI affinity measurements (R2 = 0.87) (Fig. 3b). Following NGS, the batched mating percent for each interaction in the network was calculated from the raw interaction counts, providing relative interaction strengths for each PPI in the network. We constructed barcoded SAP cassettes for six BCL2 family pro-survival proteins and nine engineered binders and measured the relative interaction frequencies of each possible interaction in a batched mating. As before, we tested proteins with binding affinities ranging from 500 pM to above 100 μM, which led to a more than a 500 fold difference in batched mating percent. In addition to the de novo binding proteins, seven natural peptide binders with diverse binding profiles were added to a batched mating22 (Fig. 3c). The interaction profile between these peptides and the five pro-survival proteins was consistent with previous work23. For example, Noxa was confirmed to bind Bfl-1 with high specificity (Fig. 3d) and Puma was confirmed to bind nonspecifically to Bcl-w, Bcl-xL, Bcl-2, and Bfl-1 (Fig. 3e). Even Bad, which had been observed to interact the least overall, gave the expected interaction profile: relatively strong binding to Bcl-xL and Bcl-2, weak binding to Bcl-w, and minimal binding to Bcl-B and Bfl-1 (Fig. 3f).
A comparison of the pairwise and one-pot methods showed a near perfect 1:1 agreement (Supplementary Fig. 6). To compare the two approaches, pairwise mating efficiency was normalized so that the mating efficiency of all tested pairs summed to one hundred, giving a relative mating percent. A paired two-sided T-test of relative mating percent and batched mating percent gave a p-value of 0.80, indicating no statistically significant difference between the two methods.
Large PPI library characterization
For large library chromosomal integrations, a “landing pad” approach24 was used to achieve over 10,000 integrants in a single transformation and allowed for multi-fragment homologous recombination integrations (Fig 4a). At the SAP cassette integration locus, both ySYNAGa and ySYNAGα were transformed with an expression cassette consisting of a GAL promoter, SceI endonuclease, and Cyc1 terminator flanked by SceI cut sites. GAL induction prior to transformation with a SAP library resulted in DNA nicking at the site of integration, which dramatically improved integration efficiency25. NGS of genomic DNA extracted from yeast libraries was used to pair each SAP variant to its distinct barcode and to count relative barcode frequencies in the naïve library.
A single-pot batched mating was used to characterize 7,000 distinct PPIs. A partial site-saturation mutagenesis (SSM) library of XCDP0714 consisting of 1,400 distinct variants was mated with five BCL2 pro-survival homologues, including the intended binding partner of XCDP07, Bcl-xL. For each variant, interaction strength (the number of times a particular variant was observed to have mated with Bcl-xL divided by the number of times that variant was observed in the naïve library) and specificity (the percent of observed matings with Bcl-xL minus the percent of observed matings with the next highest BCL2 homologue) was determined. As a proof of principal, interactions involving variants with premature stop codons were analyzed (Fig. 4b). Only 8 of 55 premature stop codons included in the library resulted in even a single mating and only 6 resulted in more than 2 matings. These six variants contained stop codons at residue 93 or later, which is beyond the central binding helix. Two variants, with stop codons at residues 113 and 114, showed improved interaction strength and specificity. These early stops likely minimally affected binding or stability of the 116-residue full-length protein and served to remove the C-terminal myc tag fusion, which may have negatively impacted binding.

(a) Transformation efficiency for large library integrations is increased with a GAL inducible SceI landing pad. This approach was used to construct a 1,400 member partial site-saturation mutagenesis (SSM) library of the Bcl-xL binder XCDP07, which was evaluated for binding with 5 BCL2 homologues using YSA. (b) Interaction strength versus specificity plot with highlighted premature stop codon variants. The diameter of each point is a function of its representation in the naïve library, which is used as a measure of confidence (c) Cartoon model of XCDP07 bound to Bcl-xL. (d) Mating percent with each BCL2 homologue for WT XCDP07 and for two point mutants that also showed enrichment in a one-sided yeast surface display screen. (e) Interaction strength and specificity of the SSM library with two confirmed affinity and specificity improving mutations highlighted.
Favorable mutations from a yeast surface display library were correctly identified using YSA along with on- and off-target binding specificities (Fig. 4d,e). In particular, two mutations at the interface periphery, L47R and A48T, were found to be favorable for interaction strength. Both mutations were enriched by FACS sorting of an XCDP07 SSM surface display library incubated with fluorescently labeled Bcl-xL and unlabeled competitor homologues14. In addition to identifying mutations that improve affinity, YSA provided detailed information about binding specificities to each target (Fig. 4d). We observed moderately improved on target specificity for L47R, mostly through relative weakening of the interactions with Bcl-w and Bcl-B. We observed that A48T more dramatically weakened all off target interactions with a 16.5% increase of on-target binding.
