

Abstract
As part of a broader collaborative network of exome sequencing studies, we developed a jointly called data set of 5,685 Ashkenazi Jewish exomes. We make publicly available a resource of site and allele frequencies, which should serve as a reference for medical genetics in the Ashkenazim. We estimate that 30% of protein-coding alleles present in the Ashkenazi Jewish population at frequencies greater than 0.2% are significantly more frequent (mean 7.6-fold) than their maximum frequency observed in other reference populations. Arising via a well-described founder effect, this catalog of enriched alleles can contribute to differences in genetic risk and overall prevalence of diseases between populations. As validation we document 151 AJ enriched protein-altering alleles that overlap with “pathogenic” ClinVar alleles, including those that account for 10-100 fold differences in prevalence between AJ and non-AJ populations of some rare diseases including Gaucher disease (GBA, p.Asn409Ser, 8-fold enrichment); Canavan disease (ASPA, p.Glu285Ala, 12-fold enrichment); and Tay-Sachs disease (HEXA, c.1421+1G>C, 27-fold enrichment; p.Tyr427IlefsTer5, 12-fold enrichment). We next sought to use this catalog, of well-established relevance to Mendelian disease, to explore Crohn’s disease, a common disease with an estimated two to four-fold excess prevalence in AJ. We specifically evaluate whether strong acting rare alleles, enriched by the same founder-effect, contribute excess genetic risk to Crohn’s disease in AJ, and find that ten rare genetic risk factors in NOD2 and LRRK2 are strongly enriched in AJ, including several novel contributing alleles, show evidence of association to CD. Independently, we find that genomewide common variant risk defined by GWAS shows a strong difference between AJ and non-AJ European control population samples (0.97 s.d. higher, p<10−16). Taken together, the results suggest coordinated selection in AJ population for higher CD risk alleles in general. The results and approach illustrate the value of exome sequencing data in case-control studies along with reference data sets like ExAC to pinpoint genetic variation that contributes to variable disease predisposition across populations.
Recent advances in genome sequencing technologies are improving our understanding of the etiology of human diseases1,2. Similarly, epidemiological studies over the past century have improved our understanding of their global distribution3–5. To date, it remains unclear the extent to which genetics may play a role in population-based differences in prevalence and/or incidence. Efforts to increase inclusion of a broader representation of populations in genomic studies will likely improve our interpretation of these observed differences6. Here, we present a study on the relative contribution of DNA sequence variants to the risk and prevalence of Crohn’s disease (CD) and rare diseases in the Ashkenazi Jewish (AJ) population.
Genetic population isolates, defined as populations that start with a small group of founders and that may experience bottlenecks altering with periods of population growth7, have facilitated the mapping of alleles contributing to human disease predisposition8–11. Tight bottlenecks scatter the relative contributions of genes and as a consequence may make it easier to discover some disease-associated genes, although other genes, where the alleles may have been depleted from the isolated population, will be harder to detect10.
