

Abstract
Species achieve evolutionary innovations through two major genetic mechanisms, namely regulatory- and structural-level mutations. The ability of populations to evolve involves a balance between selection, genetic drift, epistasis, biochemical and biophysical requirements, thermodynamic properties and other factors. This adaptive diversity begs the question as to whether a restricted pathway governs adaptations or whether multiple pathways are possible to achieve an adaptive response. By combining a unique set of tools drawn from synthetic biology, evolutionary biology and genomics, we experimentally evolved and then characterized the adaptive properties of a modern E. coli strain containing a 700 million-year-old reconstructed ancestral Elongation Factor Tu (EF-Tu) gene inserted into its genome for the first time. We then tracked the evolutionary steps taken by the ancient-modern hybrid microorganism through laboratory evolution by monitoring genomic mutations. This study reveals that lineages respond to the ancient gene by increasing the expression levels of the maladapted protein, rather than through direct accumulation of mutations in the open reading frame. In particular, these findings show that the general strategy for the bacteria to adapt to the ancient protein is to accumulate mutations in the cis-regulatory region; gene-coding mutations appear to preclude rapid adaptation upon integration of the ancient gene for our system.
Author Summary Understanding the historical forces that have shaped the evolution of past organisms over time mainly relies on analyzing the behavior of organisms that exist today. This reconciliation requires an evolutionary framework that includes explicit functional links between genomes, natural selection, molecular innovation, phenotypic diversity and adaptation; yet, creating a framework that synthesizes all of these components remains a challenge. Here we make a novel attempt at such a synthesis by combining synthetic biology with natural selection to explore the historical constraints at work in evolutionary processes. In order to study historical pathways and the mechanisms of protein evolution in a complex cellular environment, we directly engineered a synthetic gene representing a 700 million-year-old ancestor of the contemporary elongation factor protein inside a modern E. coli strain. We then traced the evolutionary steps of the microorganism harboring this ancient gene by subjecting it to laboratory evolution, directly monitoring any resulting changes within the integrated ancient gene and the rest of the host genome through whole-genome sequencing. Our results demonstrate that an ancient gene can interact with modern cellular machinery, albeit with a cost of decreased fitness, and that lineages respond to the ancient gene by increasing the transcription levels of the maladapted protein. Further development of ancient-modern hybrid model systems has the potential to provide information about fundamental evolutionary processes at work in modern microbes.
Introduction
In the context of Darwinian evolutionary theory, species that persist through time must retain a mechanism that provides functional adaptation in a changing environment. Two major mechanisms orchestrate functional innovation at the genetic level. In the first, mutations accumulating in the non-coding regions influence gene expression, leading to phenotypic change (1-5). In the second, mutational changes in gene coding regions generate structural alteration of proteins and thus lead to functional diversity (6, 7). Whether one mechanism is favored over the other is the subject of ongoing debate in the field of evolutionary biology (8-10). The primary argument against a substantive role for coding mutation-mediated evolution is the assumption that mutations in protein-coding genes have extensive detrimental pleiotropic and fitness effects (11, 12). While there is no doubt that mutations altering protein expression level play an important role in shaping phenotypic diversity, failure to recognize the role of protein level mutations as a source of biological innovation may hamper a comprehensive understanding of phenotypic variation (13-18).
In this study, we describe a novel laboratory system that allows us to experimentally evolve Escherichia coli (E. coli) bacteria in a manner that appoints regulatory vs. structural mutations for a reconstructed 700 million-year-old Elongation Factor Tu protein (EF-Tu) (S1 Fig). Known as one of the most sequence-conserved proteins in life, EF-Tu constitutes an essential component of the bacterial translation machinery by delivering aminoacylated-tRNAs to the A-site of the ribosome (19). Our experimental system exploits the unique scenario in which E. coli bacteria have a paralogous copy of the EF-Tu gene tufA, in the form of tufB, that frequently recombines with the original copy (20). Each of the EF-Tu genes have their own specific expression machinery, while EF-Tu produced through tufB accounts for one-third of the cellular EF-Tu as that produced by the tufA gene in bacteria. Their respective encoded proteins differ from one another by a single amino acid in the C-terminus (21-23). The EF-Tu produced by these two genes form 5-10% of the total protein produced in E. coli, signifying EF-Tu is a highly abundant protein in the bacterial cell (24). As such, EF-Tu promoters are routinely used to drive recombinant protein expression since they are constitutively active (25).
In our system, we replaced the tufB copy of a laboratory strain of E. coli with an ancient EF-Tu variant under the control of the endogenous tufB promoter, and we completely removed the tufA gene from the bacterial genome (S1 Fig). We hypothesized that a reconstructed ancient protein will be maladapted in a modern genome and that there were two adaptive responses to laboratory evolution. One possible adaptive route would accumulate mutations that alter the encoded protein’s structure and function in a manner that recapitulates the historical pathway by which the modern gene evolved from its ancient ancestor. Alternatively, the second route may accumulate mutations in the “weaker” promoter region of tufB where the ancient gene is engineered, thus altering the expression of the ancient gene in the modern cell. We further investigated whether in vivo analyses into the functionality of ancestral components can be used to discern effects arising from the substituted gene when screened from adaptive responses taken by the host cell to the sub-adapted genetic component (26).
The engineered EF-Tu represents an ancestral γ-proteobacterium that is inferred to be approximately 700 million-years-old, and has 21 (out of 392) amino acid differences with the modern E. coli EF-Tu protein (S2 and S3 Figs). An in vitro peptide synthesis assay demonstrates that resurrected EF-Tu can participate in a translation system in which all other necessary components for translation besides EF-Tu are provided by modern E. coli, albeit with a lower efficiency than the wild-type (27). The temperature profile of the ancient EF-Tu protein is 39.1 °C, slightly higher than the temperature optimum of the endogenous E. coli EF-Tu at 37 °C (28). To examine the adaptation between the ancestral EF-Tu protein and contemporary bacteria, we engineered the ancient gene into the modern bacterial genome. We then evolved the hybrid bacterial populations for 2,000 generations in the laboratory in multiple independent parallel lines, and examined the adaptive response through fitness, whole-genome sequencing, proteomics and biochemical assays.
