

Abstract
Sex chromosomes play a prominent role in development and evolution and have several characteristic features that distinguish them from autosomes. Across diverse taxa, recombination is typically suppressed at the sex-determining region (SDR) and proportionally elevated in the remainder of the chromosome or pseudoautosomal region (PAR). However, in most model taxa the sex chromosomes are ancient and highly differentiated from autosomes, and thus little is known about recombination dynamics of homomorphic sex chromosomes with incipient sex-determining mechanisms. Here we examine male function (pollen production) and female function (fruit production) in crosses of the dioecious octoploid strawberry Fragaria chiloensis in order to map the small and recently evolved SDR controlling both traits and to examine recombination patterns on the young ZW chromosome. The SDR occurs in a narrow 280kb window, in which the maternal recombination rate is lower than in the orthologous paternal region and the genome-wide average rate, but within the range of autosomal rate variation. In contrast to the SDR, the ZW recombination rate in the PAR is much higher than the rates of the ZZ or autosomal linkage groups, substantially overcompensating for the SDR rate. By extensively sequencing sections of the SDR vicinity in several crosses and unrelated plants, we show that W-specific divergence is elevated within a portion of the SDR and find only a single SNP to be in high linkage disequilibrium with sex, suggesting that any W-specific haplotype protected from recombination is not large. We hypothesize that selection for recombination suppression within the small SDR may be weak, but that fluctuating sex ratios could favor elevated recombination in the PAR to remove deleterious mutation on the W. Thus these results illuminate the recombination dynamics of a nascent sex chromosome with a modestly diverged SDR, which could be typical of other dioecious plants.
Author Summary In many species, the sex-determining chromosomes have distinct features relative to the rest of the genome, including unusual recombination rates. The wild strawberry Fragaria chiloensis possesses a recently evolved sex chromosome that illuminates the early stages of sex chromosome differentiation and adjustment of recombination rates. We examined the phenotypes of both male function (pollen production) and female function (fruit production) among hundreds of strawberry offspring resulting from several crosses, which allowed us to identify a narrow chromosomal region influencing both of these traits. We then examined and compared maternal and paternal recombination rates in the vicinity of this sex-determining region and all along the ZW sex chromosome. A region of suppressed recombination surrounding the sex gene(s) is relatively small, but substantial differences in recombination rate among the parents are seen on the sex chromosome outside of this region. These results help us to understand how sex-determining regions appear, are maintained, and impact the rest of the genome.
Introduction
Sex chromosomes are genomic oddities, often with distinctive inheritance patterns, effective population sizes, recombination frequencies, and structural arrangements, leading to unique evolutionary dynamics [1], [2]. These unusual features give sex chromosomes a key role in numerous biological phenomena such as speciation [3–5], disease progression [6], whole-genome duplications [7],[8], and contribution to the genetics of complex traits including sexual dimorphism [9–12]. Sex determination mechanisms across the tree of life are diverse, and include male heterogamety (XY inheritance), female heterogamety (ZW inheritance), and more complex scenarios (reviewed in [13]). Although sex phenotype is typically determined by only one or two genes, these functional element(s) frequently reside within a larger sex-determining region (SDR) which is inherited as a unit. The remainder of the sex chromosome outside the SDR, known as the pseudo-autosomal region (PAR), can also have distinct properties that distinguish it from ordinary autosomal sequence [14],[15]. One of the defining features of sex chromosomes is a specific and repeatable pattern of heterogeneity in recombination rate that has been seen across diverse taxa including vertebrates, arthropods, fungi, and flowering plants [16],[17]. That is, the SDR typically shows suppressed recombination in the heterogametic parent, while the PAR may show elevated recombination in these same individuals (reviewed in [15], [18]). The adaptive basis for these recombination patterns has been extensively examined theoretically [13],[19]. In brief, if an SDR-linked locus harbors an allele that is advantageous in one sex and disadvantageous in the other (i.e. sexually antagonistic mutations), recombinants will have decreased fitness and recombination suppression will be favored. In the case where sex is determined by two loci both carrying fertility/sterility mutations, recombinants lack both male and female fertility alleles and are thus neuters, making the suppression of recombination highly advantageous [1], [19], [20]. As recombination is suppressed, differential substitutions and transposable elements accumulate within the widening SDR, resulting in evolutionary strata of sequence divergence between X/Z and Y/W [16], [21]. Recombination is nonetheless usually required for faithful chromosome segregation, so the recombination rate of the PAR may increase in the heterogametic sex to compensate for the SDR [15],[17]. Even with elevated PAR recombination, the total sex chromosome recombination rate averaged across the PAR and SDR is usually less than or equal to the genome-wide average [15].
The most extensively studied sex chromosomes, primarily in model animals systems, are ancient and highly heteromorphic, making it difficult to understand the initial stages of sex chromosome evolution [22],[23]. However, a much greater diversity of sex determining systems exists, especially in plants [13],[18],[24]. While few of the 15,600 dioecious species of flowering plants [25] have heteromorphic sex chromosomes, and the genetics of sex determination in the vast majority are unknown [2],[26], several recent studies have revealed homomorphic sex chromosomes with small SDRs [20],[27–34]. For instance, strikingly small SDR were identified in Vitis and Populus. The Vitis sex locus has been mapped to 143kb, so if there is a suppressed-recombination SDR at all, it must occur within this window [28]. The SDR in Populus is only ~100kb yet determines both male and female function [32]. The slightly larger SDRs (1-10Mb) in plant taxa such as Actinidia [34], Carica [29], and Asparagus (A. Harkess personal communication) are still relatively small on a chromosomal scale, and vastly so relative to cytogenetically heteromorphic chromosomes. Most previous studies in plants have examined XY systems in diploids (but see [33]), leaving open the question of whether similar small SDRs are common in ZW systems or in polyploids, the latter of which gains special importance in flowering plants given the potential association between dioecy and polyploidy [8],[35]. Furthermore, homomorphic sex chromosomes may not necessarily display any recombination heterogeneity, or it may be weak, and thus the resultant patterns of sequence divergence and linkage disequilibrium may be subtle or nonexistent. Such chromosomes, which largely resemble autosomes but for the SDR, could represent the precursors of heteromorphic chromosomes, although these are rare in flowering plants [26]. Alternatively, SDRs may remain small for tens of millions of years, as in ratite birds [36], or control of sex may turn over so rapidly among distinct genomic regions that SDRs undergo minimal evolutionary change, as in the homomorphic sex chromosomes of many frogs [7], [37]. The strength of selection for expansion and divergence of the SDR likely depends on the degree of sexual dimorphism, and therefore the number of genes with alleles showing sex-specific fitness [38]. Whatever their eventual fate, homomorphic sex chromosomes, for which recombination still occurs along most or all of their length, present several advantages for research: fine-mapping candidate causal sex genes, assessing patterns of recombination heterogeneity, estimating the fitness of recombinants, and inferring chromosomal evolution in response to sex linkage [11],[39].