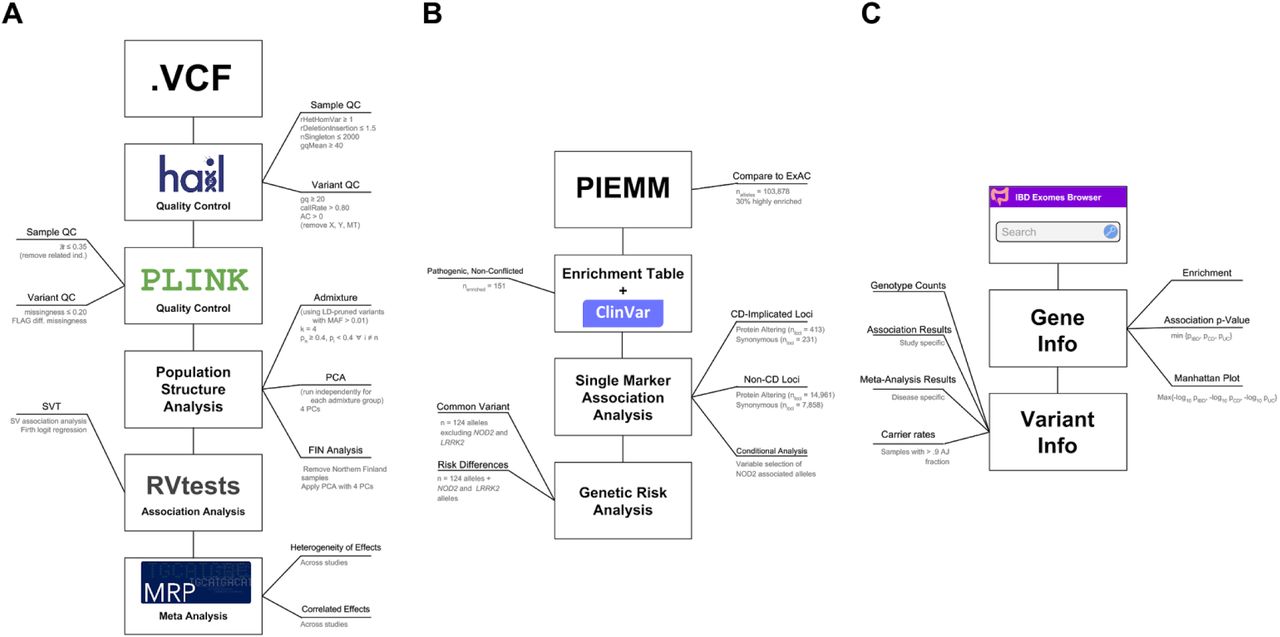
Genome-wide association studies (GWAS) and follow-up targeted sequencing efforts have been unusually successful in CD and have established a substantial role for low frequency variants across the more than 200 loci2,12 defined to date. In addition, the documented 2-4 fold enrichment of CD prevalence in the AJ population13,14, a population with an established founder effect, motivated the use of exome sequencing along with genome-wide array data to evaluate the degree to which bottleneck-enriched protein-altering alleles and unequivocally implicated common variants contribute an excess CD genetic risk to AJ, and as a consequence, to the documented increased prevalence of CD in AJ13 (Figure S1).

A) Quality control, population structure, and association analysis workflow. B) AJ specific analysis workflow. C) Results and summary statistics are uploaded to the IBD browser hosted at: http://ibd.broadinstitute.org - a website that contains a gene and variant search engine, a “Gene Info” landing page that contains a manhattan plot and additional summary statistics, and a detailed “Variant Info” page that contains additional information about the alleles identified in our exome sequencing studies.
Additionally, founder effects, and possible selection, have made some rare diseases more prevalent in the Ashkenazi Jewish (AJ) population15,16,17,18, akin to the well-documented 40 rare diseases known as “Finnish heritage diseases”, which are much more common in Finland9 and whose difference in prevalence has largely been attributed to genetics. Despite the remarkable progress in mapping genes and alleles for some of these rare diseases, precise estimates of the risk-allele frequency and the carrier rate in the AJ population isolate have not yet been determined16. Through this study we provide a frequency resource of protein-coding alleles from over 2,000 non-CD AJ samples with low admixture fraction that will serve to improve interpretation of the carrier rate of rare disease risk alleles in the AJ population (Figure S1).
Results
We generated a jointly called exome dataset consisting of 18,745 individuals from international IBD and non-IBD cohorts1,19. As the present study aimed to focus on variation observed in the AJ population in comparison to reference populations in ExAC19,20 (including non-Finnish Europeans (NFE), Latino (AMR), and African/African-American (AFR)) populations, we chose a model-based approach to estimate the ancestry of the study population using ADMIXTURE21.
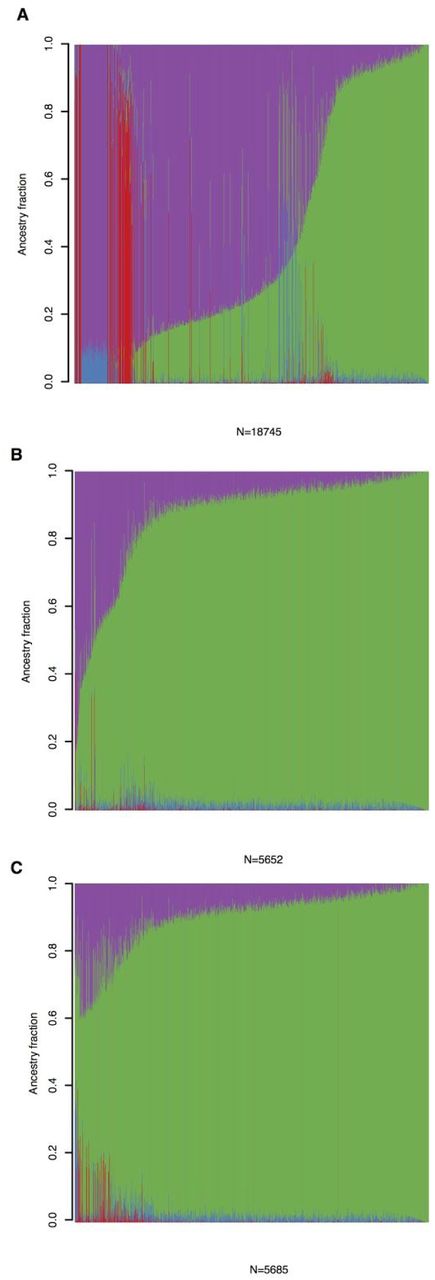
To identify AJ individuals and estimate admixture fractions we included a set (n=21,066) of LD-pruned common (MAF>1%, see Supplementary Note for additional details) variants after filtering for genotype quality (GQ>20). The 18,745 samples were assigned to four groups (K=4) using ADMIXTURE. In one of the four groups, 3,522 samples had estimated ancestry fraction > 0.9, with the majority of the samples labelled as “AJ” by contributing study sites (Figure S1). Because we were interested in computing an enrichment statistic that would not be affected by possible admixture we obtained alternate (non-reference) allele frequency estimates by restricting the enrichment analysis to the 2,178 non-IBD Ashkenazi Jewish samples that passed QC and relatedness filtering and had AJ focused ancestry fraction > 0.9 (Figure S1).