PPI network response to environmental changes
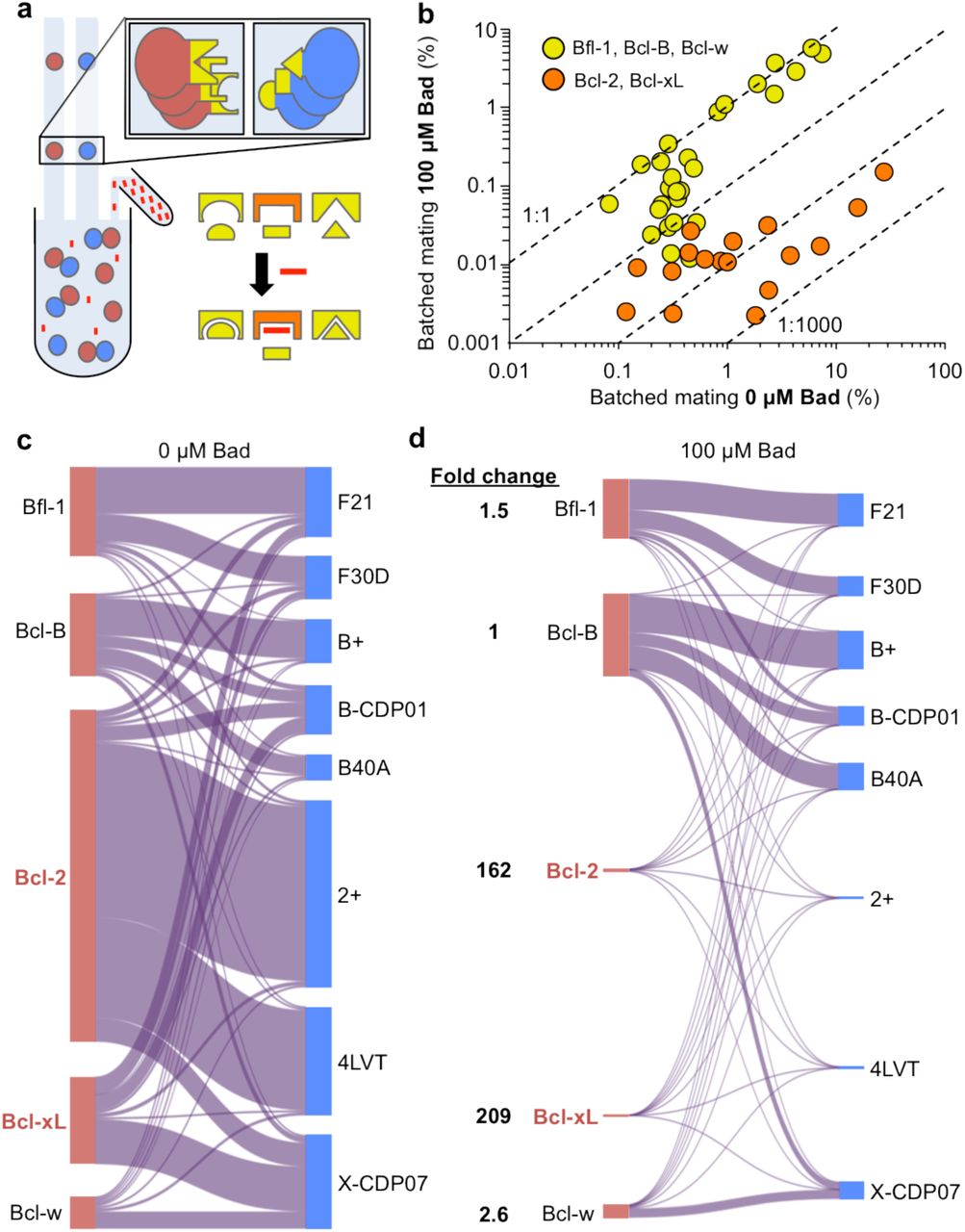
To demonstrate the characterization of a PPI network in a new extracellular environment, we added a non membrane soluble competitive binder at the start of a batched mating, which selectively inhibited PPIs up to 800 fold (Fig. 5a,b). In the interaction network of the BCL2 pro-survival proteins and their natural and de novo binding partners, one peptide, the BH3 domain of Bad, bound predominantly to Bcl-xL and Bcl-2, weakly to Bcl-w, weaker still to Bfl-1, and minimally to Bcl-B (Fig. 3f). Since Bcl-B showed no detectable interaction with Bad, batched matings with and without 100 nM Bad were normalized to one another with the assumption that interactions involving Bcl-B were not affected by on-target binding. This normalization accounted for differences in total sequencing reads between conditions and the effect of additional protein in the media causing non-specific blocking.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(a) The batched mating environment can be manipulated with the addition of arbitrary molecules (red) that disrupt interactions involving particular proteins (orange) but not others (yellow). (b) The addition of a competing peptide, Bad, to a mating between five BCL2 homologues and a panel of de novo binders results in the isolated disruption of interactions involving Bcl-2 and Bcl-xL. Dashed lines representing 1:1, 1:10, 1:100, and 1:1000 differences between conditions are given for reference. (c,d) A visualization of the PPI network (c) without and (d) with the addition of 100 μM Bad. Fold changes are given for the aggregate interactions of each BCL2 homologue.