The wild strawberries, genus Fragaria, are a model system for early sex chromosome evolution [20],[40],[41]. Evolving from a hermaphroditic ancestor just 2 mya [42], the genus has diversified into species displaying various sexual systems, including gynodioecy (females and hermaphrodites), subdioecy (females, males, and hermaphrodites), and dioecy (females and males). In the gynodioecious diploid F. vesca subsp. bracteata, at least two unlinked loci harbor male sterility alleles, only one of which, on chromosome IV (out of seven haploid chromosomes, designated with Roman numerals I-VII), is heterozygous in females [43],[44]. Fragaria vesca subsp. bracteata is an ancestor of allo-octoploid Fragaria species (2n=8x=56), having provided the chloroplast genome, a putative mitochondrial cytoplasmic male sterility (CMS) gene, and one of the four nuclear subgenomes (subgenome Av out of Av, B1, B2, and Bi) [42],[45–47]. It does not share its sex loci with the octoploid clade, though. Wild octoploid F. virginiana subsp. virginiana possesses a female-heterozygous SDR at the start of linkage group VI-B2-m (i.e., haploid chromosome six, subgenome B2, maternal chromosome), while wild octoploid F. chiloensis possesses a female-heterozygous SDR at the end of linkage group VI-Av-m [11],[27],[45],[48]. Octoploid Fragaria are highly diploidized, such that these subgenomes remain evolutionarily distinct [45]. Thus, sex determination in this genus has frequently evolved independently, and/or exhibits a rapidly shifting genomic position. Moreover, since the octoploid clade arose 1 mya [42], and SDRs are not shared among the dimorphic species they appear to be some of the youngest studied to date.
Fragaria chiloensis occurs on or near the Pacific coasts of North and South America and is divided into several subspecies. While F. virginiana is subdioecious [48], F. chiloensis is nearly completely dioecious, although some hermaphrodites do occur [49]. Thus, F. chiloensis has undergone perhaps the greatest sexual differentiation in the genus with respect to the hermaphroditic Fragaria ancestor [40],[50]. Because male and female function both map to a single maternal genomic location with no previously observed recombination between them [27], F. chiloensis has all the hallmarks of a complete, albeit incipient, ZW sex chromosome. In fact, as all other flowering plant sex chromosomes showing evidence of recent origin (homomorphic, taxonomically restricted, small SDRs, etc.) are XY, F. chiloensis appears to possess the youngest plant ZW chromosome yet characterized. However, neither the precise genomic location of the SDR, its physical size, nor the recombination profile of the SDR-carrying chromosomes could be determined from the initial SSR-based linkage map [27].
Here we characterize the F. chiloensis sex chromosome with respect to Z- or W-specific recombination rates in the PAR and SDR, as well as recombination-dependent metrics like sequence divergence and linkage disequilibrium. To achieve this goal, we first precisely define the SDR by fine-mapping both male and female function. We substantially increase the numbers of both molecular markers and progeny for a previously described cross [27] and also examine the two other crosses and several additional unrelated individuals. By analyzing SNPs both extremely close to the causal sex gene(s) and all along the sex chromosome, and by leveraging the large and highly duplicated genome as an internal control for recombination estimates of autosomes (i.e. of the autosomal VI homeologs and across the rest of the genome), we can infer the initial evolutionary adjustments in recombination rate and divergence that occur in response to sex linkage.
Results
Phenotypes
In order to define the SDR, we first phenotyped male and female function in hundreds of plants. In our largest cross, HM×SAL [27], 1285 seeds germinated overall and we obtained genotype and/or phenotype data from 1277 of these (S1 Table). We determined male function for 693 progeny (352 male sterile, 341 male fertile), and female function for 619 progeny (157 with fruit set <5% and thus “female sterile” as defined in Goldberg et al. [27]; mean flower number = 15.5). Male and female function were highly negatively correlated (R2 = 0.75; p < 10−15), such that 98% of male-sterile plants showed at least 5% fruit set, while only 50% of male-fertile plants showed at least 5% fruit set (Fig 1). Including total flower number in multiple regression analysis did not significantly improve the proportion of variance explained in the simple linear regression, suggesting little contribution of plant size to the relation between male and female function (R2 = 0.76; p < 10−6). Similar phenotype distributions were observed among two sets of 20 offspring from independent crosses (EUR and PTR, S2 Table), and among 16 additional unrelated plants (S3 Table).

Male function is defined as the ability (male fertility, blue) or inability (male sterility, pink) to produce mature pollen and is a binary trait found in approximately half of the progeny. Female fertility is defined as the proportion of flowers producing fruit, and has a continuous though bimodal distribution, shown here partitioned into bins of 5%. Male and female function are highly negatively correlated, with nearly all male-fertile offspring showing <50% female fertility, and nearly all male-sterile offspring showing >50% female fertility.
Target capture mapping
In order to initially map the SDR, we examined 42 HM×SAL offspring in a previously described target capture dataset (S4 Table) [45]. We observed 23 male-fertile offspring and 19 male-sterile offspring. Male function mapped to the end of the VI-Av-m (maternal) linkage group, as expected (LOD = 10.6; Fig 2) [27]. Specifically, the last recombination event before the male function region occurs after reference genome position Fvb6_37.391Mb, after which only a single male-sterile individual (which does not recombine anywhere on the linkage group) has a genotype mismatching sex (R2 = 0.91; p < 10−15). Thus, these results suggest the SDR occurs somewhere within the remaining 1.482Mb of the chromosome (the “SDR vicinity”), on which we observed 10 SNPs in 9 targeted regions. An alternate, much weaker potential quantitative trait locus (“QTL”) on linkage group II-Av-p (LOD = 3.6; Fig 2) was assumed to be a statistical artifact and not pursued further, because it showed no significant association after accounting for the QTL on VI-Av-m (R2 = 0.04; p > 0.1). Female function showed the same bimodal distribution as seen in the complete dataset (Fig 1), with most (59%) samples showing a fruit set of either 0 (26%) or 1 (33%). The highest QTL for female function (R2 = 0.77; p < 10-13), treated as a quantitative trait, occurred at the same location on VI-Av-m as male function, peaking at Fvb6_37.391Mb (LOD = 18.8). The best two-QTL model for female function included this region and was not significantly better than the single-QTL model.