Results
Rapid fitness improvement of the ancient-modern hybrid bacterial populations
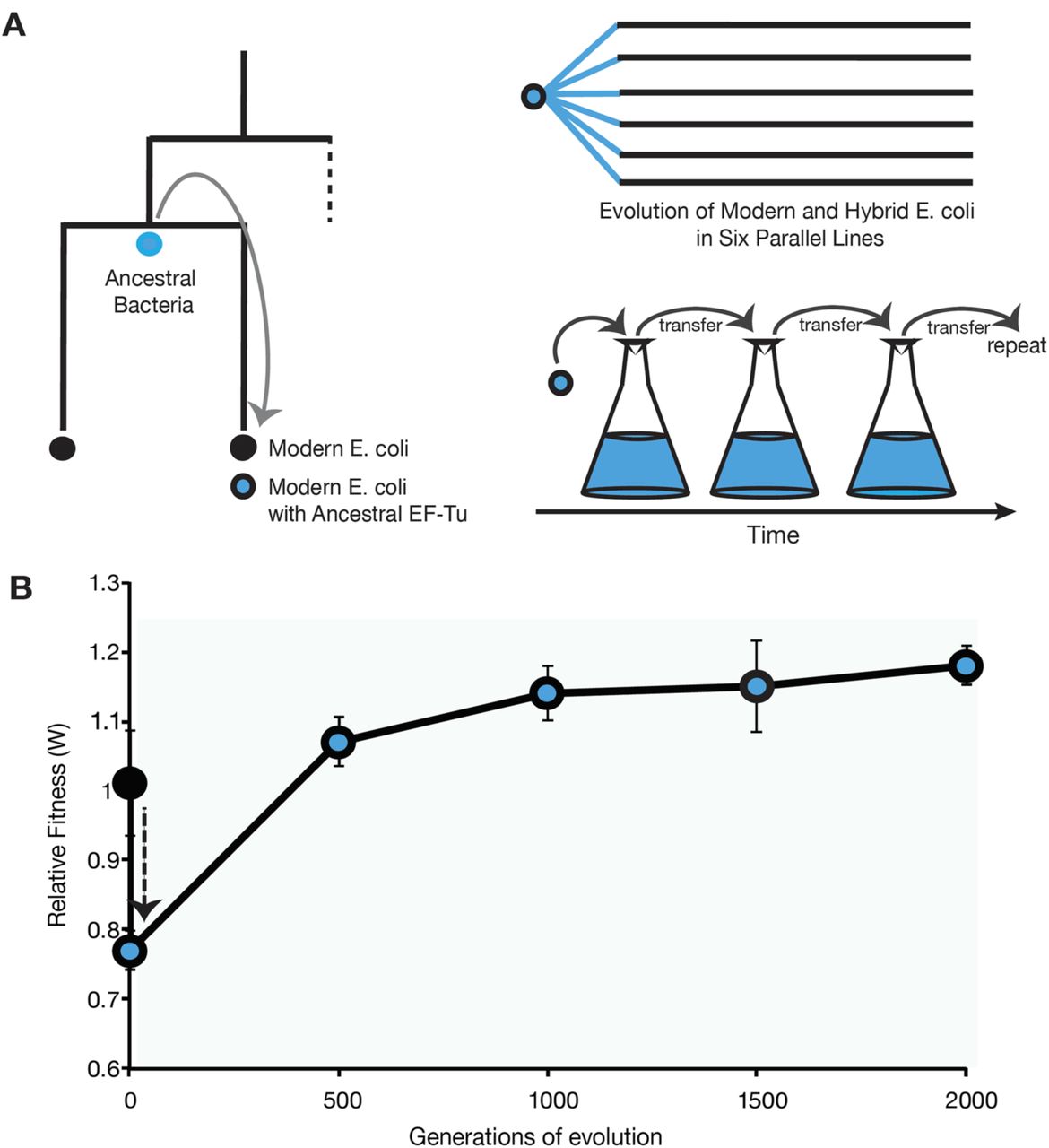
Engineering E. coli harboring a reconstructed ancestral EF-Tu gene results in two-fold increase in bacterial growth doubling-time and 20% decrease in bacterial fitness relative to the founding strain E. coli REL606 (Fig 1, S4 Fig). Adaptation to selective laboratory media by the ancient-modern hybrid populations is shown by the 20% increase in fitness values and a rapid drop in growth doubling-time compared to that of native E. coli bacteria by 500 generations. The fitness continues to increase by ~5% every 500 generations until stabilizing by generation 1500 (Fig 1, S4 Fig).

(A) Engineering a modern E. coli strain with an ancient EF-Tu gene followed by laboratory evolution of initially identical six ancient-modern hybrid populations in parallel. (B) Replacement of the endogenous EF-Tu gene with the reconstructed ancient EF-Tu allele significantly reduces the fitness of the ancient-modern hybrid relative to the original strain (black circle), indicated by the black dashed arrow. Hybrid population mean fitness rapidly improved during experimental evolution in minimal glucose medium (blue circles). Error bars show 95 % confidence interval among six replicate populations.
Whole Genome Sequencing reveals adaptation of EF-Tu’s promoter
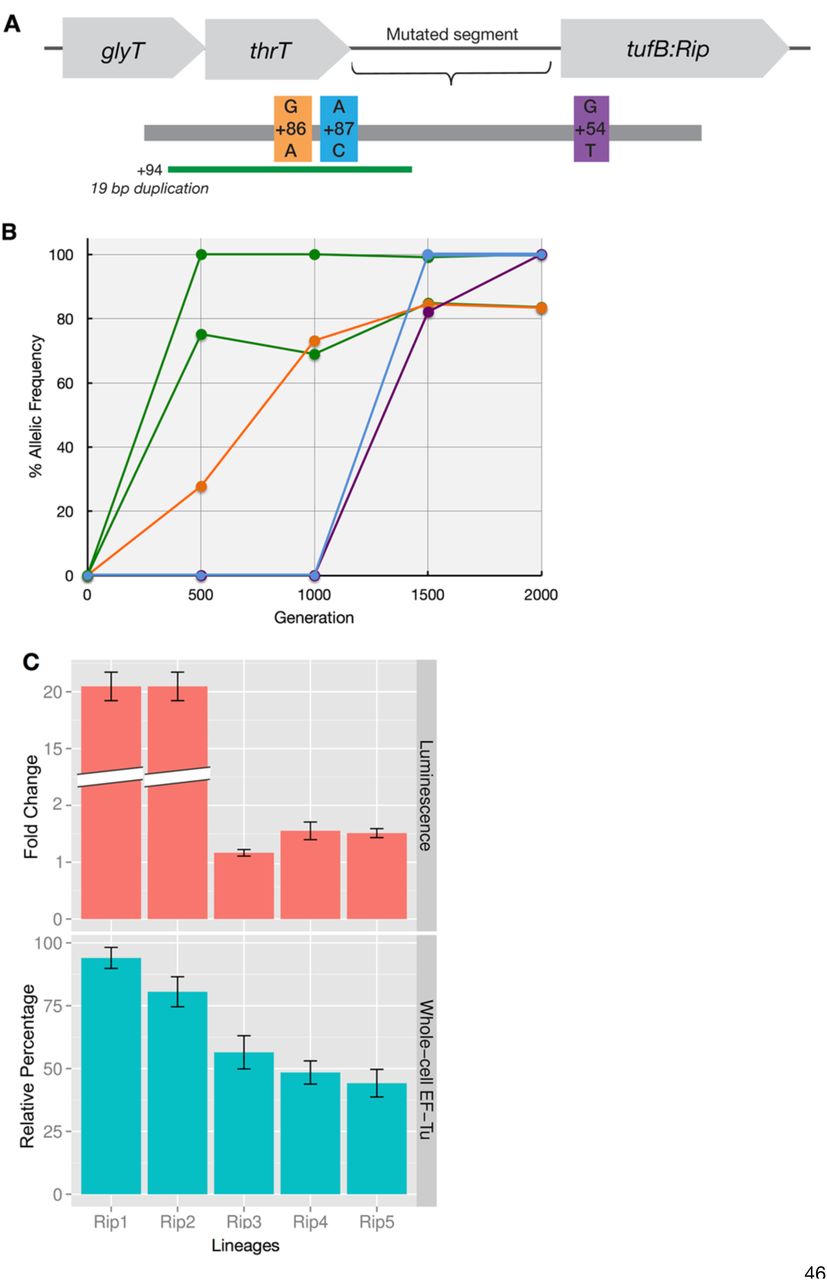
Whole genome sequencing of evolved modern (as a control) and ancient-modern hybrid E. coli populations was performed periodically throughout the evolutionary course and revealed that the accumulation of parallel mutations in a variety of genes across the populations (Table 1). In particular, five out of six independent lineages have accumulated mutations in thrT/tufB region, corresponding to a promoter region for tufA gene (29); however no mutation on the ancestral EF-Tu gene in any of the six lineages is observed. Shown in Fig 2 are the frequencies of the allelic mutations located in ancient EF-Tu gene’s promoter region, demonstrating that a majority of the mutations accumulated early during the course of the experiment.