The addition of Bad at a concentration of 100 nM resulted in strong inhibition of interactions involving Bcl-2 and Bcl-xL. Comparing batched matings with and without the addition of Bad, we observed no change for all strong interactions involving Bfl-1, Bcl-B, and Bcl-w, homologues that do not interact with Bad. Pairwise interactions involving Bcl-2 and Bcl-xL, however, were inhibited by at least 16 fold and up to 800 fold (Fig. 5b,c,d). Weak interactions with Bfl-1, Bcl-B, and Bcl-w showed reduced mating percent with the addition of Bad, which can be attributed to non-specific blocking. Considered together, all PPIs involving Bcl-xL and Bcl-2 were strongly inhibited, with normalized mating percent fold changes of 209 and 162, respectively. The weaker Bad binders, Bcl-w and Bfl-1, displayed a normalized mating percent fold change of 2.6 and 1.5, respectively. All aggregate fold changes were consistent with previous batched matings characterizing interactions between Bad and the five BCL2 homologues.
Discussion
We show that the mating of S. cerevisiae can be reprogrammed by the surface expression of arbitrary synthetic adhesion proteins that replace the function of the native sexual agglutinin proteins, Aga2 and Sag1. Using YSA, we demonstrate quantitative library-on-library characterization of up to 7000 distinct PPIs in a single pot. Additionally, we show how YSA could be used for characterizing PPI networks in different environments by adding an exogenous competitor to the mating environment. To date, tools for screening libraries of PPIs are limited by throughput, a fixed intracellular environment, or accuracy. Previous strategies for developing library-on-library screening platforms have used cell-free systems, which are far less scalable. In contrast, YSA combines the scalability of a cellular assay with the feature of environmental manipulation on a library-on-library scale.
YSA provides a high-throughput platform for screening environment-responsive PPIs and PPI-inhibiting drug candidates. Engineered PPIs that respond to environmental changes, such as pH, are valuable for biosensors 26 and drug delivery27. YSA may enable the rapid identification of functional variants using one-pot screening of design libraries rather than individual testing of protein pairs. Drug-induced PPI inhibition is a powerful therapeutic strategy for treating cancers, inflammation, and infectious diseases. YSA may streamline pre-clinical drug screening workflows by testing candidate compounds on a protein interaction network consisting of both on- and off-target PPIs, simultaneously screening efficacy and specificity.
In addition to its utility for PPI characterization, YSA provides a unique ecological model for studying pre-zygotic genetic isolation. Previous work described the large diversity in sexual agglutination proteins across yeast species and suggested that co-evolution of these proteins may drive speciation by genetically isolating haploid pairs28. Here, we have created a fully engineerable synthetic pre-zygotic barrier that can be used as a model to study complex ecological phenomena such as speciation and sexual selection, similar to the use of engineered E.coli for modeling predator-prey dynamics29.
Quantitative affinity characterization using YSA is currently limited to PPIs with KD’s ranging from 500 pM to 25 μM. It may be possible to expand this range through optimization of mating conditions including media composition, shake rate, and cell density. However, we believe that the dynamic range currently provided by YSA is sufficient for most protein engineering applications and enables high-throughput screening for many new protein engineering challenges.
Methods
DNA construction
Isogenic fragments for yeast transformation or plasmid assembly were PCR amplified from existing plasmids or yeast genomic DNA with Kapa polymerase (Kapa Biosystems), gel extracted from a plasmid digest (Qiagen), or synthesized by a commercial supplier (IDT). Plasmids were constructed with isothermal assembly18 and verified with Sanger sequencing19. MATa and MATα SAP cassette plasmids were assembled using a four piece assembly, including two backbone fragments, a SAP cassette fragment, and a barcode containing fragment (Supplementary Fig. a,b). Site-saturated mutagenesis (SSM) library DNA was prepared with overlap PCR13 using Kapa polymerase and custom NNK primers for each codon. For a complete list of plasmids used in this study, see Supplementary Table 1. Sequences for all cloning primers, fragments, and plasmids are available upon request.
Yeast Strain Construction
A MATα variant of the EBY1006 strain was constructed with mating, sporulation, tetrad dissection, and screening with selectable markers30. EBY100a was mated with a leucine prototroph W303α variant. Following sporulation, positive selection was performed for HIS, LEU, and URA and replica plating was used to identify MATα haploids auxotrophic for lys and trp (Supplementary Fig. 7a). Plating on 5-FOA was used to select strains with URA3 inactivating mutations31. Final strains were constructed with many rounds of chromosomal integration, each consisting of a single transformation, auxotrophic or antibiotic selection, and PCR to verify integration into the expected locus (Supplementary Fig. 7b). Isogenic chromosomal integrations consisted of digesting a plasmid with Pme1 and performing a standard lithium acetate transformation32. SSM libraries were transformed into yeast using nuclease assisted chromosomal integration24. Prior to transformation, parent yeast strains were grown in YPG media for five hours. Growth in galactose media induced SceI expression and caused DNA damage at the integration site (Fig. 4a). 100 μL of cell pellet, rather than 10 μL for a standard transformation, was used for each library transformation and all other reagents were scaled up accordingly. Four fragments, approximately 2 μg of each, were added to each transformation. The fragments included two mating type specific adaptor fragments, a SAP SSM library fragment, and a barcode library containing fragment (Supplementary Fig. 5c). Following the transformation, cells were washed in 5 mL YPD and resuspended in YPD to a total volume of 5 mL. 100 μL were immediately removed and a dilution series was plated on SDO-trp to quantify the total number of transformants in the library. The remaining culture was grown for 5 hours, washed twice with 5 mL SDO-trp, and grown in 20 mL SDO-trp overnight to select for transformants. 2 mL 25% glycerol aliquots were then prepared for later use.
Peptide construction and purification
DNA encoding the BH3 domain of Bad (Bcl-2 agonist of cell death protein; residues 103-131) was synthesized by IDT and inserted into a modified pMAL-c5x vector resulting in an N-terminal fusion to maltose binding protein and a C-terminal 6-histidine tag. The vector was transformed into BL21(DE3)* E. coli (NEB) for protein expression. Protein was purified from soluble lysate first with nickel affinity chromatography (NiNTA resin from Qiagen), then by size exclusion chromatography (Superdex 75 10/300 GL; GE). Purified protein was concentrated via centrifugal filter (Millipore), snap-frozen in liquid nitrogen and stored at -80°C.
Surface expression screening
Prior to mating assays, isogenic yeast surface expression strains and yeast libraries were tested for functional surface expression. To measure yeast surface expression strength, 10 μL of freshly saturated cells were washed with 1 mL PBSF, incubated in 50 μL PBSF media with 1 μg FITC-anti-myc antibody (Immunology Consultants Laboratory, Inc.) for 1 hour at 22°C, washed with 1 mL PBSF, and read with the FL1.A channel on an Accuri C6 cytometer.
Pairwise (Two-Strain) Mating Assays
MATa and MATα haploid yeast strains were grown for approximately 24 hours to saturation in 3 mL of YPD from isogenic colonies. For each mating assay, 2.5 μL of a saturated MATa culture and 5 μL of a saturated MATα culture were combined in 3 mL of YPD and incubated at 30°C and 275 RPM for 17 hours. 5 μL from the mixed culture were then added to 1 mL of water and cellular expression of mCherry and mTurquoise was characterized with a Miltenyi MACSQuant VYB cytometer using channels Y2 and V1, respectively. A standard yeast gate was applied to all cytometry data and Flowjo was used for analysis and visualization.
Yeast Library Preparation
Pre-characterized yeast libraries were prepared by combining individually transformed isogenic yeast strains with validated SAP surface expression and known barcodes determined with Sanger sequencing (Supplementary Table 3). Each individual strain was grown for approximately 24 hours to saturation in 3mL of YPD from an isogenic colony. Strains of the same mating type were then pooled with equal cell counts of each isogenic strain, measured with an Accuri c6 flow cytometer.
Uncharacterized yeast SSM libraries were constructed with nuclease assisted chromosomal integration and large volume transformation, as described above. Prior to mating, libraries were characterized using NGS to map each library variant with its 10 bp barcode and to determine relative counts of each variant in the naïve library population. One 2 mL glycerol stock was thawed, washed once with 1 mL YPD, and grown in 50 mL YPD for 24 hours. Genomic DNA was then prepared for NGS.
Library Mating Assays
2.5 μL of the MATa library and 5 μL of the MATa library were combined in 3 mL of YPD and incubated at 30°C and 275 RPM for a 17 hour mating. When characterizing interactions in the presence of Bad.BH3, the peptide was added at a concentration of 100 nM to the 3 mL YPD culture. Following each 17 hour mating, 1 mL was washed twice in 1 mL SDO-lys-leu and transferred to 50 mL SDO-lys-leu with 100 nM β-estradiol (βE) for diploid selection and induction of CRE recombinase. After 24 hours of growth, genomic DNA was prepared for NGS.
Preparation for NGS
50 mL yeast cultures were harvested by centrifugation and lysed by heating to 70°C for 10 min in 2 mL 200 mM LiOAc and 1% SDS33. Cellular debris was removed with centrifugation and the supernatant was incubated at 37°C for 4 hours with 0.05 mg/mL RNase A. An ethanol precipitation was then performed to purify and concentrate the genomic DNA and a 2% agarose gel was run to verify genomic DNA extraction. Two rounds of qPCR were performed to amplify a fragment pool from the genomic DNA and to add standard Illumina sequencing adaptors and assay specific index barcodes. For the primary PCR, different primers were used for naïve library characterization and post-mating characterization. An index barcode was added in the secondary PCR with the reverse primer. For a list of all NGS primers used in this study, see Supplementary Table 4. Both PCRs were terminated before saturation in order to minimize PCR bias. The first PCR was run for 25-30 cycles, and the second PCR was run for 5-7 cycles. The final amplified fragment was gel extracted, quantified with a Qubit and sequenced with a MiSeq sequencer (Illumina). A 600-cycle v3 reagent kit was used for naïve library characterization and a 150-cycle v3 reagent kit was used for post-mating characterization.
Sequence analysis
Pre-mated SSM libraries were sequenced in order to match each variant with a 10 bp barcode and to determine the relative population size of each variant. All sequences were first filtered for quality by requiring a perfect match for 15 bp in a constant region immediately before and after the mutated gene. Forward and reverse reads were stitched together and full SSM coding regions were translated to amino acid sequences. Sequences were then grouped by their 10 bp barcode and a consensus amino acid sequence was determined for each group. Only groups with zero or one amino acid mutation were kept. Groups representing the same amino acid mutation were then pooled. The number of sequences in each pooled group provided naïve library counts for each SSM variant and the barcodes attributed to each pooled group were used for later matching of mated diploids to SSM variants.
Post-mating sequences were first filtered for quality by requiring a perfect match for 10 bp in a constant region immediately before and after both barcodes. Barcodes from forward and reverse reads were then isolated and replaced with the protein variant they were previously found to represent. A dataframe with interaction counts for every possible pairwise interaction was generated.
Accession numbers
BioProject Accession number: PRJNA380247
BioSample Accession numbers: SAMN06642476, SAMN06642477,
SAMN06642478, SAMN06642479, SAMN06642480, SAMN06642481,
SAMN06642482, SAMN06642483, SAMN06642484, SAMN06642485
Code availability
All code is fully available on GitHub:
Contributions
D.Y., D.B., and E.K. contributed to the technical design. D.Y. and S.B. implemented the methods. D.Y. analyzed the data and wrote the manuscript with contributions from all authors.
Competing Financial Interests
The University of Washington has filed a patent application based on the findings in the article. U.S. application no. 15/407,215. D.Y, D.B., and E.K. are co-inventors.
Acknowledgements
We thank M. Dunham for technical discussions, A. Rosenberg for data analysis support and M. Parks and the UW Biofab for assistance with the construction of many plasmids and yeast strains used in this study. This work was supported by US National Science Foundation (NSF) award number 1317653. D.Y. and S.B. are supported by the NSF GRFP.
References