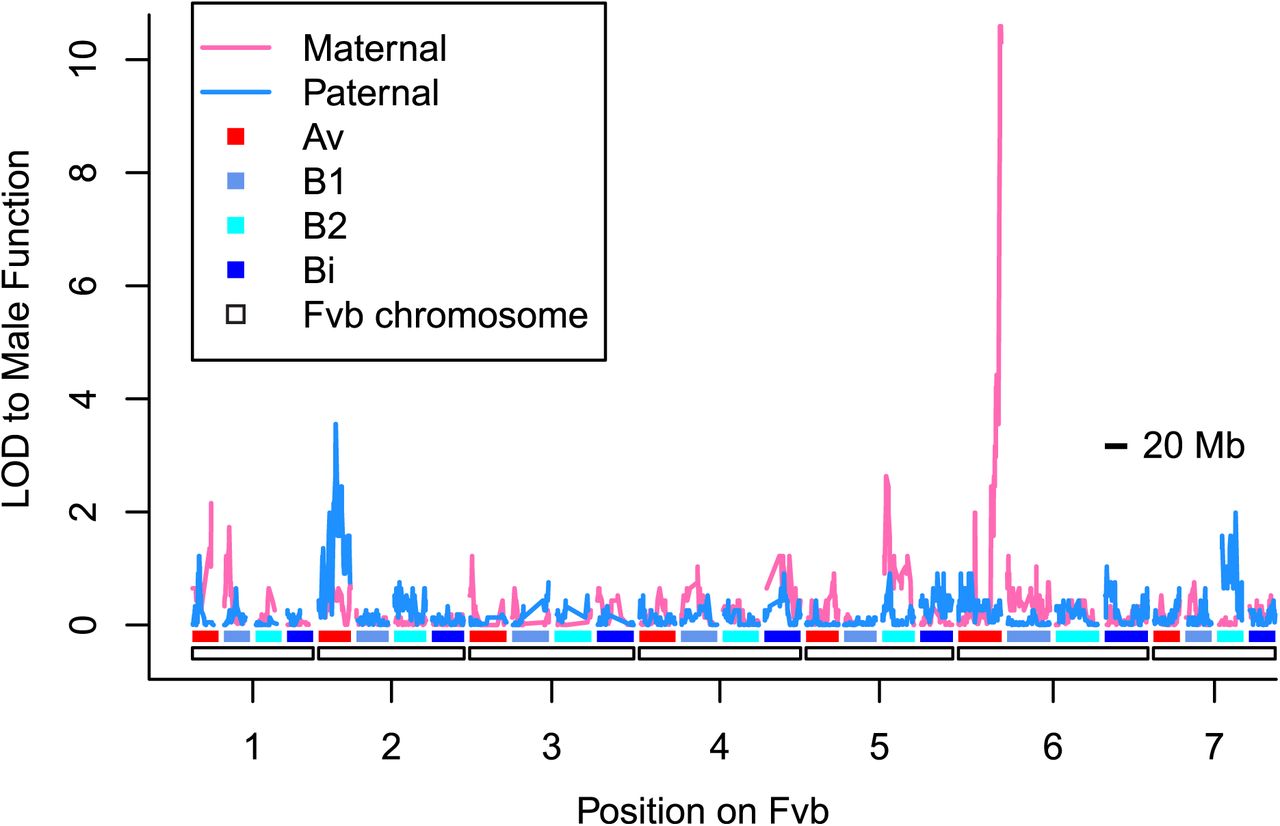
The haploid chromosomes of the Fvb reference genome are denoted by white rectangles along the x-axis. Each haploid chromosome occurs on each of four subgenomes (Av, B1, B2, or Bi), shown as red or blue shaded bars. LOD scores for maternal and paternal linkage maps are shown on the y-axis. The highest LOD score occurs on linkage group VI-Av-m, peaking at 10.6 for the last ten segregating sites in the linkage group, all aligning after Fvb6_37.391Mb. We subsequently validated this region on VI-Av-m using hundreds of additional offspring, and thus the second-highest LOD peak on linkage group II-Av-p (LOD = 3.6) appears not to be biologically meaningful.
Amplicon fine mapping
In order to further fine-map the SDR and calculate recombination rates, we genotyped HM×SAL progeny at amplicons in the SDR vicinity. Of the 1285 HM×SAL progeny, we obtained useable genotypes from 1235 offspring, including 1215 with Av-m genotypes and 1196 with Av-p genotypes (S1 Table). Although the set of useable sites varied slightly among each of three sequencing rounds, by the third round we could genotype 36 segregating sites among 24 amplicons on VI-Av-m, and 24 segregating sites among 16 amplicons on VI-Av-p. Recombinants were observed in all growing years and all sequencing rounds. We observed 32 plants that recombined on VI-Av-m, and these allowed us to further fine-map the male function region to a 280kb section between Fvb_37.428Mb and Fvb6_37.708Mb (Fig 3). Specifically, we observed near-perfect matches to male function at all sites between Fvb6_37.566Mb and Fvb6_37.682Mb, with two male-fertile downstream recombinants showing mismatched genotypes starting at Fvb6_37.708Mb, and two upstream recombinants at Fvb_37.428Mb, one of which was phenotyped as male fertile. Seven individuals (one male-sterile from the target capture genotyping described above, two more male-sterile, plus four male-fertile) had genotypes mismatching male function in this region. These individuals did not recombine anywhere in the SDR vicinity. With <1% of offspring mismatching, the correlation between genotype in this male function region and male function phenotype was very strong (R2 = 0.96; p < 10-15). The male function region resides entirely within a single scaffold of the Fvb reference genome, scf0513124_6, minimizing the possibility of genome assembly errors in this region. The original F. vesca genome annotation identified 68 genes in this male function region (S5 Table). Two additional genes that do not overlap these genes appear in the most recent annotation [51] (S5 Table).

Physical distance (Mb) along the distal end of Fvb6, encompassing portions of two genomic scaffolds (scf0513124_6 and scf0513112a), is shown on the x-axis. All amplicon markers in the SDR vicinity in VI-Av-m (pink) and VI-Av-p (light blue) are plotted, with relative linkage map position (cM, arbitrarily starting at 0) shown on the y-axis. An X marks the position of the “universal marker” SNP on VI-Av-m which shows a high correlation with sex across the full set of unrelated plants. Recombination rates can be inferred from the slopes of lines connecting VI-Av-m (pink) and VI-Av-p (light blue) markers. Three colored bars with arbitrary vertical position indicate different estimates of the SDR. The female-function region showing significantly higher correlation with fruit set than the surrounding regions is indicated with a dark purple bar. The male-function region, bounded by the recombinants which decrease the correlation with male function, is indicated with a dark red bar. The high W divergence region, within which most W-specific SNPs are observed, is indicated by an orange bar. The SDR encompasses a relatively small portion of the 39Mb chromosome. Maternal and paternal recombination rates are similar, except near the SDR, where they are lower in the mother.
The highest LOD (145.9) to quantitative female function was observed on VI-Av-m at Fvb6_37.428Mb, and correlations that were not significantly weaker than this were observed between Fvb6_37.428-37.708Mb (LOD = 143.1-145.9), precisely the extent of the male function region. Significantly worse correlations were observed upstream at Fvb6_37.391Mb (LOD = 142.4) and downstream at Fvb6_37.858Mb (LOD = 126.8), thus defining the limits of the 467kb female function region (Fig 3). A QTL analysis that included the seven linkage groups in homeologous group VI for which we had segregating markers among the sequenced amplicons of the SDR vicinity (VI-B2-m not observed) supported the single QTL on VI-Av-m. The correlation between SDR genotype and quantitative female function (R2 = 0.76; p < 10−15) was similar to the correlation between male sterility and quantitative female fertility (Fig 1).
Confirmation of SDR in additional crosses
In order to examine the species-wide relevance of the HM×SAL results, we tested whether sex mapped to the SDR in two independent crosses. In the EUR cross, we genotyped SDR-vicinity amplicons of 10 male-fertile and 10 male-sterile offspring. We observed 4 Av-m segregating sites and 3 Av-p segregating sites in EUR (S2 Table; Fig S1). Similarly, in the PTR cross we genotyped SDR-vicinity amplicons of 6 male-fertile offspring, 11 male-sterile offspring, and 3 offspring of unknown sex phenotype. We observed 12 Av-m segregating sites and 4 Av-p segregating sites in PTR (S2 Table; Fig S1). Among both EUR and PTR, all Av-m SNPs in the SDR vicinity showed a perfect match to sex, with no observed recombinants.
Recombination
In order to calculate recombination rates along the entire ZW sex chromosome (VI-Av-m and VI-Av-p) and compare them to genome-wide patterns, we re-examined all linkage groups in the published F. chiloensis target capture maps [45]. Among all linkage groups we observed 747 maternal recombination events and 731 paternal recombination events, suggesting a genome-wide recombination rate of 2.5 cM/Mb given a genome size of approximately 700 Mb [52]. In order to examine variation in recombination rates across the genome, we calculated rates for 138 segments of 5-10Mb. Recombination rates varied from 0 to 7.0 cM/Mb (median = 2.3, mean = 2.5, standard deviation = 1.4; Fig 4). There was no significant difference between maternal and paternal recombination rates genome-wide (Wilcoxon rank sum test, p > 0.1; Fig 4). Remarkably, among the 56 linkage groups, VI-Av-m (the ZW linkage group) had the second-highest observed recombination rate of 3.3 cM/Mb, only slightly exceeded by the rate of 3.4 cM/Mb observed on I-Av-p (S6 Table). A relatively high rate appears consistently across this entire VI-Av-m linkage group (Fig 4 pink circle) and is notably higher than the rate in all other VI linkage groups (Fig 5). Based on Fvb chromosome lengths, the VI-Av-m linkage group is estimated to constitute 2.3% of the F. chiloensis genome, suggesting an expected 34 recombination events, a prediction significantly exceeded by the observed 53 recombination events on VI-Av-m (53:1425 vs. 34:1444 recombination events; χ2 = 4.3, p < 0.05). Conversely, VI-Av-p (the ZZ linkage group) had one of the lowest recombination rates at 1.7 cM/Mb (Fig 4 blue circle), lower than all other VI linkage groups (Fig 5), and with only 5 out of 56 linkage groups showing less recombination (S6 Table). Thus, recombination rate is nearly 2-fold higher between ZW chromosomes than between ZZ chromosomes (53:27 vs. 40:40 recombination events, χ2 = 4.3, p < 0.05).
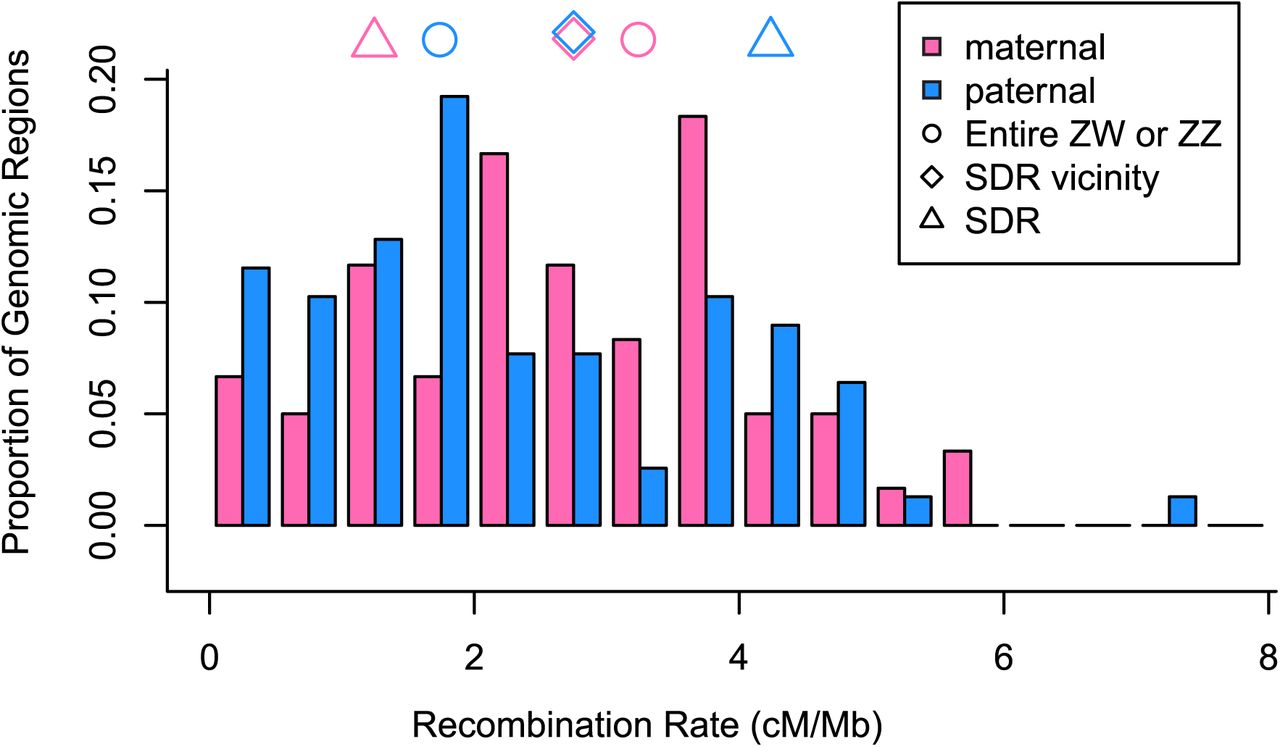
The F. chiloensis target capture linkage maps were divided into sections of 5-10Mb, free from any known rearrangements. Recombination rate was calculated for each section, as shown in this histogram of maternal (pink) and paternal (light blue) recombination rates. Symbols indicate maternal and paternal recombination rates across the entire VI-Av linkage group (circles; ZZ or ZW), across the SDR vicinity from Fvb6_37.38-38.29 (diamonds), and across the SDR from Fvb6_37.38-37.71 Mb (triangles). Recombination is notably higher in the mother than in the father across VI-Av, but this pattern reverses at the SDR.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Markers in each linkage group are plotted with physical Fvb reference genome position on the x-axis and centimorgan position on the y-axis. Recombination rates are therefore indicated by the slopes of the lines, as illustrated by the example slopes in the figure legend. Lines are color-coded based on subgenome, and are solid for maternal linkage groups and dashed for paternal linkage groups. The SDR is shown as a dark red box. For ease of visualization, markers showing radical rearrangements relative to the reference genome (<5% of all markers) have been removed. The VI-Av-m linkage group harboring the SDR has the highest recombination rate across the entire chromosome, while the VI-Av-p linkage group has the lowest recombination rate.
In order to test for recombination suppression at the SDR, we calculated maternal and paternal recombination rates from the amplicon data. Across the 1Mb of the SDR vicinity in which we observed segregating sites in amplicons, recombination rates were nearly identical on VI-Av-m and VI-Av-p (2.9 cM/Mb maternal, 2.8 cM/Mb paternal; Fig 4 diamonds). However, in the 330kb between Fvb6_37.378-37.708Mb, containing the entire male function region and most of the female function region, recombination was notably lower on VI-Av-m relative to VI-Av-p (Fig 4 triangles). Specifically, there are six Av-m recombination events in this window (1.5 cM/Mb) compared to sixteen Av-p recombination events (4.1 cM/Mb) (6:1209 vs. 16:1180 recombination events; χ2 = 4.7, p < 0.05). In comparison to genome-wide rates, the recombination rate at the SDR in VI-Av-m is lower than average, but not significantly so (given 2.5 cM/Mb mean rate, expect 10 recombination events between Fvb6_37.378-37.708Mb, 6 observed; χ2 = 2.6, p > 0.1), and well within the range observed elsewhere in the genome (30% quantile; Fig 4). Likewise, the recombination rate at the SDR in VI-Av-p is higher than the genome-wide average but not an outlier. Unfortunately, we did not observe SNPs at both boundaries of this interval in any other homeolog, and therefore we cannot directly compare recombination rates among subgenomes specifically at the SDR.
Divergence between W and Z
In order to assess sequence divergence between W and Z chromosomes, we evaluated SNPs in the vicinity of the SDR in the three crosses. In each of these, we classified SNPs as either in coupling with male sterility/female fertility (W-specific), in repulsion in the mother, or segregating in the father (Fig S1). In all three crosses, we observed more SNPs that differentiate Z from W (52 total, not correcting for shared SNPs among crosses) than SNPs which differ between two Z chromosomes (31 total, not correcting for shared SNPs among crosses). Specifically, on VI-Av in HM×SAL, we observed 18 SNPs in coupling, and 18 SNPs in repulsion, with male sterility/female fertility, and 9 and 15 SNPs on each of the paternal Z haplotypes. On VI-Av in EUR, we observed 2 SNPs in coupling, and 2 SNPs in repulsion, with male sterility/female fertility, and 0 and 3 SNPs on each of the paternal Z haplotypes. On VI-Av in PTR, we observed 7 SNPs in coupling, and 5 SNPs in repulsion, with male sterility/female fertility, and 0 and 4 SNPs on each of the paternal Z haplotypes. Intriguingly, the highest concentration of W-specific SNPs occurred within the male-sterility region, specifically in a 143kb “high W divergence” window between 37.565-37.708Mb, with 10 SNPs in coupling with male sterility/female fertility in HM×SAL, 4 in PTR, and 1 in EUR. For HM×SAL, the W-specific divergence (SNPs in coupling out of all sequenced sites) in the high W divergence window is 0.39%, substantially higher than both the 0.07% of sites segregating on a single Z haplotype in this window (observe 10:5 W:Z, expect 3.75:11.25, χ2 = 4.8, p < 0.05), and the 0.06% W-specific divergence observed across all amplicons outside of this high-divergence window. The higher SNP counts observed in the HM×SAL cross relative to EUR and PTR crosses may be due to the parents of the former originating in different populations (and thus fewer SNPs segregating in both parents, which we ignored in our mapping methodology), as well as the much larger offspring sample size (and thus higher power to map SNPs even when a large proportion of offspring have missing genotypes). Nevertheless, a single G/C SNP, which occurs within the high divergence window at position Fvb6_37594072, was in coupling with male sterility/female fertility (W allele = C; Z allele = G) in all three crosses (Fig 3; Fig S1).
Association between genotype and sexual phenotype in unrelated plants
In order to identify SNPs in linkage disequilibrium with the causal sex gene(s), we examined genetic diversity at SDR vicinity amplicons in 22 unrelated plants, 10 male-sterile and 12 male-fertile (the six parents of the crosses plus 16 additional plants). A single SNP showed a near-perfect correlation with sex: the same G/C SNP at Fvb6_37594072 for which the C allele was in coupling with male sterility/female fertility (W) in all three crosses (Fig S1). The SNP was heterozygous (G/C) in 9 out of 10 male-sterile plants (ZW), and homozygous (G/G) in 11 of 12 male-fertile plants (ZZ). Due to its widespread and consistent association with sexual phenotype, we designated this SNP the “universal marker” (Fig 3; Fig S1). Four additional SNPs (Fvb6_37565581, Fvb6_37565641, Fvb6_37567599, and Fvb6_37567962; Fig S1), all within the high W divergence window of the male function region, showed a weaker association with sex, but all were heterozygous in 80-90% of male-sterile plants and homozygous in 60-70% of male-fertile plants.
Discussion
Our study of incipient sex chromosomes in octoploid F. chiloensis illuminates how recombination rates may first begin to adjust in response to sex linkage. Sex chromosomes are characterized by having atypical recombination patterns relative to autosomes [16]. These unique recombination dynamics are the evolutionary basis for the other unusual features of sex chromosomes, such as heteromorphy, rapid molecular evolution, degradation, and determination of hybrid incompatibility [53]. Recombination rates across the genome are determined by several factors: crossover motifs and the associated epigenetic and chromatin modifications, sequence divergence among sister chromatids including rearrangements such as inversions, and specific genes that dictate pairing compatibilities [54],[55]. Selection pressures to rapidly adjust recombination rates for nascent sex chromosomes are similar to those facing neopolyploids undergoing diploidization [54], and both processes may involve similar molecular mechanisms and a similar period of adaptation during which recombination patterns are not optimal. Here we have examined recombination in the youngest and least differentiated plant ZW chromosome yet described.
A small SDR
In contrast to most older sex chromosomes, we do not observe a wide (>1Mb) SDR characterized by suppressed recombination. In fact, across most of the SDR vicinity targeted by amplicon sequencing, recombination rates were typical and nearly identical between VI-Av-m and VI-Av-p (Fig 3). Both male and female function mapped to a single 280kb chromosomal region within the region first identified by Goldberg et al. [27] at the distal end of VI-Av-m. The maximum possible extent of suppressed recombination occurs within a 330kb window centered on this SDR. Observed VI-Av-m recombination in this window is less than half that of VI-Av-p. However, the VI-Av-m recombination rate is not unusually low relative to autosomal rates. The fact that neither male nor female function was perfectly correlated with the SDR suggests that other unlinked loci or environmental effects may also influence sex in F. chiloensis. This is especially likely for female function, a quantitative trait for which the SDR explains 74% of variation, but also one known to be highly environmentally labile in other Fragaria [56]. Such minor contributors to female function variation have also been seen in the subdioecious Fragaria virginiana which has a major SDR [11], and are known to segregate in the natural hybrid F. × ananassa subsp. cuneifolia [57]. Male function in contrast was almost perfectly determined by the SDR, with <1% of HM×SAL offspring showing a mismatch, suggesting that any other factors affecting the propensity to produce pollen are negligible.
We cannot determine whether male and female function are controlled by two closely linked genes, or both by the same gene. This question is relevant to recombination patterns because the primary theoretical basis for recombination suppression assumes two genes [19]. Presumably a single gene initially controlled a single phenotype such as male function, especially given the observed gynodioecy in the closely related Fragaria vesca subsp. bracteata [58] (but see [44]). Under the classic model of sex chromosome evolution [18],[19], a second linked mutation occurs such that there are two distinct linked genes controlling male and female function. Recombination between these genes produces neuters, prompting strong selection for recombination suppression. In F. chiloensis, the two-locus model would require a remarkably small physical distance between the two loci (within the predicted 280kb SDR), but this could arise either coincidentally or else via adaptive translocation of the second locus in order to achieve tight linkage. Because baseline recombination should only occur rarely within a 280kb region (fewer than 1% recombinant offspring at the 2.5 cM/Mb genomic mean rate), selection for additional recombination suppression would not be particularly strong. Although recombination between loci controlling male and female function would theoretically produce neuters [11],[18], we observe only 7 out of 619 HM×SAL offspring showing both male and female sterility, none of which show recombination on VI-Av-m, and four of which produced only 1-2 flowers, and thus may have failed to fruit by chance. Alternatively, there may be only a single sex determinant at the SDR, as proposed for other dioecious systems in which only a single regulator of sex has been identified [59],[60]. A single sex-determining gene for Fragaria was proposed by Ahmadi and Bringhurst [61], although their genus-wide model was too simple given the distinct sex loci subsequently observed among species [11],[27], [43], [44],[48]. Ancestrally, individuals with ZZ genotypes may have been universally hermaphrodites, as in gynodioecious species with dominant male sterility allele(s) such as F. vesca subsp. bracteata [43],[44]. Variation in female function among male-fertile individuals could have been influenced by gene(s) anywhere in the genome, not necessarily on VI-Av. If males had higher fitness than hermaphrodites [62], alleles conferring female sterility (but only in male-fertile individuals) would increase in frequency or even fix, leading to the highly dioecious condition observed in F. chiloensis even without new sterility mutations on VI-Av. Selection could favor linkage of female fertility modifiers to the SDR, but such selection would be weak if these mutations had little phenotypic effect in females [63]. If a single gene controls both male and female function in F. chiloensis, there would only be selection for recombination suppression if other linked genes had alleles with differential fitness between sexes, i.e., antagonistic selection [15].