Genes that accumulated synonymous mutations in at least three out of six laboratory evolved ancient-hybrid populations, and that achieved a frequency of at least 20% between generations 500 and 2000 in the population. thrT/tufB is the intergenic region between the ancient EF-Tu gene and the thrT gene. The bottom three listed genes were specific to the single lineage in which no mutation in the thrT/tufB region was detected (referred as Rip6). Genes highlighted with an asterisk accumulated mutations in at least one population containing the wild-type EF-Tu gene (control experiment).
In vivo and in vitro experiments indicate mutations upregulate ancient EF-Tu expression
Whole-cell quantitation of EF-Tu protein levels through shotgun proteomics shows that upon insertion, ancestral EF-Tu produced by E. coli decreases by 70% compared to the native strain and increases in most of the lineages upon accumulation of mutations in the thrT/tufB region during laboratory evolution (Fig 2, S5 Fig). In order to assess the impact of mutations on expression levels, we individually cloned the evolved promoters into a plasmid expressing a reporter gene, whose fluorescent strength is directly linked to the strength of the evolved promoter region. Results demonstrate that mutations in the promoter region amplify the protein expression levels to varying degrees, ranging from 1.5-20 fold higher (Fig 2). Both in vivo and in vitro data assign an upregulatory role for these promoter-level mutations. However, the magnitude detected in vitro does not directly correlate with the values observed through LC/MS-MS measurements via whole-cell proteomics that suggest an enhancement of 1-2 fold for the mutations. This is expected, however, since the promoter assay is enzymatically-based and thus amplifies signal. Mutations that have lower impact on ancient EF-Tu protein expression relative to a 19 basepair duplication in the thrT/tufB region all appear later in the evolutionary experiment. This suggests that, to a first approximation, each population exhibits parallel patterns in response to the ancient EF-Tu (i.e., upregulation of the EF-Tu gene) but that each population may exhibit other unique mechanisms in response to the ancestral gene.
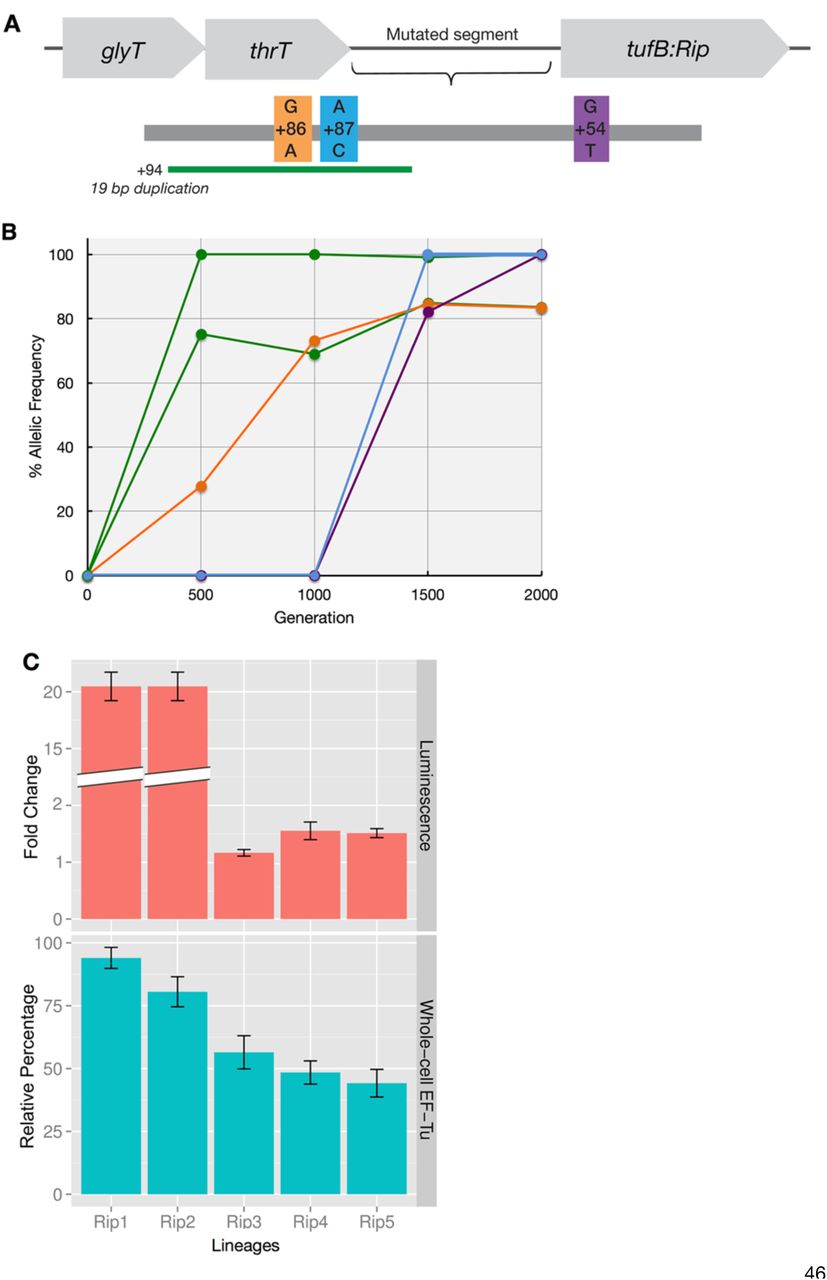
(A) The thrT/tufB promoter region in which five of six evolved hybrid populations were found to have accumulated mutations. (B) The allelic frequency of the intergenic mutations per generation per population during laboratory evolution. (C) (Top) Effect of mutations on the expression of a reporter gene with the EF-Tu promoter region measured using luciferase. (Bottom) Relative abundance of ancient EF-Tu protein among evolved hybrid strains using the peak area quantification from MS proteomics data (30, 31). Error bars obtained using Anova/t-test.
Intriguingly, none of the modern lineages evolved in parallel to the ancient-modern populations exhibited mutations in the tufB promoter region thr/tufB or on the tufB gene. When over-expressed from a plasmid, ancient EF-Tu protein increases the fitness of the unevolved hybrid strain by 10% relative to wild-type E. coli. The 20% decrease of the organismal fitness upon insertion of the ancestral EF-Tu, and the 10% increase of the unevolved ancestral-modern hybrid strain upon overexpression of the ancient EF-Tu suggests that background mutations also play a role in an organism’s ability to adapt to selective media; the fitness increase is dependent, albeit not entirely, on the ancient EF-Tu’s upregulation in evolved lineages. The total number of synonymous mutations accumulated in each lineage throughout the course of laboratory evolution is shown in S6 Figure.
Ancient EF-Tu strains no longer require a protein interaction partner to maintain fitness
Amongst the evolved six lineages, one lineage (Rip6) did not accumulate mutations in the thrT/tufB region. This lineage contains some mutations that are in parallel with the other lineages but also contains various mutations that are exclusive, namely ribosomal initiation factor protein IF2 (infB) and the transcriptional regulation protein NusA (Table 1). The nusA gene, which is known to be transcribed by the same operon as infB (32) and autoregulates infB (33), acquires a 27 base-pair deletion near the C-terminal end of the protein, mutant NusA protein nusAΔ9 (S Table 1). These two mutations appear by Generation 500 of this lineage, each with an abundance of over 90% in the population.
Upon deletion of its native nusA gene, modern E. coli experiences a statistically significant fitness decrease of 8%. In contrast to E. coli, chromosomal deletion of nusA and nusAΔ9 introduces no significant fitness change to the unevolved hybrid bacterial strains, nor to the evolved hybrid Rip6 harboring the mutant allele, respectively. In vitro protein binding assays performed through Isothermal Calorimetry measurements indicate that modern EF-Tu and NusA proteins interact with a robust binding constant (Kd) of 14.6 ± 5.2 μM whereas ancient EF-Tu binds to NusA protein two-fold weaker (Fig 3, S7 Fig). On the other hand, nusAΔ9 and ancient EF-Tu exhibit a Kd of 680 ± 66 μM, suggesting a complete loss of the interaction caused by at least twenty-fold lower binding to ancient EF-Tu. The interaction between EF-Tu and NusA had no observable effect on dipeptide formation in the ribosome. Additionally, our EF-Tu hydrolysis assays using an EF-Tu H84 variant (34, 35) indicate that NusA does not bind to 70S ribosomes (S8 Fig). Overall these data suggest that the interaction between EF-Tu and NusA does not take place in the ribosome, further, upon replacement of the modern EF-Tu with the ancient counterpart, EF-Tu-NusA interaction is lost and nusA accepts mutations that are otherwise deleterious.

{kind=link}
{kind=link}
{kind=link}
(Left) Bacterial constructs with nusA knockouts are constructed and competed against the native E. coli bacteria for fitness measurement. (Right) The interaction between the native EF-Tu, ancient EF-Tu and NusA variants are measured via in vitro Isothermal Calorimetry binding assays.
Discussion
The ability to design synthetic genes and engineer genomes has provided new methods to characterize phenotypic states and the responses of biological systems to perturbations (36-39). One such method involves replacing native genes with homologous substitutes to investigate the gene and protein functionality of a cell (40). In some cases, however, this approach is limited by divergence between host and donor lineages, which can result in adverse phenotypic responses that preclude production of functional hybrids suitable for study. Substituting the gene sequences of ancestors may lessen these adverse impacts. Moreover, new insights into the functionality of ancestral components may be possible when effects arising from the substituted sequence are screened for functional innovation in artificial or cellular systems (41-50).
In this work we described a novel experimental system that combines ancient gene reconstruction with experimental evolution and provided molecular insight into organismal-level adaptation to an ancient gene. We focused on the highly-expressed essential protein EF-Tu, and replaced its native gene with an inferred Precambrian ancestral counterpart. Alteration of contemporary EF-Tu with its ancestor resulted in a drastic decrease in cellular fitness, suggesting that the ancient gene is compatible, albeit maladapted to native bacterial cell conditions. We speculate that this observation is primarily due to the suboptimal function of the ancestral EF-Tu protein in the ribosomal machinery, as demonstrated by previous in vitro biochemical assays (27) and also due to the decreased protein levels upon replacement of the modern EF-Tu with its ancestral homolog (S5 Fig). The reconstructed ancient EF-Tu used in this study is codon optimized to function in E. coli; however, subtle nucleotide changes may still be affecting its in vivo expression levels once the gene is transcribed from the genome under the native E. coli promoter. For instance, deletion of tufB gene from the E. coli genome is known to decrease the EF-Tu abundance by 15% (24). The correlation between the cellular concentration of EF-Tu and organismal fitness (51) provides further evidence of the intertwined role between the level of gene expression and rates of cell growth (52, 53). Indeed, over-expression of ancient EF-Tu protein through an inducible plasmid in the unevolved bacterial ancient-modern hybrid resulted in 10% fitness increase, suggesting that the cellular concentration of the EF-Tu protein influences organismal fitness.
A rapid fitness increase in populations subjected to experimental evolution demonstrates the fitness constraint introduced on the system by the ancient EF-Tu. Genome sequence analyses reveal that evolved ancient-modern hybrids generate a complex array of cellular responses. A majority of the evolved lineages accumulate mutations in the promoter location of the ancient tuf gene, which leads to an increase in the ancient EF-Tu levels throughout these populations. This result assigns the accumulation of cis-regulatory mutations as one of the main mechanisms in adaptation for our laboratory generated system, contrary to what others have observed in studies, where direct accumulation of mutations were repeatedly observed on genes engineered in homologous microbes in laboratory evolution experiments (54, 55). No mutation is observed on the ancient EF-Tu gene-coding region in any of our evolved lineages.
Understanding the lack of direct accumulation of mutations on the ancient EF-Tu requires a consideration of various possibilities on the fitness effects of the potentially contributing mutations. A suggested background mutation rate of ca. 1×10−10 to 5×10−9 per basepair per generation in E. coli (56) corresponding to a mutation rate of ca. 1.8×101 mutations per EF-Tu per generation and ca. 3.6×104 per EF-Tu over the 2000 generations of laboratory evolution. This suggests that every site in the EF-Tu protein has been mutated at least once during the course of our laboratory evolution experiment. Various mechanisms can explain the lack of direct accumulation of non-synonymous mutations on the EF-Tu gene-coding region. The mutations experienced by EF-Tu were detrimental to its essential function in the translation machinery, leading to a non-viable organism. Indeed, considering the important role of EF-Tu in the cell’s translational machinery, as well as its role as a hub in a protein-protein interaction network, mutations accumulating directly on the EF-Tu gene can cause cell lethality and thus may not be readily adaptive (45, 57, 58). Relatedly, essential genes evolve slowly (59); increasing the cellular protein level may represent an emergency response from the organism to cope with a drastic alteration introduced by a maladapted protein central to translation machinery. Second, the genetic integration of the suboptimal EF-Tu may have introduced a rugged adaptive landscape on the genome during the experimental evolution, prohibiting EF-Tu from accumulating a favorable amino acid, beyond the ancestral sequence. Such behavior may suggest a key role for extensive epistasis between EF-Tu’s intergenic interactions that acts as a barrier between the combined deleterious effects, and the intermediate states of EF-Tu en route to adaptive solutions (60-64).
Intriguingly, EF-Tu gene amplification was not observed during the course of the laboratory evolution despite the fact that two genes encode for EF-Tu protein in the contemporary E. coli, a condition specific to γ-gammaproteobacterial organisms (65). It is possible such amplification could be observed in the furtherance of laboratory generations, or it is likely that the mutations that accumulated in the promoter regions override the need to have an additional copy of EF-Tu. This is an interesting outcome, given that not all the mutations lead to a final EF-Tu concentration in the cell that is equal to the modern E. coli EF-Tu concentration. Various mutations that accumulated in the promoter region converged on increasing protein expression levels, including a few mutations that evolved multiple times independently (such as the 19bp duplication in the +94/-21 thrT/tufB region in the Rip1 strain), indicating that mutations altering the expression may be more likely and therefore easier to implement in laboratory timescales of observation. It is important to note that the predominance of cis-regulatory mutations in our system does not diminish the obvious importance of structural mutations in all cases, but rather indicates that the former class of mutations can make a more extensive contribution on short timescales of adaptation than the latter, particularly for highly-conserved essential hub proteins that have many interaction partners.
One remaining ancient-modern hybrid evolved lineage, Rip6, deviates from the pattern observed in the majority of the evolved lineages because it did not accumulate mutations in the cis-regulatory region. Interestingly, Rip6 maintains an increase in fitness despite no significant change in the EF-Tu protein levels. Our experiments focused on the direct measurement of suppressor mutations specific to this lineage, particularly nusA (accumulating a 27 basepair deletion in its C-terminal, which we refer to as nusAΔ and potential biochemical relationship between these mutations/proteins and EF-Tu activity as well as overall bacterial fitness. Gene knockout and fitness studies show that the deletion of the nusA and nusAΔ9 genes does not alter the fitness of the unevolved ancient-modern or evolved Rip6 strains, respectively. On the other hand, deletion of nusA from the modern E. coli strain decreases its fitness by 8%. Could this decrease be related to perturbations of an interaction between NusA and EF-Tu proteins upon integration of the ancient EF-Tu? Isothermal Calorimetry binding measurements show that ancient EF-Tu protein binds to native NusA protein two-fold weaker than the modern EF-Tu, therefore substitution of the EF-Tu with an ancestral variant diminishes the interaction with NusA. Further, the suppressor mutation accumulated on the nusA gene in Rip6 strain does not recover the binding between the NusA and the ancient EF-Tu protein. These mutations are likely instead be a symptom of NusA no longer being functional in the context of an ancient EF-Tu. In addition, one can speculate the lack of interaction between NusA and EF-Tu is important for the adaptation of the ancient EF-Tu in the modern cell, if, especially, the interaction between EF-Tu and NusA proteins did not exist 700 million years ago. To test this hypothesis, we phylogenetically reconstructed an ancient NusA protein representing the ancestor of γ-proteobacteria, and measured the biochemical interaction between the reconstructed ancient nusA and the reconstructed ancient EF-Tu. Our Isothermal Calorimetry measurements did not indicate a detectable interaction between these two ancient proteins, supporting our hypothesis that an ancient interaction between NusA and EF-Tu did not exist. Is NusA-EF-Tu interaction important for EF-Tu’s function in the translation machinery? The dipeptide formation assay exhibits no detectible functional role for the NusA-EF-Tu interaction in the ribosome (S7 and S8 Figs). We speculate that upon replacing the endogenous protein, a secondary function for EF-Tu beyond its known functions with the ribosome is altering the strain’s fitness. More extensive experiments are required to examine and isolate EF-Tu’s alternative roles in the cell to fully explain the observed impacts on cellular fitness.
and potential biochemical relationship between these mutations/proteins and EF-Tu activity as well as overall bacterial fitness. Gene knockout and fitness studies show that the deletion of the nusA and nusAΔ9 genes does not alter the fitness of the unevolved ancient-modern or evolved Rip6 strains, respectively. On the other hand, deletion of nusA from the modern E. coli strain decreases its fitness by 8%. Could this decrease be related to perturbations of an interaction between NusA and EF-Tu proteins upon integration of the ancient EF-Tu? Isothermal Calorimetry binding measurements show that ancient EF-Tu protein binds to native NusA protein two-fold weaker than the modern EF-Tu, therefore substitution of the EF-Tu with an ancestral variant diminishes the interaction with NusA. Further, the suppressor mutation accumulated on the nusA gene in Rip6 strain does not recover the binding between the NusA and the ancient EF-Tu protein. These mutations are likely instead be a symptom of NusA no longer being functional in the context of an ancient EF-Tu. In addition, one can speculate the lack of interaction between NusA and EF-Tu is important for the adaptation of the ancient EF-Tu in the modern cell, if, especially, the interaction between EF-Tu and NusA proteins did not exist 700 million years ago. To test this hypothesis, we phylogenetically reconstructed an ancient NusA protein representing the ancestor of γ-proteobacteria, and measured the biochemical interaction between the reconstructed ancient nusA and the reconstructed ancient EF-Tu. Our Isothermal Calorimetry measurements did not indicate a detectable interaction between these two ancient proteins, supporting our hypothesis that an ancient interaction between NusA and EF-Tu did not exist. Is NusA-EF-Tu interaction important for EF-Tu’s function in the translation machinery? The dipeptide formation assay exhibits no detectible functional role for the NusA-EF-Tu interaction in the ribosome (S7 and S8 Figs). We speculate that upon replacing the endogenous protein, a secondary function for EF-Tu beyond its known functions with the ribosome is altering the strain’s fitness. More extensive experiments are required to examine and isolate EF-Tu’s alternative roles in the cell to fully explain the observed impacts on cellular fitness.
Taken together, these findings are indicative of a stringent fine-tuning by the evolved microbe towards the ancient EF-Tu. In addition to decreased EF-Tu protein levels, and the diminished capacity of ancient EF-Tu to participate in the translation system, a third factor to explain the fitness decrease in bacteria is likely to be the perturbation of EF-Tu’s interactions with other proteins in the cell. A detailed assessment to dissect the responses that are triggered due to the difference in function of the ancient component and responses due to the laboratory evolution process is necessary. One way to address this would be to use strains that have already undergone adaptation to the laboratory evolution environment and then engineer the bacterial genome with the ancestral variants. This additional step could also allow accumulation of mutations that arise due to the adaptation to the media before the foreign gene is introduced, specifically the identified intragenic mutations (such as pykF and mrdA genes) that were observed in several other laboratory evolution studies using E. coli as well as in our experimental system (66-68). Further, a detailed assessment of genetic level responses is clearly a prerequisite to quantifying the overall impact of the substituted ancestral gene. Such an improvement in genome engineering design would facilitate performing a genome-wide assessment to separate the mutations triggered by the ancient genotype/phenotype from the mutations that are simply due to the adaptation to the laboratory environment.
Engineering native genomes with ancient genes was recently identified as a challenging experimental approach due to the functional incompatibility of some ancestral genes in modern organisms (42). The substitution of a 700 million-year-old gene into a modern host demonstrates that, despite large changes in the microenvironment, some portion of the gene sequence topography for the host E. coli spanning this time interval (and possibly farther into the past) may continue to include functional variants of modern genes that are not yet found in extant organisms.
On the basis of the data obtained from substitution with the ancestral EF-Tu, is it possible to reveal or retrace the actual historical mechanisms or pathways followed in nature by the various lineages used to construct the ancestral EF-Tu sequences? The short answer appears to be no, since there were no mutations that accumulated on the ancestral gene itself. All mutations appear to relate to the function of the host cell in response to a suboptimal cellular component. Nevertheless, the observed outcome for this particular gene in this particular host demonstrates the possibility that insightful historical information may be accessible through this method. The most consistent drivers of historic mutational change may be macroscopic variables that cannot be readily incorporated into laboratory-scale synthetic evolution experiments, such as the myriad influences of ancient ecological and environmental perturbations induced by novel innovations (69-71). The EF-Tu protein phenotype, though tightly coupled to the optimal growth temperature of its host organism (28), is not likely to have exhibited any changes within its host bacteria over the last 700 million years of its evolution that may in turn be coupled to any other externally measurable changes to the larger biological or geological system. Nevertheless, the system points to the possibility of uncovering, in a relative sense, how recent some protein-protein interactions (such as those between EF-Tu and NusA) might be. Future studies that focus on a particular macroscopic driver, in which this driver is an integral part of the laboratory evolution experimental design, may yield insights into how evolutionary trajectories may have functioned in the past.
Conclusion
Engineering bacterial genomes with phylogenetically reconstructed genes complements the current technique of genome level alterations of gene and gene clusters with extant homologs, and provides insights into molecular mechanisms of adaptation by accessing historical states of currently existing proteins. The synthetic system described here provides insights into adaptation of foreign genes in bacterial genomes, particularly the role of regulatory vs. structural mutations in adaptive evolution. We designed a method that introduced an ancient variant that is optimal in some ways (presumably essential primary functions such as tRNA binding and/or ribosome binding), and suboptimal in ways that evolved later in the course of evolution (binding to ancillary proteins or fine-tuning epistatic interactions with tRNAs and ribosomes). Our results show that lineages respond to the ancient gene by increasing the expression levels of the maladapted protein, rather than through direct accumulation of mutations on the ancestral gene. We expect our method to uniquely reveal those components and represent a model system for a new area of the design and study of functional synthetic genes in modern microbial systems.
Methods
Bacterial Strains and Culture Conditions
All experiments were done at 37 °C unless stated otherwise. Luria-Broth (LB) media was used as base media for liquid cultures and agar plates, Davis minimal media (DM) was used as base media for experimental evolution and competition assays (supplied with 25 mg/mL of glucose as the sole carbon source), tetrazolium arabinose (TA) agar plates were used as a base media for competition experiments. When required, LB and DM media was supplemented with kanamycin, chloramphenicol and tetracycline antibiotics. All dilutions took place in sterile saline solution. All LB and DM cultures were incubated on a rotary shaker at 200 rpm and 150 rpm, respectively. The gene encoding the ancestral EF-Tu was codon optimized for expression in E. coli, chemically synthesized by DNA 2.0 and cloned into a pET15b plasmid as reported previously (72).
Construction of the ancient-modern hybrid strain
Integration of the ancient EF-Tu gene (Rip) into the chromosome of E. coli strain REL606 (73) was carried out via the λ-red homology recombineering approach as described by Datsenko and Wanner (74). First, linear DNA containing homology sequences of upstream and downstream of tufA gene was amplified by PCR, via (5’ GTGGTTGCGAAAATCATCGCTAGAATTCCGGGGATCCGTCGACC 3’ and 5’ TGTAATTAGCCCAGAACTTTAGCAACTGTAGGCTGGAGCTGCTTCG 3’), and pKD13 plasmid as template, and then transferred in REL606 cells through electrophoration, together with the temperature sensitive pKD46 plasmid. Recombinants were isolated from LB agar plates containing 50 μg/μL kanamycin at 37 °C, grown in in liquid LB medium containing 50 μg/μL Kanamycin and their genomic DNA was isolated using Promega Wizard Genomic DNA Purification Kit. Confirmation PCR was performed using genomic DNA isolated from colonies as a template, with primers aligning to the chromosome outside of the recombination site (5’ CAGGCCGTAATTGAAGCCCGTGGTAAATAAGCC 3’) and (5’ GAATAATTTATTCGTTCTGACAGTACGAATAAG 3’). Once the successful replacement of tufA gene with the kanamycin marker was confirmed via sanger sequencing, the strain was transformed with linear DNA containing homology sequences of upstream and downstream tufB flanking the ancient EF-Tu DNA construct SOE-d to a chloramphenicol marker originally amplified from the A007 loxP-Cm-loxP plasmid (Gene Bridges GmbH) via Gibson Assembly. The transformants were selected on LB plates containing 25 μg/μL chloramphenicol and 50 μg/μL kanamycin at 37 °C and the correct insert was screened with primers aligning to the chromosome outside of the recombination site using Fwd primer 5’ TCCGTGTCTTAGAGGGACAATCGATG 3’and Rev primer 5’ GCAATTAGCTCAGAACTTTTGCTAC 3’. Once confirmed, both the kanamycin and the chloramphenicol markers were removed using pCP20 and 706-Cre plasmids (Gene Bridges GmbH), respectively, followed by the confirmation of the deletions by genomic PCR analysis. Plasmids pKD46 and pCP20 were cured by growing the cultures at 42 °C, the final ΔtufA, ΔtufB:Rip construct was moved into a fresh ancestral strain via bacteriophage P1 transduction. Freezer stocks of the REL606 ΔtufA, ΔtufB:Rip were prepared by mixing 50% sterile glycerol and overnight liquid cultures originated from a single colony, in 1:2 ratios. All stocks were stored in −80 °C. Isogenic Ara+ variants of the REL606 ΔtufA, ΔtufB:Rip were obtained through gene-gorging protocol (75)(plasmid pJEB12 is kindly donated by Jeff Barrick).
Deletion of the nusA gene from the bacterial chromosome
The nusA gene from the chromosome of REL606, ancestral REL606 ΔtufA, ΔtufB:Rip and evolved REL606 ΔtufA, ΔtufB:Rip strain from lineage Rip2 were replaced with a FRT-kan-FRT fragment in the presence of pKD46 helper plasmid as described by Datsenko and Wanner using primers 5’ TCCTGCGTGAAGATATGCTG 3’ and 5’ TCACTTCTTCGCCGATTTCT 3’. PCR amplification of the recombination region and sanger sequencing of this amplified region confirmed the correct replacement and the removal of the selection cassette. The cassette was then removed from the chromosome via pCP20, followed by the curation of pKD46 and pCP20 plasmids at 42 °C.
Growth assays
Saturated overnight cultures were preconditioned by dilution into sterile saline by a 1:100, then again by 1:100 into the DM media, followed by an overnight growth. Preconditioned cultures were diluted 1:100 into the assay medium and 100 μL of this culture was transferred into a 96-well microplate. OD readings were taken at 420 nm every 15 minutes with continuous shaking between readings.
Experimental Evolution
Experimental evolution took place through serial transfers in DM medium supplemented with glucose as carbon source, for 2000 generations (~6.6 generations per day) as described previously (76). Relative fitness was measured using standard Lenski evolution competition assays every 500 generations via competing the evolved populations and selected strains against the wild-type E. coli strain REL606 or REL607, and calculated the fitness based on the Malthusian parameter [m=(cdx* f^x)/cd0, where cd0 = count of the competitor on day 0, and cdx = count of the competitor on day x, f = growth of the population over time (x-0). In our minimal media competitions, f = 100 because our transfers involve 100-fold dilution.
Fitness measurement of the ancestral strain in the presence of over-expressed EF-Tu
Ancient EF-Tu was cloned into a pASK-IBA43 (IBA Life Sciences) vector inducible under a tetracycline promoter using the following primers Forward 5’ GTTGGAATTCATGTCTAAAGAAAAGTTTGAACGTAC 3’ and Reverse 5’ CGGGATCCTCAAGCGATGATTTTCGCAACCAC 3’, between the Xho and Nde sites leading to plasmid pASK-IBA43 harboring the ancient EF-Tu gene. Ligation was confirmed using Forward primer 5’ GAGTTATTTTACCACTCCCT 3’ and Reverse primer 5’ CGCAGTAGCGGTAAACG 3’. The plasmid was transferred to REL606 ΔtufA, ΔtufB:Rip cells via electroporation and selected on LB agar plate with chloramphenicol. Five representative colonies were picked, preconditioned in LB media containing 250 μM anhydrous tetracycline for 24 hours, followed by a 1:100 dilution into DM media containing glucose. Over-expression of the EF-Tu protein was confirmed through SDS-PAGE gel analysis in comparison to ancestral cells that harbored no plasmid and non-induced plasmid. A REL607 strain was acclimated to the competition environment by separate grown under the same environmental conditions as REL606 ΔtufA, ΔtufB:Rip harboring pASK-IBA43 with the ancient EF-Tu gene. The competitors were then mixed in equal stoichiometric ratios by diluting into fresh DM medium with glucose containing 250 μM anhydrous tetracycline. Samples were plated on tetrazolium arabinose agar plate every 4 hours during the 24-hour competition. The competitions were carried out two times to ensure the precision of fitness estimates.
Whole genome sequencing
Whole genome sequencing was completed for 2000 generations for eight lineages harboring ancient EF-Tu, as well as the wild-type strains. To prepare the sequencing library, we isolated 3 mg of genomic DNA from bacteria grown in 10 mL LB overnight, fragmented and tagged the isolated DNA with specific Illumina adapters using Nextera DNA sample preparation kit. We purified the product using Zymo DNA Clean and Concentrator Kit, dual-indexed the libraries with TruSeq Dual Indexed Sequencing primer sets and ensured the products were pure using an Agilent 2100 BioAnalyzer. We combined the sets of compatible barcodes (11plex) into a single lane on Illumina HiSeq 2500 Rapid Run flow cell (v1) after QC. Sequencing was in a paired end 2 x 100 basepair format (PE100) using TruSeq Rapid SBS reagents. The Breseq (0.23) software was used for the generation and the analysis of the mutations (77).
Luciferase Assay
TufB and pBBRlux plasmid cloning
The wild-type and mutant (evolved) promoter region of tufB gene (EF-Tu protein) was cloned into the pBBRlux plasmid as adapted from Lenz et al. (78). Phusion High-Fidelity DNA polymerase, dNTPs, restriction enzymes (high fidelity), and T4 ligases were all obtained from New England Biolabs. DNA purification materials were purchased from QIAGEN. Promoters were amplified using PCR primers 5’-CAGAATGAAAATCAGGTAGCCGAGTTCCAG-3’ and 5’-TAGTGATTGCAGCGGTCAGCGTTGTTTTAC-3’ and resulted in a 403 basepair product from REL606 E. coli in the 4155251-4155654 region of the genome. Restriction sites were subsequently added to the ends of the tufB promoter with the following primers: Forward 5’-GATACTAGTCAGAATGAAAATCAGGTAGCCGAGTTCCAG-3’ and Reverse 5’-TATGGATCCTAGTGATTGCAGCGGTCAGCGTTG TTTTAC-3’ (underlying restriction sites correspond to SpeI and BamHI, respectively). The EF-Tu promoter was cloned upstream of the luciferase operon in the pBBRlux plasmid in order to drive transcription. pBBRlux provides chloramphenicol (CMP) resistance.
Scintillation Counts
Four experimental constructs: 19 basepair duplication, +94/-21 (Rip and Rip2), +87/-28 (A+87C) (Rip3), +54/-61 (G+54T) (Rip4) and +86/-29 (G+86A) (Rip5) and two control constructs: P (no promoter), PAtuf (wild-type, or unevolved ancestor, tufB promoter) were transformed into chemically competent E. coli (REL606) cells and incubated at 37 °C for 24 hours on chloramphenicol (CMP) agar plates. A single colony was cultured in LB media containing CMP at 37 °C for 24 hours. A 100 μL aliquot of the overnight culture was diluted one thousand-fold prior to being transferred into a 50 mL Erlenmeyer flask containing 9.9 mL of DM25 media. Cells were grown for ~8.25 hours, or ~5 doublings as monitored by plating (this represents the end of log growth since these cultures reach stationary phase after ~6.6 generations in DM25) and then pelleted. The supernatant was aspirated until 100 μL of media remained, and the pellet was then resuspended in the remaining 100 μL. Scintillation counting was used to quantify the amount of light signal generated by the luciferase pathway. For all six constructs, three readings per sample were averaged for each of the two replicates assayed.
Bacterial Enumeration
For each construct, a 10μL aliquot was serially diluted 50 thousand-fold and 50 μL was plated on agar petri dishes containing CMP. Extrapolation was utilized to determine the total amount of cells in each scintillation assay. Three plates per flask were averaged.
Luciferase Assay Statistical Analysis
The luciferase expression per cell was normalized by:

Luciferase expression for each construct was subtracted by the amount of luciferase signal from P to eliminate any leaky expression from the pBBRlux vector without promoter and presented as fold-change relative to the amount of luciferase signal from Patuf. A one-way ANOVA with α = 0.05 and a post-hoc Tukey HSD Test were performed against Patuf to determine significant differences.
Protein biochemistry
Cloning, expression and purification of modern EF-Tu and ancient EF-Tu proteins
Both of the EF-Tu genes were ligated into pET15b plasmid between BamH1/EcoR1 sites, containing an N-term His-Tag with ampicillin-resistance. For expression, the plasmids were transferred in a BL21(DE3) strain, the cells were grown in LB media until OD600 reading reached 0.6-0.8 and then induced with 1 mM IPTG for 4 hours at 37 °C. The cells were lysed using Bugbuster protein extraction reagent (EMD Millipore) containing benzonase. For purification of the His-tagged protein from the supernatant, the cell debris and membrane fractions were spun at high speed prior to adding onto nitrilotriacetic acid (Ni-NTA) resin gravity-flow columns (Qiagen, Hilden, Germany) at 4 °C that was pre-equilibriated with lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8). The Ni-NTA gravity-flow column was washed twice with lysis buffer containing 20 mM imidazole. His-tagged protein was eluted using elution buffer (50 mM NaH2PO4, 300 mM NaCl, 200 mM imidazole, pH 8).
Cloning, expression and purification of NusA proteins
Both the wild-type nusA and the evolved nusA genes were amplified from their host bacterial genome using Forward primer 5’-GTGAAGGTGTCGACGCTGCGTGCGCT-3’ and Reverse primer 5’-AGCGCACGCAGCGTCGACACCTTCAC-3’. The amplified DNA was purified through gel extraction, removed from salt and then cloned into a pET15b vector (Novagen) using 5’ GGCGACATATGAACAAAGAAATTTTGGC 3’ and 5’ GGAGCTCGAGTTACGCTTCGTCACCGA 3’ primers in between BamH1 and XhoI sites. The plasmids were transferred into a BL21(DE3) strain for expression and induced by IPTG. Cells were lysed by French Press in Buffer A (20 mM Tris-HCl at pH 7.5, 50 mM MgCl2, 200 mM NaCl, 5% glycerol). 25 μM GDP was added in Buffer A for EF-Tu purification. After centrifugation for 30 min at 16,000 rpm (F21-8x50 rotor, Thermo), the supernatant was applied to Ni-NTA column and elute with gradient Buffer B (Buffer A supplied with 500 mM imidazole). To prepare EF-Tu and NusA for Isothermal Calorimetry experiment, the proteins were dialyzed in Buffer C (20 mM Tris-HCl at pH 7.5, 50 mM MgCl2, 100 mM KCl) for 16 hours at 4°C.
Isothermal Calorimety Analysis
The Isothermal Calorimetry data was measured on a Microcal ITC200 System (GE Healthcare). The syringe was loaded with 42 μL of 0.6-1 mM NusA and the sample cell was filled with 10uM EF-Tu. NusA protein was titrated (2.5 μL for each) into EF-Tu with 120 s intervals and the first injection was 0.25 μ L. The stirring speed was set at 1000 rpm. Control blank experiment was performed by titrating NusA protein into Buffer C (20 mM Tris-HCl at pH 7.5, 50 mM MgCl2, 100 mM KCl).
LC-MS/MS Analysis
Sample preparation
Whole cell lysate was generated from each ancestral and evolved strains using Bug Buster reagent (EMD Millipore) and following manufacturer’s instructions. Total protein was quantified via BCA assay using Pierce BCA protein assay kit (Thermo Fisher Scientific). 30 μg of whole cell lysate was submitted to the Proteomics and Metabolomics Facility at Colorado State University. Samples were processed for in-solution trypsin digestion as previously described (79). Briefly, protein was precipitated out of solution in the presence of 4 volumes of 100% −20°C acetone and then resolubilized in 8M urea, 0.2% ProteaseMAXtm surfactant trypsin enhancer (Promega, Madison, WI). Samples were reduced and alkylated with 5mM dithiothreitol and 5mM iodoacetamide. Trypsin (MS Grade, Thermo Pierce, San Jose, CA) was added at an enzyme to substrate ratio of 1:50 and incubated at 37 °C for 3-hours. Trypsin was deactivated with the addition of 5% trifluoroacetic acid and desalted using C18 OMIX tips (Agilent Technologies, Santa Clara, CA) using manufacturer’s instructions. Peptide eluate was dried in a vacuum evaporator and resuspended in 3% acetonitrile/0.1% formic acid at a concentration of approximately 1 μg/μL. Relative Quantitation of EF-Tu proteins were carried out using spectral counting approach. Approximately 2 μg of tryptic digest for each sample was injected using an EASY nanoLC-II system (Thermo Scientific, San Jose, CA). Peptides were purified and concentrated using an on-line enrichment column (EASY-Column, 100 μm ID x 2cm ReproSil-Pur C18). Subsequent chromatographic separation was performed on a reverse phase nanospray column EASY-Column, 3 μm, 75 μm ID x 100mm ReproSil-Pur C18) using a 180-minute linear gradient from 10%-55% buffer B (100% ACN, 0.1% formic acid) at a flow rate of 400 nanoliters/min. Peptides were eluted directly into the mass spectrometer (Thermo Scientific Orbitrap Velos). The instrument was operated in Orbitrap-LTQ mode where precursor measurements were acquired in the Orbitrap (60,000 resolution) and MS/MS spectra (top 20) were acquired in the LTQ ion trap with normalized collision energy of 35%. Mass spectra were collected over a m/z range of 400-2000 Da using a dynamic exclusion limit of 2 MS/MS spectra of a given peptide mass for 30 s (exclusion duration of 90 s). Compound lists of the resulting spectra were generated using Xcalibur 2.2 software (Thermo Scientific) with a S/N threshold of 1.5 and 1 scan/group.
Data Analysis - Spectral Counting
Database searching Tandem mass spectra were extracted, charge state deconvoluted and deisotoped by ProteoWizard version 3.0. All MS/MS samples were analyzed using Mascot (Matrix Science, London, UK; version 2.3.02). Mascot was set up to search the Uniprot_e_coli_custom_reverse database (Updated August 2014, 8750 entries) (80) assuming the digestion enzyme trypsin, allowing up to 3 missed cleavages. Mascot was searched with a fragment ion mass tolerance of 0.80 Da and a parent ion tolerance of 20 PPM. Oxidation of methionine M (+15.99) and carbamidomethyl of cysteine C (+57) were specified in Mascot as variable modifications. Scaffold (version Scaffold_4.3.4, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 69.0% probability to achieve an FDR less than 0.1% by the Scaffold Local FDR algorithm. Protein identifications were accepted if they could be established at greater than 99.0% probability to achieve an FDR less than 1.0% and contained at least 2 identified peptides (81, 82). Protein probabilities were assigned by the Protein Prophet algorithm (83). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Binary comparisons were created in separate Scaffold files comparing wild-type E. coli REL606 and unevolved ancestor harboring the ancient protein and the evolved lineages tested (biological replicates n=3) to Strain/Treatment Group (each n=3). Biological samples were organized into Categories based on strain type. Each Category had 3 biological replicates. Normalization of spectral counts was not applied based on these criteria: An equal amount of sample from each replicate was loaded into the mass spectrometer and there was no deviation in processing and the number of spectra between samples is closely similar (% CV < 5% between biological replicates). Spectral counting uses the sum of the MS/MS spectra assigned to each protein as a measure of abundance (84). A t-test was performed on Total Spectral Counts for each MS sample using the embedded algorithm in Scaffold v 4.3.4. Proteins with P-values less than 0.05 are excluded in calculation of fold changes compared to E. coli REL606.
Acknowledgements
We gratefully acknowledge Neerja Hajela for providing the E. coli REL606 strain, members of the Rich Lenski laboratory for providing feedback during the earlier phase of this work, Deepak Unni for his assistance during the Breseq analyses, Jennifer Zhang for her assistance in the laboratory, Istem Fer for her assistance with R, and Brian Hammer for providing the pBBRlux plasmid. Zachary Blount and Seth Childers provided helpful discussion and comments on this manuscript.
References
- 1.↵
- 2.
- 3.
- 4.
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.
- 15.
- 16.
- 17.
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.
- 38.
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.
- 44.
- 45.↵
- 46.
- 47.
- 48.
- 49.
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.
- 62.
- 63.
- 64.↵
- 65.↵
- 66.↵
- 67.
- 68.↵
- 69.↵
- 70.
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵




