

ABSTRACT
Higher educational attainment (EA) is known to have a protective effect regarding the severity of schizophrenia (SZ). However, recent studies have found a small positive genetic correlation between EA and SZ. Here, we investigate possible causes of this counterintuitive finding using genome-wide association results for EA and SZ (n = 443,581) and a replication cohort (1,169 controls and 1,067 cases) with high-quality SZ phenotypes. We find strong genetic overlap between EA and SZ that cannot be explained by chance, linkage disequilibrium, or assortative mating. Instead, our results suggest that the current clinical diagnosis of SZ comprises at least two disease subtypes with non-identical symptoms and genetic architectures: One part resembles bipolar disorder (BIP) and high intelligence, while the other part is a cognitive disorder that is independent of BIP.
INTRODUCTION
Schizophrenia (SZ) is the collective term used for a severe, highly heterogeneous and costly psychiatric disorder that is caused by a complex interplay of environmental and genetic factors1–4. The latest genome-wide association study (GWAS) by the Psychiatric Genomics Consortium (PGC) identified 108 genomic loci that are associated with SZ5. These 108 loci jointly account for ≈3.4% of the variation on the liability scale to SZ5, while all single nucleotide polymorphisms (SNPs) that are currently measured by SNP arrays capture ≈64% (s.e. = 8%) in the variation in liability to the disease6. This suggests that most of the genetic variants contributing to the heritability of SZ have very small effects and that they have not been isolated yet. This could be due in part to the fact that the clinical disease classification of SZ spans across many different syndromes (e.g., catatonia, paranoia, grandiosity, difficulty in abstract thinking, thought blocking, social withdrawal, hallucinations) that may not have identical genetic architectures. Therefore, identifying additional genetic variants and understanding through which pathways they influence specific SZ syndromes is an important step in understanding the etiologies of the ‘schizophrenias’7. However, GWAS analyses of specific SZ syndromes would require very large sample sizes to be statistically well-powered, and the currently available datasets on deeply phenotyped SZ individuals are not large enough yet for this purpose.
Here, we use an alternative approach that combines data for SZ with another cognitive phenotype that can be studied in very large GWAS samples—educational attainment (EA). The relationship between SZ and EA is peculiar: There are contradictory results on the relationship between SZ and EA from phenotypic and genetic data that can be used as an avenue to further our understanding about SZ. Phenotypic data seem to suggest a negative correlation between EA and SZ8. For example, SZ patients with lower EA typically show an earlier age of disease onset, higher levels of psychotic symptomatology, and worsened global cognitive function8. In fact, EA has been suggested to be a measure of premorbid function and predictor of outcomes in SZ. Moreover, it has been forcefully argued that retarded intellectual development during childhood and bad school performance should be seen as core features of SZ and early indicators of the disease that precede the development of psychotic symptoms9,10. Furthermore, credible genetic links between SZ and impaired cognitive performance have been found11.
In contrast to these findings, recent studies using GWAS results identified a small, but positive genetic correlation between EA and SZ12,13. Here, we explore possible reasons for this contradictory result using the largest, non-overlapping GWAS samples on cognitive traits to date, totaling 443,581 individuals of European descent (the vast majority of observations coming from EA). For follow-up analyses, we use data from an independent replication sample that has exceptionally detailed measures of SZ symptoms, the GRAS (Göttingen Research Association for Schizophrenia) data collection4,7,14.
As a first step, we used the proxy-phenotype method (PPM) to illustrate the genetic overlap between EA and SZ. As a side-result, this approach may isolate novel empirically plausible candidate genes for SZ, comparable to similar studies using PPM that have demonstrated this for cognitive performance15, Alzheimer’s disease, intracranial and hippocampal volume13, depression and neuroticism16. PPM is a two-stage approach that increases statistical power by using genetic association results from a large, independent sample for a related phenotype to limit the multiple testing burden for the phenotype of interest15. Previous evidence suggests a strong genetic overlap between EA and SZ, which implies that EA could be used as a proxy-phenotype for SZ because EA can be studied in much larger samples13,16. However, compared to the present work, these previous studies used substantially smaller and partially overlapping samples and did not have access to an independent cohort that could be used for replication and follow-up analyses.
There are several possible reasons why EA-associated SNPs may also be associated with SZ. One possibility is that a set of genes that is generally important for all brain-related phenotypes is driving this enrichment. This hypothesis suggests that the set of genetic loci that our proxy-phenotype analysis identifies should be generally enriched for association with all brain-related phenotypes, but not for non-brain-related outcomes. To investigate this possibility, we test genetic loci that are jointly associated with EA and SZ for enrichment across 21 additional traits (Supplementary Note).
Second, enrichment could also be a generic consequence of EA-associated SNPs exhibiting above average linkage disequilibrium (LD) with neighboring SNPs. This would increase the probability that these SNPs “tag” other genetic variants that are associated with SZ, or any other disorder12. To investigate this possibility, we propose a measure that tests for enrichment beyond what is expected for each EA related SNP given its LD with its neighbors (Supplementary Note).
A third possible cause of strong enrichment and weak genetic correlation is heterogeneity in SZ—i.e., sub-types of the disease having different biological causes and varying genetic correlations with EA. Heterogeneity in the disease may also be a reason why previous studies did not succeed in predicting specific syndromes of SZ using a “normal” polygenic score (PGS) that was derived from large-scale GWAS on SZ, which implicitly assumed that all SZ-associated SNPs influence all syndromes in the same way4,17. If heterogeneity in the disease is causing the observed enrichment of EA with SZ, the sign concordance pattern of SNPs with both traits may contain relevant information that is pertinent to specific SZ syndromes. We tested this by constructing PGS in our replication cohort with high-quality SZ phenotypes that take the sign concordance of SNPs for EA and SZ into account (Supplementary Note). As a robustness check, we repeat this analysis excluding patients diagnosed with schizoaffective disorder.
A fourth possible cause of enrichment is that other phenotypes are genetically correlated with both EA and SZ. Previous studies indicated a particularly strong positive genetic correlation between SZ and bipolar disorder (BIP), which may influence the genetic overlap of both diseases with related phenotypes such as EA, childhood intelligence (IQ), and neuroticism12,13,18. We use genome-wide inferred statistics (GWIS) that allow controlling for the genetic correlation between SZ and BIP to investigate how “unique” SZ (controlling for BIP) and “unique” BIP (controlling for SZ) are related to EA, childhood IQ, and neuroticism18.
A fifth possible cause may be assortative mating, which has been demonstrated both for EA19 and SZ20. We use simulations to explore if independent assortative mating for the two phenotypes may induce a spurious genetic overlap.
This list of potential causes for the genetic overlap between EA and SZ may not be exhaustive and several of these factors may be at work simultaneously.
RESULTS
Proxy-phenotype analyses
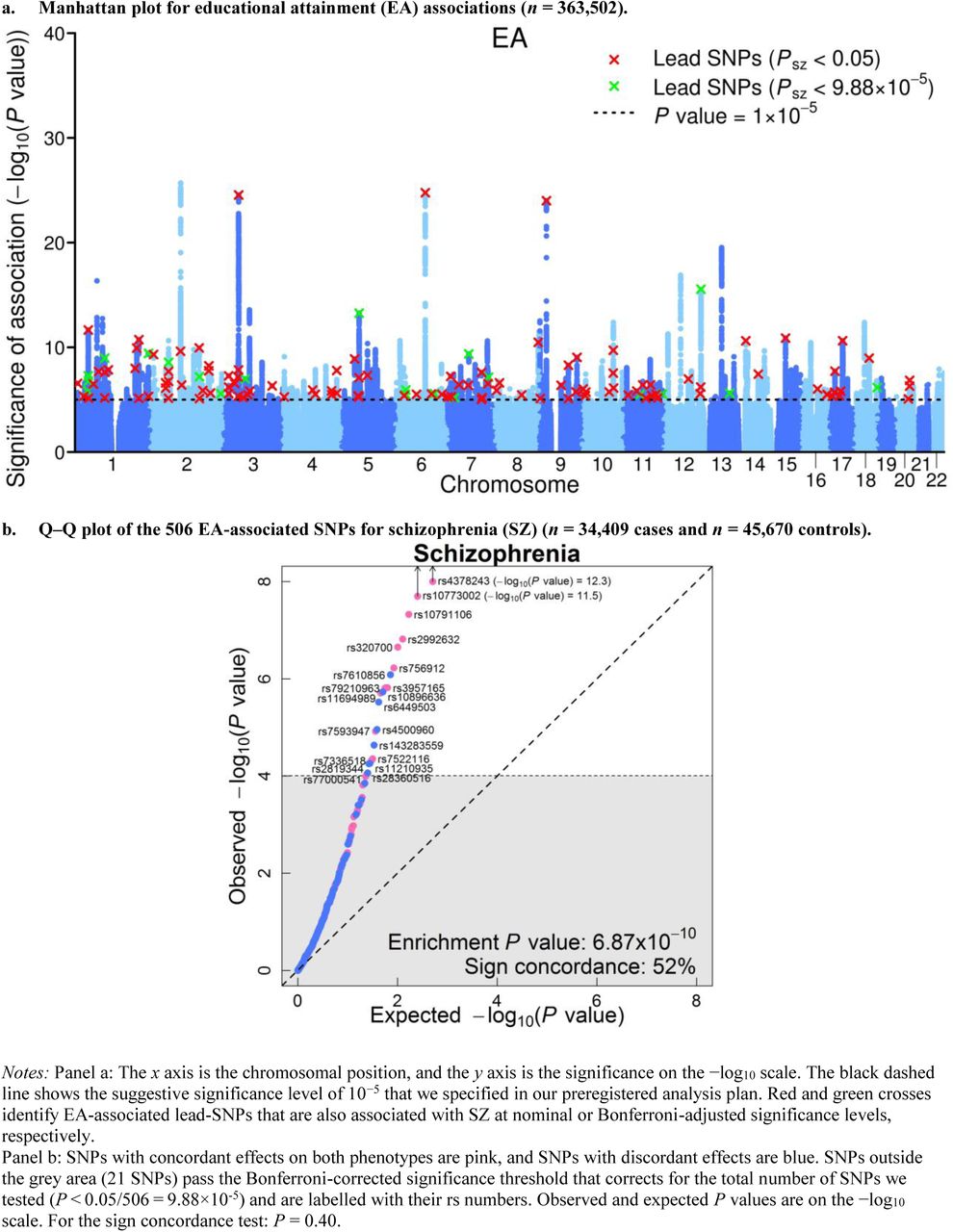
Figure 1 presents an overview of the proxy-phenotype analyses. The first-stage GWAS on EA (Supplementary Note) identified 506 loci that passed our predefined threshold of PEA < 10−5 (https://osf.io/dnhfk/); 108 of them were genome-wide significant (PEA < 5 × 10−8, see Supplementary Table 5.1). Of the 506 EA lead-SNPs, 132 are associated with SZ at nominal significance (PSZ < 0.05), and 21 of these survive Bonferroni correction  . LD score regression results suggest that the vast majority of the association signal in both the EA13 and the SZ5 GWAS are truly genetic signals, rather than spurious signals originating from uncontrolled population stratification. Figure 2a shows a Manhattan plot for the GWAS on EA highlighting SNPs that were also significantly associated with SZ (red crosses for PSZ < 0.05, green crosses for PSZ = 9.88 × 10−5).
. LD score regression results suggest that the vast majority of the association signal in both the EA13 and the SZ5 GWAS are truly genetic signals, rather than spurious signals originating from uncontrolled population stratification. Figure 2a shows a Manhattan plot for the GWAS on EA highlighting SNPs that were also significantly associated with SZ (red crosses for PSZ < 0.05, green crosses for PSZ = 9.88 × 10−5).


A Q-Q plot of the 506 EA lead SNPs for SZ is shown in Figure 2b. Although the observed sign concordance of 52% is not significantly different from a random pattern (P = 0.40), we find 3.23 times more SNPs in this set of 506 SNPs that are nominally significant for SZ than expected given the distribution of the P values in the SZ GWAS results (raw enrichment P = 6.87 × 10−10, Supplementary Note). The observed enrichment of the 21 EA lead SNPs that pass Bonferroni correction for SZ  is even more pronounced (27 times stronger, P = 5.44 × 10−14).
is even more pronounced (27 times stronger, P = 5.44 × 10−14).
Bayesian credibility of the results
The effect sizes of these 21 SNPs on SZ are small, ranging from Odds = 1.02 (rs4500960) to Odds = 1.11 (rs4378243) after winner’s curse correction (Table 1). However, Bayesian calculations with reasonable prior beliefs (e.g., 1% or 5%, Supplementary Note) suggest that most of these 21 SNPs are likely or virtually certain to be truly associated with SZ.
Prediction of future genome-wide significant loci for schizophrenia
Of the 21 variants we identified, 12 are in LD with loci previously reported by the PGC5 and 2 are in the major histocompatibility complex (MHC) region on chromosome 6 and were therefore not separately reported in that study. Three of the variants we isolated (rs7610856, rs143283559, rs28360516) were independently found in a recent meta-analysis of the PGC results5 with another large-scale sample21. We show in the Supplementary Note that using EA as a proxy-phenotype for SZ helped to predict the novel genome-wide significant findings reported in that study, which illustrates the power of the proxy-phenotype approach. Furthermore, two of the 21 variants (rs756912, rs7593947) are in LD with loci recently reported in a study that also compared GWAS findings from EA and SZ using smaller samples and a less conservative statistical approach22 (Supplementary Note). The remaining 2 SNPs we identified (rs7336518 on chr13 and rs7522116 on chr 1) add to the list of empirically plausible candidate loci for SZ.
LD-aware enrichment across different traits
Figure 3 and Supplementary Table 5.2 show the LD-aware enrichment of the SNPs that are jointly associated with EA and SZ across 22 traits. We find significant joint LD-aware enrichment of this set of SNPs for SZ, BIP, neuroticism and childhood IQ, and for inflammatory bowel disease and age at menarche. However, we find no LD-aware enrichment for other brain-traits that are phenotypically related to SZ, such as depressive symptoms, subjective well-being, autism, and attention deficit hyperactivity disorder. We also do not find LD-aware enrichment for most traits that are less obviously related to the brain (e.g., BMI, coronary artery disease) and our negative controls (e.g., fasting insulin, birth weight, birth length). Furthermore, one of the novel SNPs we isolated shows significant LD-aware enrichment both for SZ and for BIP (rs7522116).

Replication in the GRAS sample
A PGS based on the 132 loci jointly associated with both EA and SZ (SZ_132) adds ΔR2 = 7.54% – 7.01% = 0.53% predictive accuracy for the SZ case-control status to a PGS (SZ_all) derived from the GWAS on SZ alone (P = 1.7 × 10−4, Table 2, Model 3). The SZ_132 score also significantly adds (P = 3.4 × 10−4) to the predictive accuracy of the SZ case-control status when all other scores we constructed are included as control variables. In addition to SZ_132, PGS for SZ (SZ_all) and for BIP (BIP_all ) also predict case-control status, jointly reaching an adjusted ΔR2 of ≈ 9% (Table 2, Model 9 and Supplementary Note).
Polygenic prediction of schizophrenia measures in the GRAS sample
We find that the number of years of education is phenotypically correlated with later age at prodrome, later onset of disease, and less severe disease symptoms among SZ patients in the GRAS sample (Supplementary Note, Supplementary Table 8.1 and Supplementary Fig. 1). The EA_all score is associated with years of education (P = 2.6 × 10−6) and premorbid IQ (P = 2.3 × 10−4) among SZ patients (Supplementary Note and Table 3). Consistent with earlier results4, we find that none of the SZ measures can be predicted by the normal SZ PGS (SZ_all, Supplementary Table 8.2). Importantly, by utilizing GWAS results from both EA and SZ, we show that it is possible to predict specific features of SZ (Global Assessment of Functioning (GAF), Clinical Global Impression of Severity (CGI-S), and Positive and Negative Syndrome Scale (PANSS)) from genetic data. In a multiple regression analysis23 that allows a “ceteris paribus” interpretation of the included variables, we find that the EA_all score is associated with less severe disease outcomes only if we condition on the effects of the Concordant and Discordant scores. And conditional on the EA_all score, the Concordant and Discordant scores are associated with more (less) severe positive and negative symptoms as measured by the PANSS scale, respectively (Table 3). The best predictive accuracy of SZ readouts using these scores is currently observed for GAF (R2 = 1.38%). Of note, several of the symptoms measured by PANSS are also symptoms of BIP. The degree and composition of symptoms varies with the phase at evaluation (manic or depressive) and the general disease severity. We repeated these analyses excluding patients who were diagnosed with schizoaffective disorder (SD) and found similar results, implying that the genetic heterogeneity in SZ that we identify is not only due to SD (Supplementary Note, Supplementary Table 8.4.a).
Controlling for the genetic overlap between schizophrenia and bipolar disorder
None of the EA-associated lead SNPs (PEA < 5 × 10−8) are significantly associated with “unique” SZ(min BIP) after Bonferroni correction (Supplementary Table 9.1, Supplementary Note). The sign concordance of the EA lead SNPs with “unique” SZ(min BIP) was 44.5% (P = 0.046). Supplementary Figure 2 shows a Q-Q plot of the EA lead-SNPs for “unique” SZ(min BIP). Although we find 1.6 times more EA-associated SNPs with PSZunique < 0.05 than expected by chance (raw enrichment P = 0.02, Supplementary Note), the enrichment is much weaker than in the main SZ GWAS results that did not control for the genetic overlap between SZ and BIP. The genetic correlations between EA SZ(min BIP), and IQ and SZ(min BIP) are negative and significant (rg = -0.16, P = 3.88×10−04 and rg = -0.31, P = 6.00×10−03 respectively), which is in line with the idea of SZ being a cognitive disorder9. Furthermore, the genetic correlations of EA and IQ with BIP(min SZ) remain positive and get somewhat stronger (rg = 0.31, P = 2.87×10−07 and rg = 0.33, P = 3.18×10−02 respectively) compared with the ordinary BIP GWAS results. However, controlling for the genetic overlap of SZ and BIP does not affect the genetic correlations with neuroticism (Figure 4).

Simulations of assortative mating
Our simulations for assortative mating were based on relatively extreme assumptions that increased our chance of finding spurious enrichment of EA loci for SZ. The results suggest it is unlikely that assortative mating is a major cause for the genetic overlap we observe between EA and SZ (Supplementary Fig. 3).
Biological annotations
Biological annotation of the 132 SNPs that are jointly associated with EA and SZ using DEPICT identified 111 significant reconstituted gene sets (Supplementary Table 10.1). Pruning these resulted in 19 representative gene sets including dendrites, axon guidance, transmission across chemical synapses, and abnormal cerebral cortex morphology (Supplementary Table 10.2 and Figure 5a). All significantly enriched tissues are related to the nervous system and sense organs (Figure 5b). Furthermore, “Neural Stem Cells” is the only significantly enriched cell-type (Supplementary Table 10.3). DEPICT prioritized genes that are known to be involved in neurogenesis and synapse formation (Supplementary Table 10.4). Some of the genes, including SEMA6D and CSPG5, have been suggested to play a potential role in SZ24,25. For the two novel candidate SNPs reported in this study (rs7522116 and rs7336518), DEPICT points to the FOXO6 (Forkhead Box O6) and the SLITRK1 (SLIT and NTRK Like Family Member 1) genes, respectively. FOXO6 is predominantly expressed in the hippocampus and has been suggested to be involved in memory consolidation, emotion and synaptic function26,27. Similarly, SLITRK1 is also highly expressed in the brain28, particularly localized to excitatory synapses and promoting their development29, and it has previously been suggested to be a candidate gene for neuropsychiatric disorders30.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DISCUSSION
We explored the genetic overlap between EA and SZ using the largest currently available GWAS sample on human cognitive traits to date. Using EA as a proxy-phenotype, we identified 21 genetic loci for SZ and showed that this approach helps to predict future GWAS hits for SZ. We isolated two additional candidate genes for SZ, FOXO6 and SLITRK1. Our results show that EA-associated SNPs are much more likely to also be associated with SZ than expected by chance. However, these genetic loci do not influence both traits with a systematic sign pattern that would correspond to a strong positive or negative genetic correlation.
The results of our follow-up analyses are most consistent with two hypotheses that complement each other: First, the genetic overlap between EA and SZ is to some extent induced by pleiotropic effects of many genes that affect not only EA and SZ but also other traits such as BIP and IQ. Second, different syndromes of SZ (e.g., low cognitive performance and psychosis) seem to be driven by different genetic effects. The clinical diagnosis of SZ aggregates over these different syndromes. In particular, our results suggest that the current clinical diagnosis of SZ comprises at least two disease subtypes with nonidentical symptomatology and genetic architectures: One part resembles bipolar disorder (BIP) and high intelligence, while the other part is a cognitive disorder that is independent of BIP. Consistent with this idea, we find that PGS that take the sign concordance of SNPs with EA and SZ into account begin for the first time to predict specific SZ features from genetic data (R2 between 0.4% and 1.4%), while this was not possible with “ordinary” PGS for SZ.
Other mechanisms that we explored, in particular LD-patterns of the EA-associated SNPs and assortative mating, do not seem to be major drivers of the genetic overlap between EA and SZ. Furthermore, the loci we identified in our PPM analysis do not seem to be associated with all brain-related phenotypes, suggesting some degree of phenotype-specificity of the results. We note that the enrichment for age at menarche of the SNPs that are jointly associated with EA and SZ may be related to the final stage of brain development which coincides with the onset of puberty31–34.
The highly complex genetic architecture of the “schizophrenias” that our results point to implies that most patients will have individual-specific genetic loads for either subtype of the disease, contributing to individual differences in symptoms. The genetic heterogeneity we identified could imply that treatments will vary in their effectiveness across disease subtypes.
Overall, our study corroborates that EA is a useful proxy-phenotype for psychiatric outcomes. Specifically, combining GWAS results from EA and SZ led to the identification of two seemingly distinct subcategories of SZ. Even though each of them may still harbor highly heterogeneous disease subgroups, the new subcategories can pave the way for further biological subgroup analyses. Therefore, a psychiatric nosology that is based on biological causes rather than pure phenotypical classifications may be feasible in the future. Studies that combine well-powered GWAS from several diseases and from phenotypes that represent variation in the normal range such as EA are likely to play an important part in this development. However, deep phenotyping of large patient samples will be inevitable to link GWAS results from complex outcomes such as EA and SZ to specific biological disease subgroups.
AUTHOR CONTRIBUTIONS
P.D.K. designed and oversaw the study and conducted proxy-phenotype analyses. V.B. and M.M. carried out analyses in the GRAS sample. V.B. conducted bioinformatics and computed the LD-aware enrichment tests, which were developed by M.N. C.A.P.B. conducted simulation analyses. M.N. computed GWIS results and genetic correlations. R.K.L. assisted with biological annotation and visualization of results. P.D.K., V.B., M.M., and H.E made especially major contributions to writing and editing. All authors contributed to and critically reviewed the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no conflict of interests.
ONLINE METHODS
All reported statistical results are based on two-sided tests, unless indicated otherwise. Our proxy-phenotype analyses and our replication strategy followed a pre-registered analysis plan (https://osf.io/dnhfk/). The full description of all materials and methods is provided in the Supplementary Note.
GWAS on educational attainment
The EA sample excluded all cohorts that participated in the GWAS on SZ described below, yielding a sample size of n = 363,502 individuals of European descent13. The GRAS replication sample was not part of the GWAS on EA, either.
GWAS on schizophrenia
The SZ sample consisted of n = 34,409 cases and n = 45,670 controls, diagnosed with SZ or schizoaffective disorder5. We excluded the GRAS data collection from the GWAS on SZ.
Proxy-phenotype look-up
Analyses were carried out using 8,240,280 autosomal SNPs that passed quality controls in both GWAS and additional filters described in the Supplementary Note. We selected approximately independent lead SNPs from the EA GWAS results using the clumping procedure in PLINK35. We looked up the SZ results of all approximately independent EA lead-SNPs that passed the pre-defined significance threshold of PEA < 10−5.
We tested if the observed sign concordance between EA and SZ is different from 50% using the binomial probability test36. “Raw” enrichment factors and “raw” enrichment p-values of the EA lead-SNPs on SZ were calculated by taking the actual distribution of P values in the SZ GWAS result files into account but ignoring the LD scores12,37.
LD-aware enrichment across different traits
We developed an enrichment test that corrects for the LD score of each SNP (Supplementary Note). We conducted this test for the 132 SNPs that are jointly associated with EA and SZ in our proxy-phenotype analyses (PEA < 10−5 and PSZ < 0.05). LD scores were obtained from the HapMap 3 European reference panel. We investigated SZ and 21 additional traits for which GWAS results were available in the public domain. Some of the traits were chosen because they are phenotypically related to SZ (e.g., BIP), while others were less obviously related to SZ (e.g., age at menarche) or served as negative controls (e.g., fasting insulin). If one of the 132 candidate SNP was not available in the reference panel or the GWAS results of the other traits, we tried to use a good proxy, yielding 79 to 105 available SNPs per trait.
Phenotypic correlations
We explored the correlations between the number of years of education with 7 quantitative measures of SZ in the GRAS sample of SZ cases: Age at prodrome, age at disease onset, premorbid IQ, GAF, CGI-S, and PANSS positive and PANSS negative scores.
Replication and Bayesian credibility of results
Our replication uses a PGS in the GRAS data collection, which is based on the 132 independent EA lead-SNPs that are also nominally associated with SZ (PEA < 10−5 and PSZ < 0.05). This PGS (called SZ_132) was constructed using the regression coefficient estimates of the SZ GWAS as weights. In addition to this polygenic replication strategy, we further probed the credibility of our results using a heuristic Bayesian calculation.
Polygenic prediction of schizophrenia symptoms in the GRAS sample
We predicted the number of years of education and 7 quantitative measures of SZ in the GRAS sample of SZ cases. For each phenotype, we separately compared the predictive performance of several PGS: Scores constructed from the full GWAS result on SZ, EA, BIP, and neuroticism (called SZ_all, EA_all, BIP_all, Neuro_all, respectively); scores constructed using only the 132 SNPs that are jointly associated with EA and SZ (called EA_132 and SZ_132, using EA and SZ GWAS coefficients as weights, respectively); and two scores that split the SZ_all score into two parts based on sets of SNPs that either have concordant or discordant effects on EA and SZ (called Concordant and Discordant). Genetic outliers of self-reported non-European descent (n = 13 cases) were excluded from the analysis.
Controlling for the genetic overlap between schizophrenia and bipolar disorder
We estimated GWIS18 to obtain SNP regression coefficients that are unique to SZ, which are corrected for the genetic overlap between SZ and BIP. The SZ samples used in the GWIS are not overlapping with the samples used in the EA GWAS and they exclude our replication sample (GRAS). BIP GWAS results were obtained from the PGC38. We refer to the set of obtained summary statistics as “unique” SZ(min BIP). We then repeated the look-up of the EA-associated lead SNPs in those summary statistics as described above. Similarly, we obtained GWIS results for “unique” BIP(min SZ) using the same method and data. We computed genetic correlations of these GWIS results with EA, childhood intelligence, and neuroticism using bivariate LD score regression12 and compared the results to those obtained using ordinary SZ and BIP GWAS results.
Simulations of assortative mating
We conducted simulations to test if strong assortative mating on EA and SZ can induce a spurious genetic overlap between the two traits.
Biological annotation
To gain first insights into possible biological pathways that are indicated by the genetic loci identified by our PPM analysis, we applied DEPICT13,39 using a false discovery rate (FDR) threshold of ≤ 0.05. To identify independent biological groupings, we used the Affinity Propagation method based on the Pearson distance matrix for clustering40.
Data availability
GWAS meta-analysis results for EA and SZ as well as GWIS results for unique” SZ(min BIP) and “unique” BIP(min SZ) can be downloaded from the SSGAC website (https://www.thessgac.org/). For information about the GRAS data collection, contact the principal investigator of the study: Hannelore Ehrenreich (ehrenreich{at}em.mpg.de).
Code availability
Computer code used to generate LD-aware enrichment and GWIS results can be downloaded from the SSGAC website (https://www.thessgac.org/).
ACKNOWLEDGMENTS
This research was carried out under the auspices of the Social Science Genetic Association Consortium (SSGAC), including use of the UK Biobank Resource. We thank all research consortia that provide access to GWAS summary statistics in the public domain. Specifically, we acknowledge data access from the Psychiatric Genomics Consortium (PGC), the Genetic Investigation of ANthropometric Traits Consortium (GIANT), the International Inflammatory Bowel Disease Genetics Consortium (IIBDGC), the International Genomics of Alzheimer’s Project (IGAP), the CARDIoGRAMplusC4D Consortium, the Reproductive Genetics Consortium (ReproGen), the Tobacco and Genetics Consortium (TAG), the Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC), the ENIGMA Consortium, and the Childhood Intelligence Consortium (CHIC). We would like to thank the customers and employees of 23andMe for making this work possible as well as Joyce J. Tung, Nick. A. Furlotte, and David. A Hinds from the 23andMe research team. This study was supported by funding from an ERC Consolidator Grant (647648 EdGe, Philipp D Koellinger), the Max Planck Society, the Max Planck Förderstiftung, the DFG (CNMPB), EXTRABRAIN EU-FP7, the Niedersachsen-Research Network on Neuroinfectiology (N-RENNT), and EU-AIMS. Michel G Nivard was supported by Royal Netherlands Academy of Science Professor Award to Dorret I Boomsma (PAH/6635). Additional acknowledgements are provided in the Supplementary Online Materials.
Footnotes
↵13 These authors jointly directed this work.
↵a The actual N per SNP was not provided in the SZ GWAS summary statistics.
↵b It is typically assumed that GWAS data for European populations contain ≈1,000,000 independent loci. However, the quality-control procedures for GWAS summary statistics in studies like ours decreases the number of independent loci to <1,000,00041,62. In fact, clumping the post-QC GWAS results for SZ without a P value threshold, an R2LD<0.1, and a LD-window of 1,000,000 kb with the 1000 Genomes phase 1 version 3 European reference panel42 leads to only 223,065 independent loci. Thus, assuming 500,000 independent loci in these calculations is conservative.
↵c URL: https://www.ebi.ac.uk/gwas/api/search/downloads/full
REFERENCES