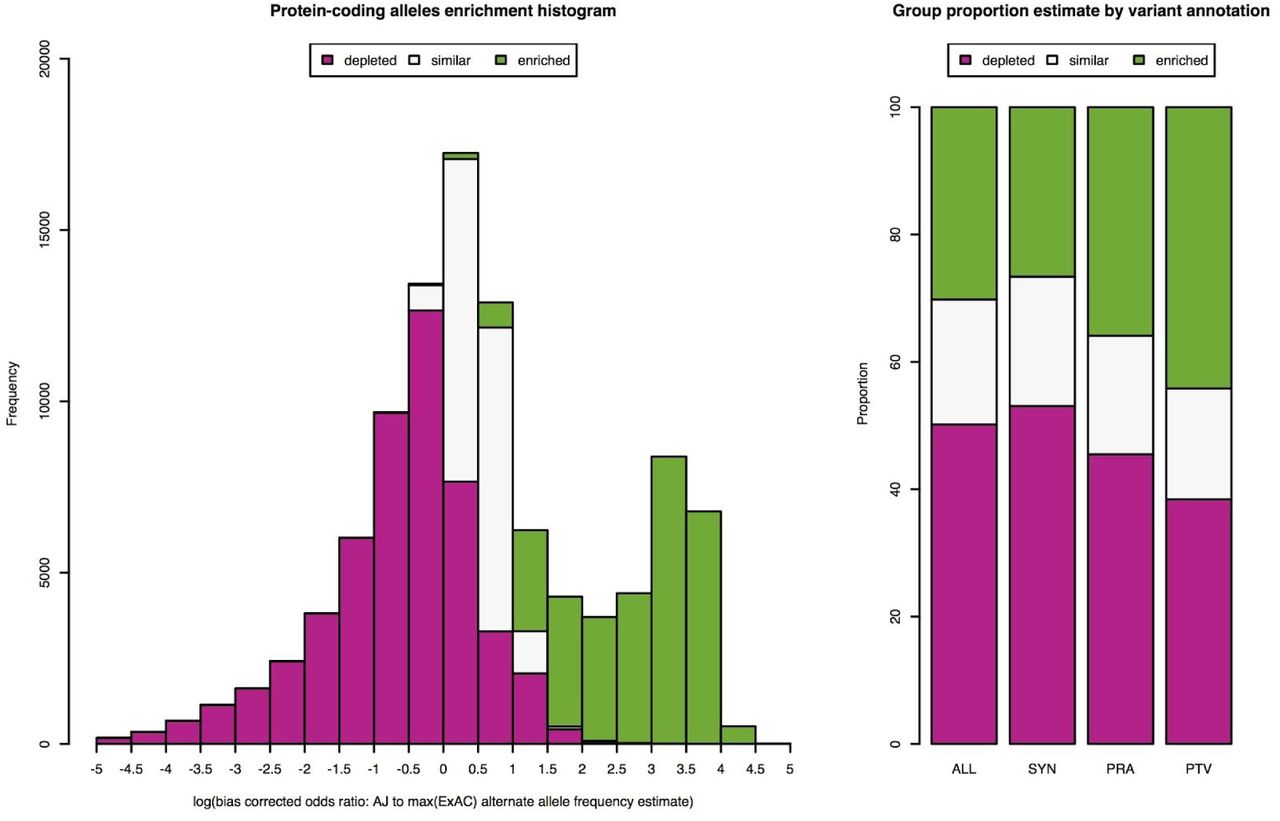
To estimate parameters of enrichment in the AJ population, including proportion of enriched alleles and degree of enrichment, we used the observed alternate allele counts and total number of alleles to take into account uncertainty in estimated allele frequencies from AJ and NFE (n=33,370), AFR (n=5,203), and AMR (n=5,789) available from ExAC release 0.3 dataset (ntotal=60,706). We focused on protein-coding alleles with estimated allele frequency of at least 0.002 in AJ (nalleles=103,878), and applied a three group Bayesian mixture model, we refer to as the Population Isolate Enrichment Mixture Model (PIEMM) (see Supplementary Note), to classify the observed alleles into three groups: “depleted”, “similar”, or “enriched”. We estimate that 30% of the analyzed protein-coding alleles have a mean 7.6-fold increased frequency compared to reference populations with markedly different proportion of alleles belonging to the “enriched” group depending on variant annotation: 44% for predicted protein-truncating variants (PTV); 36% for predicted protein-altering variants (PRA); and 27% for synonymous variants (Figure 1, Figure S2, p < 10−16 across comparisons of PTV and PRA to synonymous variants, two-proportion test, Supplementary Note).

A) Admixture plot, with 4 groups (K=4), for all samples exome sequenced. On the y-axis shown are ancestry mixture fractions for each of the samples (on x-axis) exome sequenced. B) Admixture plot for samples with self-reported Ashkenazi Jewish ancestry. C) Admixture plot for final samples used in the AJ analysis. All plots are ordered by ancestry fraction mostly loading for AJ samples (green). Group mostly loading for NFE samples is highlighted in magenta, East-Asian in red and African-American in blue.

A) Density plot of estimated log enrichment statistic, defined as the log of the bias corrected odds ratio comparing the allele frequency in AJ population to the maximum allele frequency estimated from NFE, AFR, and AMR populations in ExAC. For each histogram bin we show a barplot of the expected number of alleles belonging to the three different groups we analyzed: 1) “depleted” (magenta); 2) “similar” (light gray); and 3) “enriched” (green). B) Bar plots of estimated proportion of alleles belonging to the three different groups we analyzed for all protein-coding (ALL), synonymous (SYN), protein-altering (PRA), and protein-truncating variants (PTV). An estimate of 30% of protein-coding alleles observed in AJ with a mean shift of 7.6-fold increase in allele frequency compared to other reference populations.
As validation of our approach to identify alleles that contribute to differences in genetic risk to disease, we intersected the list of protein-coding alleles identified in the AJ exome sequencing study with reported pathogenic and non-conflicted alleles (n=42,226) in ClinVar22 resulting in 151 alleles found both in ClinVar and with posterior probability greater than 0.5 of belonging to the enriched group (Table S1). In OMIM, 49 of the 151 alleles included documentation of a disease subject with AJ ancestry (Table 1). This set of enriched alleles includes 9/14 alleles described in the American College of Medical Genetics and Genomics 2008 screening guideline study for the AJ population23. In the setting of autosomal recessive disorders these differences in population allele frequencies may contribute to an order of squared enrichment difference in genetic risk and prevalence between populations (see Supplementary Note). For instance, a 12-fold enriched frameshift indel, p.Tyr427IlefsTer5, in HEXA, contributes a 144-fold enrichment in genetic risk in AJ to non-AJ population to Tay-Sach’s disease. This large cohort of adult Ashkenazi exome database further supports recent publications of founder mutations for rare pediatric disorders including: FKTN and Walker Warburg syndrome24; CCDC65 and Primary ciliary dyskinesia25; TMEM216 and Joubert syndrome26; C11orf73 and Leukoencephalopathy27; PEX2 and Zellweger syndrome28; VPS11 and Hypomyelination and developmental delay29; and BBS2 and Bardet-Biedl syndrome30. This resource will undoubtedly assist in prioritizing variants for gene discovery to further identify founder mutations in AJ (Table S2).
HGVS and Gene is the allele nomenclature in ClinVAR and gene symbol. Enrichment corresponds to the comparison of allele frequency in AJ (AJ AF) to maximum frequency among three population groups (max EXAC AF): 1) NFE; 2) AMR; and 3) AFR. Curated trait is based on the trait description in the Online Mendelian Inheritance in Man (OMIM). Inheritance corresponds to the inheritance description in OMIM (AR: autosomal recessive, AD: autosomal dominant, risk factor: not specified genetic risk factor). Alleles are sorted in decreasing order by AJ AF.
To assess whether protein-coding alleles enriched in AJ population contribute to differences in CD genetic risk we performed case-control association analyses. Because individuals with partial AJ ancestry will still carry bottleneck-enriched alleles, here we included all samples with estimated AJ ancestry fraction of at least 0.4 (Figure S2), which resulted in a dataset of 4,899 AJ samples (1,855 Crohn’s disease and 3,044 non-IBD). To improve ability to detect association we performed a meta-analysis with CD and non-IBD case-control exome sequencing data from two separate ancestry groups: 1) non-Finnish European (NFE) (2,296 CD and 2,770 non-IBD); and 2) Finnish (FINN) (210 CD and 9,930 non-IBD samples) from a separate callset described in a previous publication31 for a total of 4,361 CD samples and 15,744 non-IBD samples.
Study-specific association analysis was performed with Firth bias-corrected logistic regression test32,33 and four principal components as covariates using the software package EPACTS34 (Figure S4). We combined association statistics in a meta-analysis framework using the Bayesian models in Band et al35. We used the correlated effects model, obtained a Bayes factor (BF) by comparing it with the null model where all the prior weight is on an effect size of zero, reported p-value approximation using the BF as a test statistic, and assessed whether heterogeneity of effects exist across studies for downstream QC (see Supplementary Note). We separately assessed CD observed vs. expected associations for enriched protein-altering (pra) and synonymous (syn) alleles in protein-coding genes in CD implicated GWAS loci (ngwas,pra=413; ngwas,syn=231), and outside implicated GWAS loci (nnon-gwas,pra=14,961; nnon-gwas,syn=7,858, Figure 2).

When applying MCMC algorithms to a data set it is customary to show the performance of the algorithm across all stages of the experiment (including the burn-in). We show that the PIEMM algorithm generates stable proportion estimates for the two groups: “enriched” (A-C) and “depleted” (D-F) for parameters: π (A, D), the proportion of alleles belonging to the group; μ (B, E), the shift of the distribution belonging to the group; and σ (C, F), the standard deviation of the distribution belonging to the corresponding group. For each group we demonstrate the parameter estimate during the burn-in (100 iterations, gray circles) and non burn-in (900 iterations, black circles) stage of the experiment used to obtain the parameter estimates reported in the manuscript.

Density plots for each of the PCs is shown (diagonal) separately for IBD cases (red) and controls (blue). Pairwise scatter plots are shown for PC1-PC4 separately for IBD cases and controls.

Q-Q plots of Crohn’s disease association for: A) AJ enriched protein-altering (protein-truncating and missense) and B) synonymous alleles in GWAS regions; and AJ enriched C) protein-altering and D) synonymous alleles outside of GWAS regions. For each Q-Q plot variants with a corresponding p-value less than or equal to a threshold where expected number of false discoveries is equal to one are annotated. The black dashed line is y = x, and the grey shapes show 95% confidence interval under the null. The gray dashed line represents the observed density of -log10 p-values.
We identified ten AJ enriched CD risk alleles (p<0.005): the previously published risk haplotypes in LRRK2 and NOD2 (LRRK2: p.N2081D; NOD2: p.N852S, p.G908R, p.M863V+fs1007insC)36,2, in addition to newly implicated alleles (NOD2: p.A612T, p=2.8x10−9; c.74-7T>A, p=1.4×10−4; p.L248R, p=6.4×10−4; p.D357A, p=0.0011; LRRK2: p.G2019S, p=0.0014, a Parkinson’s disease risk allele37). To assess whether the new NOD2 enriched alleles are conditionally independent of the previously established associated NOD2 alleles we performed conditional haplotype association analysis in PLINK and Bayesian model averaging38 for variable selection, both of which suggested independent effects for all alleles (Figure S5, Table S3).