Although the causal sex gene(s) remain unidentified, as in most dioecious plants, several promising candidates are apparent among the 70 known genes in the SDR (S5 Table). One gene encodes a mitochondrial-targeted pentatricopeptide repeat protein, a molecular family commonly implicated in male fertility restoration when cytoplasmic genes cause sterility [64]. There are two methyltransferases, another gene family which may control sex in other plants [32]. A cluster of five cystinosin homologs, out of only six in the entire Fvb genome, occurs in this window. Cystinosin mutations cause male sterility in humans [65], indicating a possible role for cystine transport in Fragaria sex function as well. Although we can’t determine how close the universal marker is to the causal genes(s), there are 13 genes within 25kb of this SNP, including four F-box proteins, an abhydrolase domain-containing protein, and a glycerol-3-phosphate dehydrogenase. An exciting future direction will be to test SDR genes for sexual function or sexually antagonistic alleles.
Recombination at the PAR
In addition to suppressed recombination at the SDR, sex chromosomes frequently show elevated recombination in the PAR [15]. In the case of F. chiloensis, the PAR represents over 99% of the 39Mb VI-Av chromosome. Thus the PAR recombination rate is approximated by examining the entire chromosome, over which VI-Av-m recombination is nearly twice as high as VI-Av-p recombination. Recombination rates are not as high on the other F. chiloensis VI linkage groups, and in fact are lower among all 56 F. virginiana linkage groups [45], suggesting that the high VI-Av-m rate in F. chiloensis is newly evolved. Although elevated PAR recombination has been attributed to compensation for suppressed SDR recombination [15], in F. chiloensis the PAR greatly over-compensates for the SDR. Thus, the adaptive value of this elevated recombination rate, if any, does not appear to involve compensation. Instead, perhaps there is a benefit for certain alleles to recombine away from the SDR, at least in the early stages of sex chromosome evolution. One intriguing possibility is that the effective population size of the W chromosome is relatively low due to fluctuating, male-biased sex ratios, which are common in dioecious plants [66], and which could explain the high W-specific divergence. Population proportions of male-sterile plants may occasionally decrease under ecological conditions that favor hermaphrodites, which could lead to accumulation of deleterious mutations on the W, unless these are eliminated by recombination. Thus, high recombination in the Z-W PAR could be favored as a way to avoid Muller’s ratchet [67].
Although examination of recombination rate is standard in studies of sex chromosomes [18], statistical detection of recombination suppression or elevation is not always straightforward. Until recently, most studies of plant recombination heterogeneity lacked a physical map (e.g. [68]-[70]), and thus could only compare the relative densities of markers at linkage map positions. Our approach of integrating linkage maps with reliable reference sequences, which is increasingly common [29],[34],[71], affords much more precision. Nevertheless, such recombination estimates carry several caveats. The first caveat is the challenge of designating an appropriate control. For example, to test if the recombination rate in an SDR is unusual, it could potentially be compared to the orthologous region in the homogametic parent, the PAR, the homeologous chromosomes, the autosomes, or the genomes of outgroup species, with variable results [72],[73]. By considering all of these comparisons, we can achieve a more nuanced view of recombination heterogeneity. For example, although maternal recombination is lower at the SDR than the PAR (Fig 4), this is due at least as much to elevated recombination at the PAR as to suppressed recombination at the SDR. A second caveat to all our recombination analyses is that physical distances are estimated from the Fvb reference genome. Major rearrangements between F. chiloensis and F. vesca subsp. bracteata, such as large insertions or deletions, would affect our estimates. However, we minimized the effects of this limitation in several ways: by identifying and avoiding rearrangements in the target capture map, by focusing on the subgenome (Av) which is most closely related to F. vesca subsp. bracteata, and by making direct comparisons between maternal and paternal maps under the assumption that most large rearrangements would be present in both parents. Thus, our conclusions are robust to both of these caveats.
Evolutionary dynamics at the SDR vicinity
The F. chiloensis SDR represents the narrowest fine-mapping of any Fragaria sex locus to date. It is distinct from the F. virginiana subsp. virginiana SDR which also occurs on a VI chromosome [11],[48]. Furthermore, the F. chiloensis SDR is distinct from one of the male-function-influencing loci F. vesca subsp. bracteata, the Rr locus which intriguingly occurs less than 1 Mb away at Fvb6_34.839-36.607Mb [44], but confers recessive male sterility (rr). Perhaps the VI chromosome, especially the distal end between Fvb6_34-38Mb, is particularly suited to controlling sex phenotype [11]. The myriad sex loci among closely related Fragaria species suggest a dynamic evolutionary process that may involve repeated independent sterility mutations, gene translocations, and/or shifting genetic control of phenotype. In fact, turnovers driven by sexually antagonistic genes and/or escape from mutational load also can produce “ever young” small SDRs [74] and can account for XY to ZW transitions as well. These are expected to be particularly common in species with undifferentiated sex chromosomes, and some autosomal pairs are more likely than others to be recruited [74],[75]. Such dynamics have been proposed to be responsible for the variation in position of the SDR and heterogamety in willows [60].
In contrast to the variable basis of sex determination between species, the SDR is highly consistent within F. chiloensis. The same SDR was identified in two additional F. chiloensis crosses from populations up to 1000 km away, suggesting it may be the most common, or only, SDR in this species, at least in North America. A single “universal marker” was in coupling with male sterility/female fertility phenotype in all three crosses, and this SNP also showed near-perfect association with sex across a set of unrelated plants. Of the two exceptions, one plant was the sole specimen from South America which may have a distinct sex determining mechanism. The other was from the same population as one of the crosses (PTR), suggesting either that the SDR does not perfectly control sex in all cases, or that the universal marker is not in perfect linkage disequilibrium with the causal gene(s). One consequence of recombination suppression should be that a W-specific haplotype behaves as a single unit in population genetics, with multiple SNPs in linkage disequilibrium with sex. The fact that only a single universal marker was observed, despite our sequencing ~2% of the 280kb SDR, suggests that there are few SNPs (less than 100) showing such high linkage disequilibrium with sex. In turn, this observation suggests that the true SDR may be substantially smaller than the 280kb mapped region, although we cannot definitively demonstrate this.
We observe a particularly high concentration of maternally-heterozygous SNPs in a small genomic section corresponding to the second half of the SDR (Fig 3, Fig S1). Thus, divergence between W and Z is particularly high in this window, especially for SNPs in coupling with male sterility/female fertility and therefore W-specific within crosses. Note that, other than the universal marker, these SNPs are not W-specific across populations, but some may be W-specific within populations. Our study design doesn’t allow us to examine divergence shared by all Z chromosomes but not the W, since such Z-specific SNPs will occur in both parents and thus will not segregate in the offspring. Female-specific mutations could accumulate at the SDR for several reasons: suppressed recombination, selection against recombinants, or low effective population size at the SDR due to fluctuating sex ratios, such that deleterious mutations are not purged by selection [29],[32]. As discussed above, the accumulation of deleterious W-specific mutations could potentially explain why high recombination in the PAR is adaptive. These processes could lead to even greater Z-W differentiation. Alternatively, given that heteromorphic sex chromosomes seem to have only rarely become established in dioecious flowering plants, these may not necessarily evolve in F. chiloensis, and Z-W divergence could remain subtle indefinitely, especially if recombination eliminates W-specific changes.
Conclusions
We have characterized an incipient ZW sex chromosome in a dioecious wild strawberry, pinpointing a single SDR controlling both male and female function within a narrow genomic window, and examining recombination and divergence patterns in Z-Z and Z-W pairs. Along with other recent studies of homomorphic plant sex chromosomes [20],[27–34], this work suggests that small, modestly divergent SDRs may be typical of many dioecious plants. By extensively analyzing recombination, this study reveals rate heterogeneity patterns both expected and surprising, which could nonetheless be typical of such young sex chromosomes. Our work is also novel in that F. chiloensis has a ZW system and that it is a higher-order polyploid, thus extending our understanding of small plant SDRs beyond XY diploids. These subtle plant SDRs may be affiliated with relatively low sexual dimorphism, and thus little selection pressure on genes to become tightly linked to sex [38], consistent with few observed sex differences in F. chiloensis [50] (but see [8]). When compared with classic, highly heteromorphic sex chromosomes of other taxa [22],[23],[71], our results speak to the diversity of genomic patterns in sexual species. However, even when the SDR represents a tiny fraction of the genome, linkage to sex may have important consequences for loci all along the sex chromosome, as exemplified here by the elevated PAR recombination rate.
The evolutionary processes that generate and modify sex chromosomes across taxa are now partially understood, but remain to be fully characterized. Theoretical assumptions, like that elevated PAR recombination serves to compensate for suppression at the SDR, or that SDRs evolve from two linked genes controlling male and female function, may not hold true for all taxa. The patterns of recombination, divergence, and linkage disequilibrium observed in the F. chiloensis ZW chromosomes can be further explored by measuring fitness, gene function, and population genetic parameters. This species thus presents an ideal opportunity for continued study of the emergence of sex chromosomes.
Materials and Methods
Samples
The primary cross examined in this study (here designated HM×SAL) represents an expanded set of F1 offspring from an intraspecific cross involving a female F. chiloensis subsp. lucida (GP33) and a hermaphroditic F. chiloensis subsp. pacifica (SAL3) used in previous work [27]. The maternal parent was obtained from the USDA National Clonal Germplasm Repository (accession no. PI 612489; ‘HM1’) which was originally collected from Honeyman Memorial State Park, Oregon (43.93N, 124.11W). To emphasize its population of origin, shared by several other plants in the current study (S3 Table), we here use the designation of the maternal parent as HM1 (although note previously this cross was referred to as GP33xSAL [27]). The paternal parent was collected from Salishan, Oregon (44.91N, 124.02W) and is a low fruit-producing hermaphrodite (10% of hand-pollinated flowers yielded fruits in the greenhouse). We hand pollinated pistillate flowers of HM1 with the pollen from SAL3. We planted a total of 1695 seeds in sets across five years (between 2009 and 2015) and 75% of which germinated. We transplanted ~ 2-month old seedlings into 3-inch-square pots filled with a 2:1 mixture of Fafard #4 (Conrad Fafard) and sand. In the first growing period after germination in each year the plants received a total of 513 mg of granular Nutricote 13:13:13 N:P:K fertilizer (Chisso-Asahi Fertilizer). All plants were grown under mild temperatures (15720° night/day) and 10- to 12-hr days throughout the majority of the flowering period. To initiate flowering plants were exposed to cooler temperatures (8°/12° night/day) with an 8 hr day with low light. Additional liquid fertilizer and pest control measures were applied as needed to maintain the health of the plants to flowering.
We also examined the parents and F1 offspring of two other independent F. chiloensis crosses, in order to test the universality of the location of the SDR identified in HM×SAL and to identify SNPs universally in coupling with male sterility/female fertility (i.e., on the W haplotype of the SDR). The parents of the first cross (EUR) were a female (EUR13) and a male (EUR3; 0% fruit set) F. chiloensis subsp. lucida collected from Eureka, California (40.762 N, 124.225 W). The parents of the second cross (PTR) were a female (PTR14) and a low fruit-producing hermaphrodite (PTR19; 10% fruit set) F. chiloensis subsp. lucida collected from Point Reyes, California (38.0683 N, 122.971 W). We raised 105 and 65 F1 offspring from the two crosses, respectively, following the same planting and growth regimes as in HM×SAL cross. As the goal of these crosses was validation of the sex-linked SNP results from the HM×SAL cross, rather than fine-mapping, we examined substantially fewer offspring than for HM×SAL. Twenty offspring from each cross are studied further here.
In addition, we examined 16 unrelated plants from six populations (the four source populations of cross plants and two other populations) across the geographic range of F. chiloensis (S3 Table). The plants were collected from the wild as clones or obtained from the National Clonal Germplasm Repository and raised under the same planting and growth conditions as described above.
Sex phenotyping
We scored plants of HM×SAL for both male and female function following Spigler et al. [11]. We scored male function on at least two flowers per plant two separate times. Male function was scored as a binary trait: “male fertile” if the plant had large, bright-yellow anthers that visibly released pollen, and “male sterile” if the plant had vestigial white or small, pale-yellow anthers that neither dehisced nor showed mature pollen. Any anthers that were in question were examined microscopically for the presence of mature pollen. To ensure full potential fruit set, we hand pollinated all plants three times per week with outcrossed pollen. We then estimated female fertility for each individual as the proportion of flowers that produced fruit by dividing the total number of fruits by the total number of pollinated flowers produced as in Spigler et al. [11]. In order to measure to extent to which male sterility co-occurs with female fertility, we calculated the association between male function and female function using simple linear regression [76]. Because female fertility may be influenced by plant size (here estimated as the total number of flowers [or buds] produced), we also included total flower number as a second independent variable in multiple regression [76]. In QTL analysis, female function was examined as a continuous quantitative trait. For progeny in the EUR and PTR crosses and the unrelated individuals male function was scored similarly as HM×SAL, but female function was scored only as binary.
DNA extraction and quantification
We extracted DNA from fresh or dry leaf tissue. Fresh tissue extractions using a modified CTAB method were done on 42 F1 offspring from the 2009 mapping population for target capture sequencing to construct initial high density linkage map, as described previously [45]. For the remaining HM×SAL offspring, we collected leaf tissue from seedlings or adult plants and stored on silica gel. Similarly, for the EUR and PTR F. chiloensis crosses, we collected, dried and extracted DNA from leaves of a total of 20 offspring representing 10 females and 10 pollen-bearing morphs (hermaphrodites or males) from each cross. To extract DNA from dry tissue samples, we used the Norgen biotek plant/fungi high-throughput 96 well DNA isolation kit (Norgen Biotek, ON, Canada) and also used DNA extraction service provider Agbiotech Inc (CA, USA). We added an additional 100ul 10% SDS and 10ul β-mercaptoethanol to the lysis buffer to improve DNA yield from all the dry tissue samples. The final DNA elution was sodium acetate/ethanol precipitated to wash and concentrate the DNA. Picogreen assays were performed using a Tecan plate reader to measure DNA concentration for each sample, and we diluted DNA to 50ng/ul using DEPC treated water to use in microfluidics PCR on the Fluidigm access array system (see below).
Target capture mapping
We previously described a target capture linkage map of 2542 segregating markers generated using 42 HM×SAL offspring [45]. All markers align to a distinct position in the haploid F. vesca reference genome (v. 2.0, here designated “Fvb” as in [45]), and also have a linkage map position in cM on a paternal (“p”) or maternal (“m”) linkage group pertaining to one of the four octoploid subgenomes (Av, B1, B2, or Bi). In order to map sex phenotypes, we first coded male function as a binary trait (male sterile=0; male fertile=1) and added it to the linkage map with OneMap [77], as in Tennessen et al. [43]. Because female function is defined as a proportion between 0 and 1, rather than a binary trait, we identified female function quantitative trait locus (QTL) using R/qtl [76], [78]. Specifically, we first used the scanone option to identify all possible female function QTL, using a minimum LOD threshold of 3. Then we checked whether more than one additive QTL was independently supported using the scantwo option; a model with two additive QTL would need a LOD at least 3 greater than the LOD of the single best QTL in order to be significant. The region of the Fvb genome where both male function and female function QTL mapped, presumably harboring the SDR and possibly also adjacent portions of the PAR, was designated the “SDR vicinity” and examined with additional genotyping as described below.
In order to estimate genome-wide recombination rates, we first re-examined the published linkage maps of HM×SAL [45] to confirm all recombination events by eye and eliminate putative recombination events that could be caused by genotyping error. As in previous studies [43], [45], a pair of adjacent recombination events in the same individual on either side of a single marker was considered to be a genotyping error rather than a real double recombination event. We also identified all inconsistencies in marker order when compared with the Fvb reference genome. If an inconsistency could be explained by a single incorrect genotype, it was attributed to genotyping error, otherwise it was considered to be a genomic rearrangement (either a genome assembly error or a real translocation, the distinction is irrelevant here). We assumed that physical distances on the octoploid chromosomes could be approximated by Mb distances in the Fvb reference genome, and thus we estimated recombination rates genome-wide and per each linkage group as cM/Mb. Although the 0.7 Gb octoploid Fragaria genome has likely undergone some gene loss and other rearrangements relative to the 0.2 Gb diploid reference [45],[52], these discrepancies should have a minimal effect when comparing relative rates among linkage groups, and should also be rarest on the Av subgenome containing the sex chromosome because it originates from F. vesca subsp. bracteata. In order to further examine variation in recombination rate across the genome of F. chiloensis, we divided all 56 linkage groups in the genome into segments of 5-10Mb that were free from rearrangements, and calculated recombination rates for each of these segments.
Amplicon generation and sequencing
In order to fine-map the SDR vicinity and examine recombination rates, we genotyped sites in SDR vicinity using Illumina sequencing of targeted amplicons [79]. The Fluidigm microfluidic approach is an effective method for mapping SNP markers in a large sample of polyploids [80],[81], and by sequencing amplicons we can unambiguously assign markers to a genomic position. We performed sequencing in three sequential rounds, refining the combination of targeted amplicons each time based on amplicon performance and fine-mapping results (S7 Table; S8 Table). Some samples were included in multiple rounds due to poor-quality genotypes or in order to further fine map recombinants. Initially, we designed primers for 48 amplicons of approximately 300-600bp (we found that larger amplicons performed poorly), with a Fvb reference genome position within the SDR vicinity. Most amplicons (41) contained at least one SNP (on any subgenome) identified in the target capture map. We used the Fluidigm 48.48 Access Array IFC (Integrated Fluidic Circuits) to generate amplicons in parallel. These steps were performed by IBEST at the University of Idaho. The forward and reverse primers were attached with common sequence tags (CS1 & CS2) to enable the addition of P5 and P7 Illumina adapters along with dual index multiplex barcodes during the four primer PCR reactions. All primer pairs were validated using DNA from parent HM1 to verify the expected product size and ascertain the on-target products to account for a minimum of 50% of the total yield (by mass) for each primer pair. For validation PCR and optimization we followed the Fluidigm Access Array System user guide for Illumina (https://www.fluidigm.com/documents; Access Array 48.48-4-primer-amplicon-tagging). We used the Agilent bioanalyzer (Agilent Technologies Inc.) to determine the expected amplicon size and yield. We redesigned primers to meet the validation criteria as necessary. After validation, we generated amplicons in microfluidic reactors, tagging each amplicon with unique dual index barcodes. We multiplex sequenced 48 pools of amplicons (one per individual) in 10-12 access array plates on one-half lanes of the Illumina MiSeq, yielding 300-bp paired-end reads. For the first round we sequenced 478 HM×SAL offspring and the two parents. For the second round of sequencing, we retained 19 of the original 48 primer pairs and replaced 29 with newly designed primer pairs. We used these to sequence 469 HM×SAL offspring, the two HM×SAL parents, and the parents and 20 offspring each from the EUR and PTR crosses. For the third round of sequencing, we retained 39 primer pairs and replaced 9 with newly designed primer pairs, and we used these to sequence 545 HM×SAL offspring, the two HM×SAL parents, the same EUR and PTR individuals from the second round, and 18 additional unrelated plants (16 of which yielded useable genotypes and were analyzed here). In total, 1302 unique plants were included in amplicon sequencing. Including the target capture samples, a total of 1274 HMxSAL offspring were genotyped.
Amplicon genotyping and fine mapping
We called genotypes using the POLiMAPS approach [45]. In brief, we first processed Illumina reads with dbcAmplicons (https://github.com/msettles/dbcAmplicons), trimmed them with Trimmomatic [82], aligned them to the amplicon regions extracted from Fvb reference genome with BWA [83], and generated pileup files with SAMtools [84]. We called genotypes in the pileup file using the same coverage criteria as in the target capture map [45], i.e. minimum depth of 32, alternate allele must occur in an individual at least twice and at ≥ 2.5% frequency, only SNPs heterozygous in a single parent are considered. However, because sample sizes were larger and there were more missing genotypes than in the target capture map, we adjusted the additional criteria for HM×SAL amplicons, allowing up to 200 missing genotypes per site and requiring 40 observations each of homozygotes and heterozygotes for a SNP to be called as real. For EUR, PTR, and the set of unrelated plants, all with much smaller sample sizes, we allowed up to 10 missing genotypes and required 4 observations each of homozygotes and heterozygotes. We converted genotypes to OneMap format and generated linkage maps in OneMap with a LOD criterion of 3.
We identified recombination events and used these to estimate recombination rates in the SDR vicinity and to fine map sex QTL. Because male function showed a near-perfect match to genotype, and the few exceptions were not recombinants in the SDR vicinity, we simply defined the male function region as the section of chromosome between the closest recombination events both upstream and downstream that caused mismatches between genotype and male function. Because female function is a continuous trait showing a weaker correlation with genotype, we calculated LOD scores for all sites in the SDR vicinity. We also calculated the correlation between genotype and female function across the SDR vicinity and used the R/cocor package [75],[85] to compare these correlations and thus to define the female function region as the region in which correlations were not significantly different from that at the site with the best LOD score. Because male function could thus be more precisely mapped than female function, we considered the male function region to comprise the maximum potential extent of the SDR. We examined genes within the male function region by consulting both the original annotation of the Fvb genome [45],[86] and a recent re-annotation by Darwish et al. [51]. As with the whole-genome analysis from the target capture map, we estimated recombination rate as cM/Mb, with physical distance estimated from the Fvb reference genome. This assumption is particularly justified for the Av subgenome because it originates with F. vesca subsp. bracteata [45].
Acknowledgements
The authors thank C. Kustek, K. Mazzaferro, L. Stanley, K. Schuller, R. Dalton, H. Wipf, E. York for greenhouse, field or laboratory assistance, B. Rosa-Neves for help error-checking the linkage maps, the Ashman and Liston labs for comments that improved the manuscript. This work was supported by the University of Pittsburgh, the National Science Foundation (DEB 1020523, DEB 1241006, and RET/REU supplements to TLA; DEB 1020271 and DEB 1241217 to AL).
References
- 1.↵
- 2.↵
- 3.↵
- 4.
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.
- 31.
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵