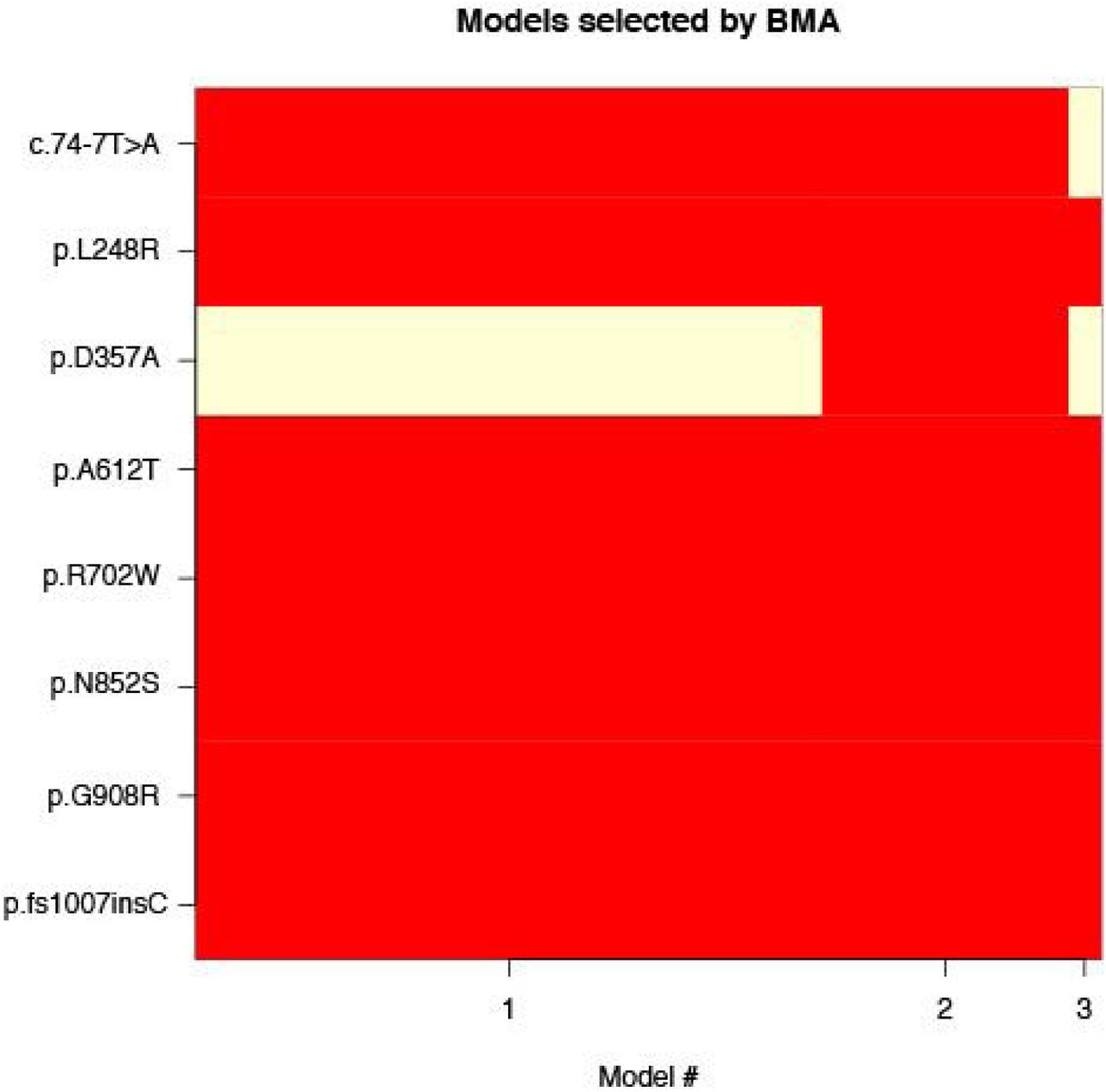
Eight protein-altering alleles with evidence of association and their corresponding membership for the models with high probability (> 0.01) after applying BMA, which accounts for the model uncertainty inherent in the variable selection problem by averaging over the best models in the model class according to approximate posterior model probability. Model #1 corresponds to the model where 7/8 alleles have a non-zero effect (p.D357A is not included) with approximate posterior model probability of .692. Model #2 corresponds to the model where 8/8 alleles have a non-zero effect with approximate posterior model probability of .273. Model # 3 corresponds to the model where 6/8 alleles have a non-zero effect (p.D357A and c.74-7T>A are not included) with approximate posterior model probability of .035.
HGVS nomenclature for each allele (HGVS) and corresponding p-values are shown for independent effects given the haplotype formed by residual variants.
Despite the functional relationship between LRRK2 and NOD239, we do not observe deviation from additivity between LRRK2 and NOD2 (p=0.273). Deviation from additivity has been reported for p.fs1007insC, p.G908R, and p.R702W in NOD240,41. We assessed whether any independent evidence of deviation from additivity exists for the newly associated single nucleotide substitutions. In agreement with previous reports, deviation from additivity existed with estimated genotype odds ratios of 1.84 for heterozygous and 7.39 for compound heterozygous/homozygous genotypes (p=0.0038, analysis of deviance, ANOVA), and found significant evidence of deviation from additivity for the newly reported alleles (p=0.00357, odds ratio = 7.53). We found no evidence of deviation from additivity for the associated protein-altering alleles in LRRK2 (p=0.418).
Given the presence of genetic variants in NOD2 and LRRK2 that contribute to differences in genetic risk in AJ population, we next asked whether unequivocally established common variant loci associations may also contribute to differences in genetic risk. We performed polygenic risk score (PRS) analysis using reported effect size estimates from 124 CD alleles including those reported in a previously published study12 and four variants in IL23R from a recent fine-mapping study42, and excluding variants in NOD2 and LRRK2. We observed an elevated PRS for AJ compared to non-Jewish controls (0.97 s.d. higher, p<10−16; Figure 3A; number of non-AJ controls=35,007; number of AJ controls=454). We observed a similar trend for the CD samples (0.54 s.d. higher; p<10−16; Figure 3B; number of non-AJ CD cases=20,652; number of AJ CD cases=1,938).

NJ: non-Jewish; AJ: Ashkenazi Jewish; CD: Crohn’s disease; PRS: polygenic risk score. A) Density plot of CD polygenic risk scores in 454 AJ (green) and 35,007 NJ (purple) controls. AJ controls have elevated CD polygenic risk score that NJ controls (0.97 s.d. higher, p<10−16). B) Density plot of CD polygenic risk scores in 1,938 AJ (green) and 20,652 NJ CD (purple) cases (0.54 s.d. higher, p<10−16). For both density plots the scores have been scaled to NJ controls, thus resulting in an NJ control PRS density of mean equal to 0 and variance equal to 1 (see Online methods). C) Ranked (decreasing order) CD associated variants by estimated contribution to the differences in genetic risk between AJ and NJ. Associated variants with estimated contribution greater than or equal to 0.01, computed as 2*log(odds ratio)*(AJ frequency - NJ frequency), assuming additive effects on the log scale, are highlighted in green. Associated variants with estimated contribution less than or equal to −0.01 are highlighted in purple. Forward slashes represent a break in variants highlighted.
To quantify the relative contribution of CD-implicated alleles to the difference in genetic risk between AJ and non-AJ populations we estimated the expected PRS value of an individual and expected difference in PRS between two populations by simply using summary statistics including the frequency of the minor allele in the two populations and the corresponding odds ratio (Supplementary note, Figures S6-S8).

A) Expected values of disease prevalence in non-AJ (black) and AJ (red) populations. The value of β0 = −20.5 was chosen to approximate the prevalence of disease in the non-AJ population at around 0.005. B) Relationship between expected prevalence ratio and choice of β0. The chosen value of β0 = −20.5 corresponds to a ratio of 1.506.

A) Probability distribution of prevalence in non-AJ (black) and AJ (red) populations given the calculated mean and variance of risk score given by the logit model. B) Equivalent probability distributions given the calculated mean and variance of the risk score in the probit model. C) A comparison of risk score values in logit and probit models. The green line corresponds to a linear fit for the entire shown region, and the red line corresponds to a linear fit for the linear range around zero.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Vertical line represents the estimated expected difference in genetic risk between AJ and non-AJ European population (0.397). For varying levels of expected difference in genetic risk we compute the expected fold-difference in prevalence. The shaded region marks the range of prevalence ratios obtained by varying β0 in a region of reasonable estimates (−24 to −20).
We applied the approach to all CD implicated alleles and observed that variants in GWAS loci annotated as IRGM, LACC1, NOD2, MST1, ATG16L1, GCKR, NKX2-3, and LRRK212 contribute substantially (>0.01) to the increased genetic risk observed in AJ. It is possibly relevant that variants contributing to increased risk in AJ correspond to autophagy/intracellular defense genes (IRGM, ATG16L1, LRRK2) and those contributing to increased risk in non-AJ correspond to anti-fungal/Th17/ILC3 genes43 (IL23R, IL12B, CARD9, TRAF3IP2, IL6ST, CEBPB; Figure 3C).
Two factors impact our ability to obtain precise estimates of the contribution of genetic risk factors to the documented differences in disease prevalence between populations. First, contemporaneous epidemiological data are largely unattainable. This confounds our ability to obtain any informative estimates due to documented variability in the occurrence of CD over time3,44. Second, there has been substantial uncertainty in reported CD prevalence estimates45,46. Here, to interpret the impact of shifts in genetic risk score on differences in prevalence, we used the logit risk model41 and evaluated a new estimate of disease probability, pnew, assuming an initial disease probability, p0, and multiple values for the differences in genetic risk. Assuming log-additive effects, and a log-risk model, we estimate that the observed differences in genetic risk between the AJ and non-AJ populations may contribute an expected 1.5-fold increase in disease prevalence in a population with environmental risk factors corresponding to AJ and baseline genetic risk corresponding to non-AJ populations (Figure S6-S8). To address the extent to which non-additive effects in NOD2 may impact the observed prevalence we assumed shared heterozygous and compound heterozygous/homozygous odds ratios of 1.84 and 7.39, respectively. We estimate a 1.6% increase in difference in prevalence attributed to the deviation from additivity, suggesting a small effect on differences in population prevalence (Supplementary Note).
Discussion
By drawing on data from 5,685 Ashkenazi Jewish exomes we provide a systematic analysis of AJ enriched protein-coding alleles, which may contribute to differences in genetic risk to CD as well as numerous other rare diseases. We identified protein-altering alleles in NOD2 and LRRK2 that are conditionally independent and contribute to the excess burden of CD in AJ. We found evidence that common variant risk defined by GWAS shows a strong elevated difference between AJ and non-AJ European population samples, independent of NOD2 and LRRK247, suggesting a coordinated selection in AJ for higher CD risk alleles48–50.
We made a couple of unique observations by studying CD in the AJ population. First, studying recently bottlenecked populations enables powerful discovery of genetic variation that markedly differ in frequency and as a consequence contributes to differences in genetic risk across population groups. Second, NOD2 and published common variant associations contribute substantially to the genetic risk of CD, making it more difficult to identify other genes whose ancestral alleles failed to pass through the bottleneck, consistent with predictions from Zuk et al10.
Finally, we provide an exome frequency resource of protein-coding alleles in AJ along with estimates of enrichment. Our approach and this resource will likely catalyze our understanding of the medical relevance of enriched alleles in population isolates.
Author contributions
M.A.R, D.P.B.M, M.J.D participated in the study design. M.A.R, J.K., M.K., and M.J.D analyzed whole exome data. M.A.R, H.H, T.H, and B.A analyzed the SNP chip data. B.M.N, A.G, D.G, B.G, I.P, G.A, N.B, A.P.L, E.S, N.P, Ben Weisburd, K.J.K, E.V.M, B.P, L.B, P.S, J.C, Graham Heap, T.A, V.P, A.W.S, S.T, Dan Turner, P.S, M.F, K.K, M.P, Aarno Palotie, S.R.B, D.G.M, R.H.D, Mark S. Silverberg, J.D.R, R.K.W, A.F, H.S, R.J.X, A.P, J.H.C, D.P.B.M provided reagents, methods, and tools for analysis. C.S. managed the project. C.J and J.H.C provided detailed analysis of rare diseases in AJ. All authors commented on the manuscript. J.D.R, Mark J. Daly, S.R.B, R.H.D, Mark S. Silverberg, J.H.C, and D.P.B.M are members of the NIDDK IBD Genetics consortium. Manuel A. Rivas, J.K., B.A, and Mark J Daly wrote the manuscript.
S2. Online resources
IBD Exomes Browser: http://ibd.broadinstitute.org
Clinvar table: https://github.com/macarthur-lab/clinvar/blob/master/output/clinvar.tsv
ExAC sites VCF: ftp://ftp.broadinstitute.org/pub/ExAC_release/release0.3/
Acknowledgments
M.J.D. is supported by grants from the following: the National Institute of Diabetes and Digestive and Kidney Disease (NIDDK) and the National Human Genome Research Institute (NHGRI; DK043351, DK064869 and HG005923); the Crohns and Colitis Foundation (3765); the Leona M. & Harry B. Helmsley Charitable Trust (2015PG-IBD001); the Stanley Center; and Amgen (2013583217). R.J.X. is supported by grants from Amgen (2013583217) and CCFA (3765). J.D.R. is funded by grants from NIDDK (DK064869 and DK062432). G.A. is supported by NIH R01 grant AG042188. N.B. is supported by NIH grants AG618381, AG021654, AG038072, and the Glenn Center for the Biology of Human Aging. A.F. and B.P. are supported by the DFG (Deutsche Forschungsgemeinschaft) Cluster of Excellence “Inflammation at Interfaces” and the DFG grant FR 2821/6-1. I.P. is supported by the Leona M. and Harry B. Helmsley Charitable Trust and New York Crohn’s Foundation. IBD Research at Cedars-Sinai is supported by grant PO1DK046763, and the Cedars-Sinai F. Widjaja Foundation Inflammatory Bowel and Immunobiology Research Institute Research Funds. D.P.B.M. is supported by DK062413, AI067068 and U54DE023789-01; grant 305479 from the European Union; and The Leona M. and Harry B. Helmsley Charitable Trust and the Crohn’s and Colitis Foundation of America. S.R.B. is support by an NIH U01 grant (DK062431). J.H.C. is supported by grants from NIH (U01 DK062429, U01 DK062422, R01 DK092235, SUCCESS), and the Sanford J. Grossman Charitable Trust. H.S. is supported by Equipe ATIP–Avenir 2012 grant and INSERM–ITMO SP 2013. E.V.M. is supported by an NIH F13 award (AI122592-01A1). Researchers at UCL are supported by the Charles Wolfson Foundation Trust. RKW is supported by a VIDI grant (016.136.308) from the Netherlands Organization for Scientific Research (NWO). R.H.D. is supported by NIH grant U01 DK062420 and the Inflammatory Bowel Disease Genetic Research Chair at the University of Pittsburgh. We thank Dr. Jonathan Bloom for proposed edits and comments on the manuscript. We thank the Broad IT team for assistance with the IBD exomes browser.
References




