

Abstract
Hybridization is recognized as a powerful mechanism of speciation and a driving force in generating biodiversity. However, only few multicellular species, limited to a handful of plants and animals, have been shown to fulfill all the criteria of homoploid hybrid speciation. This lack of evidence could lead to the misconception that speciation by hybridization has a limited role in eukaryotes, particularly in single-celled organisms. Laboratory experiments have revealed that fungi such as budding yeasts can rapidly develop reproductive isolation and novel phenotypes through hybridization, showing that in principle homoploid speciation could occur in nature. Here we report a case of homoploid hybrid speciation in natural populations of the budding yeast Saccharomyces paradoxus inhabiting the North American forests. We show that the rapid evolution of chromosome architecture and an ecological context that led to secondary contact between nascent species drove the formation of an incipient hybrid species with a potentially unique ecological niche.
One Sentence Summary Chromosomal rearrangements and hybridization between two yeast lineages drive hybrid speciation after secondary contact.
Introduction
Hybridization is a major force in evolution because it can prevent population divergence by maintaining gene exchange 1. However, hybridization can also lead to speciation by combining genomes that have been evolving independently and that, when combined, provide advantageous phenotypes to the hybrid individuals and populations 2,3. The genomic composition of the hybrid individuals may also lead to reproductive isolation with their parental lineages 4. The identification of homoploid hybrid speciation events, which occur without change in chromosome number – contrary to what is observed in allopolyploid and homopolyploid speciation – requires that we show the reproductive isolation of the hybrid with the parental species, the identification of traces of past hybridization in the genome, and that the isolating mechanisms are at least partly derived from hybridization 5. Despite extensive investigations in sexually reproducing microbes, no case of such homoploid speciation has been reported in these eukaryotes 5. Yet, laboratory experiments have shown that hybridization can contribute to rapid species formation in yeast 6.
Here we examine the genomics and the ecological bases of species formation in a yeast natural system. The budding yeast Saccharomyces paradoxus is the closest known relative of the model yeast Saccharomyces cerevisiae. S. paradoxus is a free-living saprophyte mostly found in the sap and on the bark of deciduous trees and their associate soil 7-10. S. paradoxus has a nearly worldwide distribution but contrary to S. cerevisiae, there is no evidence for its domestication by humans 8,11,12. Accordingly, the biogeography 12 and the genome evolution 13 of this unicellular fungus are mostly influenced by natural processes. Four genetically and phenotypically distinct lineages of S. paradoxus have been identified so far and correspond to populations from Europe, Far East Asia, America and North-East America 14-16. In North America, three lineages of S. paradoxus occur in partial sympatry. One of these lineages, corresponding to the European population (Lineage SpA), has a sparse distribution and strains of this lineage are mostly isogenic, due to a recent colonization event 15. The two other lineages in North America (SpB and SpC) are indigenous and are incipient species 17 differentially distributed along a southwest to northeast gradient (Figs. 1a, S1). SpB strains display enhanced fitness at high temperature and survival to a freeze-thaw cycle when compared to SpC strains, consistent with adaptations to climatic conditions 16. Despite the overlapping distributions and partial post-zygotic reproductive isolation between SpB and SpC 17, no first-generation hybrid has been identified so far, suggesting, along with with the monophyly of these lineages, that speciation has been initiated.

a, Sampling locations (circles) and distribution (inner frame) of SpB (red), SpC (dark blue; one strain in Pennsylvania) and the cryptic lineage SpC* (purple). Location abbreviations are listed in Table S1. b, Eleven SpC* strains form a monophyletic group within the SpC lineage. Phylogenetic tree of 161 strains rooted with S. cerevisiae (14,974 SNPs). SpA is the European lineage (green). Open symbols indicate replicates and spores from a same strain. c, STRUCTURE diagrams (6,881 randomly sampled SNPs, assuming K=3 and 6 populations) reveal 33 admixed SpB strains between three populations (<80% of assignation; red circles) and only three between SpC and SpC* (PE and SK strains; purple circles). d, Relative growth (log of colony size; warm colors) measured at different temperatures and in limiting nutrient conditions for seven carbon and eight nitrogen sources for 151 strains. Ten strains (grey) did not pass the selection step. e, SpC* shows enhanced growth at high temperature, similarly to SpB. Bold letters indicate significant differences among lineages (Tukey test: p≤0.05). The number of strains per lineage is indicated. Error bars indicate interquartile ranges. f, Fraction of cells surviving a freeze-thaw cycle. SpC* and SpC show reduced survival as compared to SpB (p≤0.05). g, Metabolic profiles in limiting nutrient conditions summarized by a principal component analysis (axes 2 and 3 displayed). SpC* is intermediate between SpC (amino-acid preference) and SpB (carbon preference).
Incipient speciation in wild yeast
We sequenced the genomes of 161 strains from the north east of North America to uncover the evolutionary history of this ongoing speciation event, using the European lineage (SpA) recently introduced in America as outgroup (see sup information section 1; Table S1). A phylogeny based on 14,974 filtered polymorphic sites confirms that SpB and SpC form distinct lineages with 2.09±0.01 % nucleotide divergence (Fig. 1b) and we observed no first generation hybrid among lineages, as supported by overall low heterozygosity (~0.1%; Fig. S2). The monophyletic SpB clade shows population substructure (Figs. 1b-c), consistent with its broad geographic distribution (Figs. 1a, S1). This analysis also revealed a third clade that is sister to the SpC lineage, which we call SpC* (Fig. 1b). SpC* consists of eleven SpC strains (22% of all sequenced SpC) showing limited genetic admixture with other SpC strains (Fig. 1c) and a narrow geographic distribution (Figs. 1a, S1).
Because SpC* strains are mostly found near the southwest limit of the SpC distribution, this new lineage may be locally adapted and thus show increased fitness over SpC in the relatively warmer temperatures encountered in this region 18. We measured the growth of 182 strains along a temperature gradient from 10 to 35°C (Fig. 1d) and found that SpC* strains show a growth advantage over SpC (+3-42% colony growth) similar to the SpB advantage over SpC, particularly at high temperature (+5-36%; p< 0.001; Tukey test; Tables S2-S3; Fig. 1e). We previously reported that populations of S. paradoxus showed variation for resistance to freeze-thaw cycles that is correlated with their latitude, with higher resistance in the south where these cycles are more frequent 16. We found that like SpC, SpC* is highly sensitive to a freeze-thaw cycle (8.9% and 5.2% survival, respectively), while SpB has significantly higher survival (74.4%; p < 0.05; Tukey test; Tables S2-S3; Fig. 1f). We also examined other growth conditions that may reflect the large diversity of substrates on which yeasts were isolated7,16,19. Because SpC and SpB were recently shown to perform differently in maltose, lyxose and glucosamine 21, we first examined growth in various carbon sources that are known to shape sap microbial diversity 20. We also examined growth performance on different nitrogen sources, including several amino acids, which are known to be a major nitrogen source in tree sap 22. We found that the three lineages have contrasted metabolic profiles, as suggested by the growth advantage of SpB and SpC on specific carbon and nitrogen sources, respectively (Tukey test: p<0.001; Figs. 1g, Tables S2-S3). Overall, SpC* strains show a metabolic performance profile intermediate between SpB and SpC. Assuming that yeasts have a have high dispersal potential like many other eukaryotic microbes23, these results suggest that the overall performance of SpC* in specific climatic conditions and substrates is a potential cause for its limited ecological distribution in the middle of the SpB-SpC range.
The monophyly of SpC* and its contrasted phenotypes imply limited gene flow with SpC, despite these two lineages have overlapping distributions and co-occur on neighboring trees in the region of sympatry (Fig. S1). This indicates that SpC* is reproductively isolated from SpC and could therefore represent an incipient species. Budding yeasts of the genus Saccharomyces have no obvious pre-zygotic isolation mechanisms in the laboratory24, which allows to measure progeny viability between SpC*, SpB and SpC (Fig. 2). Our results indicate that survival of spores from SpC×SpC* crosses is significantly lower than for SpC×SpC or for SpC*×SpC* (p<0.001; Tukey test) and on the same order of magnitude as what is observed for SpB×SpC, and SpB×SpC* (Fig. 2, Tables S4-S5), despite the nearly 10-fold difference in terms of molecular divergence (0.27% vs. 2.09%).

Post-zygotic reproductive success (RS) within and between lineages as estimated by progeny viability. Circle diameter is proportional to median RS (scale on bottom right; 75 percentiles of RS in white). The number of crosses per category is indicated. SpC* shows partial reproductive isolation with SpC and SpB, to an extent that is similar to crosses between SpC and SpB.
Genome dynamics following hybridization
We found that the SpC* lineage genome is a mosaic of SpC and SpB genotypes, with genomic islands of SpB-like alleles (i.e. fixed within SpB) present in SpC* strains but absent in lineage SpC (Fig. 3a). These SpB-like regions correspond to 12 large regions (28-123kb) of high diversity within the SpC+SpC* clade (He > 0.005; p <0.0001; Figs. S3a-c), overlapping with 23 large regions (22-119kb) for which divergence between SpB and SpC+SpC* populations is low (FST < 0.75; p <0.0001; Figs. S3d-g) relative to the genome-wide average (Fst ≈ 1). These SpB-like regions most likely result from the introgression of SpB into SpC, as shown by a windowbased phylogenetic analysis25 of 24 strains with high-quality genomes (Figs. S3h, S4a) and by a site-wise clustering analysis18 of the 161 strains (HC and LC combined; Fig. 3a). The monophyly of the SpC* clade is maintained even when these introgressed regions are removed (Fig. S4b), suggesting that divergence, although very limited, has accumulated between SpC and SpC* outside of the introgressions. The SpB-like regions represent 2.2-5.8% of the SpC* genome, with 1.6% shared among all SpC* strains. The common set of introgressed regions could be the remnants of a single ancestral hybridization event between the SpB and SpC lineages (marked as H0; Fig. 3a) and the polymorphic introgressed regions could result from the ongoing loss of SpB-like regions in SpC* or from secondary introgression events (Table S6).

a, SpB-like (red) and SpC-like (blue) genotypes (site-wise clustering approach, the SpA outgroup was removed) in 5kb windows along the genome of 147 S. paradoxus strains reveal that SpC* strains are a mosaic of the two lineages, with a vast majority of SpC genotypes. SpC* strains share a common set of SpB-like regions likely acquired during an original hybridization event between SpB and SpC (H0). Polymorphic SpB-like regions distinguish six SpC* groups (see Table S6) that result from the ongoing loss of SpB-like regions or from secondary introgressions. b, Lineage-specific karyotypes of 19 strains with high-quality genomes (SpA removed). Telomere exchanges (thin lines), translocations (thick lines) and inversions (colored portions in chromosomes) as compared to S. cerevisiae, mapped on circular diagrams of the 16 chromosomes (I-XVI). A translocation (VItXIII) between chromosome VI and the right arm of chromosome XIII (240kb) is fixed within SpC* and absent elsewhere. The 42kb inversion in chromosome VI overlapping with a SpB-like region (iVI) was likely transmitted from SpB to SpC* and is absent among the SpC high-quality genomes (see also Fig. S7).
We examined chromosome size variation within and between the SpC and SpC* populations (Fig. S5) and found that the SpC* karyotype was significantly different from both SpC and SpB based on chromosome migration profiles (p < 0.001; Tukey test; Fig. S6). Using de novo genome assembly, we found extensive variation in chromosome configuration among lineages (Fig. 3b) that supports the chromosome profile analysis (Figs. S5-S6). The patterns of inversion and smallscale exchange (≤ 20kb) among telomeric regions that are fixed within lineages confirm the overall S. paradoxus phylogeny (Fig. S7a). One exception is a 42kb inversion within chromosome VI (iVI; Fig. 3b). This inversion is only shared between SpB and SpC* and is absent in SpC (Figs. S7a, Table S1). Another exception is the fusion between chromosomes VI and the right arm of chromosome XIII (VItXIII) that is absent in SpC but fixed in SpC* (Figs. 3b, S7b). This fusion segregates at low frequency in SpB (3.3%; referred as SpBf strains; Figs. S7c). A phylogeny based on the iVI region reveals that SpBf is the sister group of SpC*, suggesting that SpC* inherited iVI and VItXIII from SpBf strains (Figs. S7d). Chromosomal rearrangements are often under-dominant and are thus unlikely to fix within a population26, which could explain the low frequency of VItXIII in SpB.
Chromosomal changes and reproductive isolation
Given the low level of divergence between SpC and SpC* outside of the introgressed regions, reproductive isolation is unlikely driven by molecular incompatibilities such as mismatch repair6, although those could be important in SpB×SpC and SpB×SpC* hybrids. We thus hypothesized that chromosomal changes originating from hybridization contribute to the reproductive isolation between SpC* and SpC. Accordingly, these changes would miss-segregate in the progeny of the F1 hybrids and correlate with spore viability27 (Fig. 4a). We tested this hypothesis by sequencing 325 haploid F2 strains that we obtained from the sporulation of eight SpC × SpC* diploid F1 hybrids (Fig. 4b, Table S7). We identified large genomic regions (100-200kb) where SpC* genotypes segregate unevenly in meiotic products with low spore viability (25%; Fig. 4c), corresponding to either SpB-like regions (IIL, VIr and XIIr; Fig. 4d) or to regions in linkage with the translocation (LT regions; Fig. 4e). We also observed systematic ploidy increase in the translocated arm of chromosome XIII (XIIIr; Fig. 4e). We confirmed that the genomic regions involved in at least two major chromosomal rearrangements that differentiate SpC* from SpC, namely the inversion in chromosome VI (iVI) and the right arm of chromosome XIII (XIIIr), segregate unevenly in the surviving spores and contribute to reduced spore viability, as predicted from their simple Mendelian segregation (Fig. 5a). The iVI inversion also co-localizes with a SpB-like region that is preferentially transmitted in surviving spores (Fig. S8a). Both the frequency of the iVI inversion typical of SpC* (Fig. 5b) and the ploidy of the XIIIr region (Fig. 5c) significantly decrease with spore survival (1,000 random permutations; p<0.05), showing that surviving spores systematically inherit these regions more than expected under normal segregation. The breakpoint of ploidy change in XIIIr coincides with the fusion point with chromosome VI in SpC* (Fig. S8a), indicating that VItXIIIr causes lethal aneuploidy in SpC × SpC* haploid hybrids by the sorting of XIIIr27 (Fig. 5a). We observed no significant ploidy variation on chromosome VI, suggesting that the over-representation of the iVI inversion in the spores and of the associated SpB-like region results from their linkage with XIIIr (LT region; Fig. S8b). These results show that the chromosomal polymorphism segregating at low frequency within SpB (VItXIIIr) was transmitted by hybridization with SpC and now causes reproductive barriers between SpC* and SpC. This transmission led to or co-occurred with the formation of the new lineage SpC* and contributed to reproductive isolation between SpC* and its parental lineage SpC. Because we observed similar patterns of abnormal segregation in other genomic regions (Fig. 5c), we reasoned that additional chromosomal differences between SpC and SpC* also contribute to reproductive isolation (Figs. S6; see also supplementary material, section 27 for discussion). We estimated that taken together, chromosomal differences may lead to lethal aneuploidy in 45% of spores, hence contributing to up to 80% of actual reproductive isolation between SpC and SpC* (Fig. 5a).

a, Expected patterns of ploidy increase (left) and SpB-like genotype frequency variation (right) in genomic regions involved in decrease of SpC*×SpC spore viability. b, Genome sequencing of 384 spores from eight SpC*×SpC crosses (SpC* types indicated) pooled according to the number of surviving spores per tetrad (n=1-4). c, Mean SpBlike genotype frequency in tetrads with low viability (n=1) in discrete 20kb windows along the genome. Strong segregation biases toward SpC (blue) or SpC* (red) genotypes are observed in some SpB-like regions (IIL, VIr, XIIr) and regions linked to chromosomal translocations (LT). d, Frequency of SpC* genotypes (red-blue heatmap, 5kb windows) per pool of spores. Dark regions (intensity proportional to divergence between SpC and SpC* parents) correspond to SpB-like regions. e. Heatmap of ploidy variation (warm color scale, 5kb windows) suggests aneuploidy in hybrid progeny caused by chromosomal translocations. The rightmost breakpoint in XIIIr corresponds to the fusion point with chromosome VI (VItXIIIr, purple). A similar pattern in chromosome II (IIr) suggests the contribution of another chromosomal change (XtIIr, yellow; see Figs. S6, S8). The iVI inversion has no visible effect on ploidy. Ploidy was calculated as sequencing coverage per pool normalized by coverage in a control tetrad (n=4; removed).

a, Translocation VItXIIIr (purple) and inversion iVI (red) typical of SpC* and translocation XtIIr (yellow) typical of SpC, could theoretically lead to reduced reproductive success (RS), increased ploidy in translocated regions (N) and increased frequency of the inversion (FiVI) in surviving spores. The diallelic table represents the expected cumulated effects of translocations on spore survival considering parental (P) or non-parental (NP) associations of chromosomes. Spores (circles) are either diploid (N=2; purple and yellow), haploid (N=1; black) or have no copy of the translocated region (N=0; white), which is presumably lethal (grey frames). Red and blue stars indicate the iVI configurations from SpC* and SpC, respectively. b, The iVI inversion (SpC* configuration; red line, frequency assessed by PCR) is more often transmitted than the SpC configuration (blue line) in tetrads with few surviving spores (number of tetrad per spore category in parenthesis). The purple line indicates the proportion of surviving spores harboring both configurations. c, The right arms of chromosomes II (IIr; yellow) and XIII (XIIIr; purple) and the fusion region of VItXIIIr (purple) show systematic ploidy excess (384 SpC*×SpC hybrid spores; see Fig. 4), which is negatively correlated with the number of surviving spores per tetrad. Mean (points) and standard deviation (bars) among crosses per tetrad category (number of pools per spore category in parenthesis). d, SpB-like regions on chromosomes II (IIL) and VI (VIr) are more likely to be transmitted, while the SpB-like region of chromosome XII (XIIr) is most likely to be lost in surviving spores and this bias in transmission is negatively correlated with the number of surviving spores per tetrad. The following information is given for panels b-d: values expected under a 2:2 segregation (black line) and correlation coefficient (r). Statistical significance (“*”: p≤0.05; “**”: p≤0.01; “***”: p≤0.001) and 95% statistic range (frame) were estimated after 1,000 random permutations among spore categories within crosses (n=8).
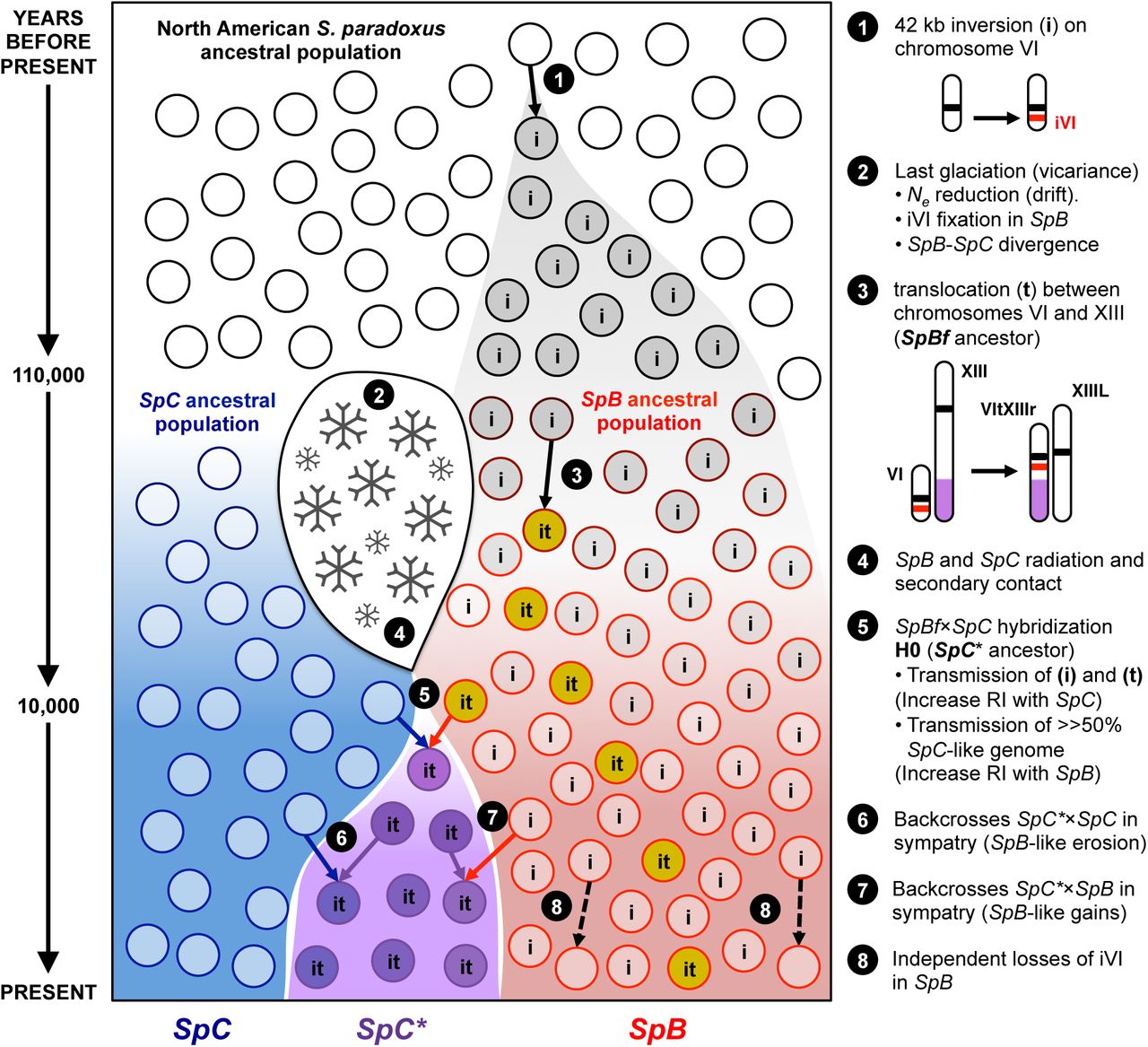
Timing of speciation
The distributions of SpB and SpC mirror those of many post-glacial taxa in this region (Table S8)17. Therefore, we hypothesized that the present population pattern is the consequence of an allopatric separation of the SpC and SpB lineages during the last glaciation (110,000-12,000 years ago) with a secondary contact after the glacial retreats (Fig. 6). The pattern of polymorphism revealed by Tajima’s D statistics is overall negative for the three groups, which suggests recent population expansion, with much greater effects in SpC than SpC* and SpB populations. Because SpC* strains mostly come from locations where SpB and SpC are found in sympatry, the initial hybridization event most likely occurred in the secondary contact zone between the two post-glacial lineages SpB and SpC (Figs. 1a, S1). The common set of introgressed regions would then be the remnants of a single ancestral hybridization event between the SpB and SpC lineages (H0). Assuming that SpB and SpC have originally been separated before the onset of the last glaciation and using polymorphism and divergence data, we found that SpC and SpC* diverged about 10,000 years ago (Fig. S9, Table S9). This date is again consistent with the fact that climate warming facilitated secondary contact and hybridization. Although no independent data point can be used to confirm these estimates, these assumptions correspond to 1.72 generations per day on average, which is consistent with all previous studies (Table S10). Under the same assumptions, we estimated the timing of the introduction of SpA into North America about 300 years ago, and the divergence between American and European lineages at about 176,000 years ago (Fig. S9, Table S9). These again are consistent with previous scenarios15,28 and estimations24 and support the proposed model for the SpB and SpC divergence and secondary contact (Fig. 6).

Genomic and phenotypic analyses support the following scenario (see details in points 1-8): an ancestral S. paradoxus population (black circles) occupied the North-East American forest before the last glaciation (110,000 years ago). Climate change (ice sheet formation; 2) lead to vicariance of the ancestral population in two populations that progressively diverged for nucleotide polymorphisms (red and blue circles). Chromosomal changes (black arrows; 1, 3 and 8) segregated or eventually became fixed by drift, giving rise to the SpB and SpC ancestral populations. Secondary contact after the ice sheet retreat 10,000 years ago (4) and hybridization between SpB and SpC (5) followed by backcrosses (6 and 7) lead to the formation of the new lineage SpC* (purple circles).
Discussion
We report a fully documented case of homoploid hybrid speciation in a eukaryotic microbe. In the yeast S. paradoxus, the incipient species (SpC*) occupies the contact zone of its parental lineages SpB and SpC and displays intermediate phenotypes typical of ecological gradients in this region, indicating that an hybrid zone may have been found in the region 10,000 years ago29. The unequal contributions of SpB and SpC to the SpC* genome suggest several non-exclusive scenarios for the mosaic SpC* genome formation. First, backcrosses between the newly-formed hybrids and parental lineages could have been more frequent with SpC, for instance if SpC strains were more abundant than SpB in the hybrid zone. This would have led to the slow erosion of the SpB haplotypes in the original hybrid. Such mechanism could be further enhanced by incompatibilities between parental genomic backgrounds30 that purged the SpB genome. This is supported by non-fixed SpB-like regions, which segregate unevenly in the surviving SpC×SpC* progeny (Fig.5d). However, backcrosses with SpB cannot be ruled out because the SpB-like region of chromosome II may have been acquired more recently by crosses with SpB (Fig. S9, Table S9). Another possibility is that the introgression of SpB-specific elements in the SpC genome may have conferred a fitness advantage to SpC* in the hybrid zone and this would have maintained these genomic regions specifically3,31. Accordingly, rounds of mitotic recombination32 or repeated selfing within the ancestral hybrid strains6 combined with natural selection could have contributed to the erosion of the SpB regions without the need for backcrosses with the parental lineages.
Studies in experimental evolution33 and on the genomics of industrial and domesticated fungi34 showed that frequent chromosomal rearrangements and genomic instability of hybrids between species may lead to reproductive isolation in artificial conditions. Extensive chromosomal polymorphism also segregates in natural yeast populations24,35, making them a great potential for initiating reproductive barriers and ecological novelties when the ecological opportunities are present. The natural system we report is comparable with S. cerevisiae × S. paradoxus hybrids generated in the laboratory6. These hybrids were also marked by extensive chromosomal rearrangements and strong reproductive isolation with parental species, but did not have the distinctive ecological divergence that has evolved in SpC*. The actual extent of reproductive isolation between SpC* and its parental lineages is likely underestimated because ecological divergence is a potential factor in generating prezygotic barriers in the wild36. Yet we demonstrated that alone, chromosomal differences could be the main cause of postzygotic reproductive isolation. Because these differences were inherited at the very beginning of hybridization and are fixed within lineages, they were likely the bases for initiating the ongoing genomic divergence and speciation. Our observations provide strong evidence that the processes of homoploid hybrid speciation can actually lead to incipient speciation in natural populations of fungi. These processes, which were shown to play key roles in shaping plant2 and animal4 natural diversity, are therefore also shaping eukaryotic microbial diversity. The historical and ecological contexts into which this event took place suggest that changes of microbial species distribution caused by climate changes, in conjunction with the limited possibility for pre-zygotic isolation and the plasticity of their genomes, make speciation by hybridization a potential mechanism of diversification in the forest microbiota37.
Online Method
Strain sampling, genome sequencing and phenotypic characterization
Strain sampling was described previously for most strains7,19. Thirty-two additional strains were sampled in Quebec, New-Brunswick, Maine and Massachusetts in 2013 as described previously7,10. DNA was extracted following standard protocols (QIAGEN DNAeasy). Whole genome sequencing was performed according to Illumina procedures. An initial set of 24 strains was sequenced at high-coverage (HC) and 137 at low coverage (LC) on two separate lanes of HiSeq 2500 Illumina. The HC libraries were prepared using the New England Biolabs® (n = 8), Lucigen® (n = 8) and TruSeq Illumina® (n = 8) protocols with no noticeable difference in quality. Reads can be retrieved from NCBI under the BioProject number PRJNA277692 (BioSamples SAMN03389655-SAMN03389678) and number PRJNA277692 (BioSamples SAMN03389659 - SAMN03389817). Reads were mapped onto the S. paradoxus reference genome (CBS432 38) using Bowtie2 39 and duplicated reads removed using Picard (http://broadinstitute.github.io/picard/). Preliminary SNP calling on these HC libraries was performed using the BCFtools from SAMtools 40 with default parameters. The LC libraries were prepared following the Illumina Nextera XT protocol. All 161 libraries (HC+LC) were used together for an overall SNP and indel calling using FreeBayes41 with default parameters and variants were filtered using VCFlib 41. The global phylogenetic reconstruction was performed with filtered variants in complete deletion using PhyML 42 with the model TN93 and a LRT for branch support. Population structure was examined using STRUCTURE 43 with a subset of markers randomly sampled in the genome and an assumed number of clusters between 1 and 6.
Phenotypic analyses
The ability of SpC (n=5) and SpB strains (n=5) to utilize specific carbon sources and nitrogen sources was initially examined using BIOLOG plates (assay plates PM1, PM2 and PM3, catalog numbers 12111, 12112, 12121) following the manufacturer protocol with the modifications described in the supplementary material (section 12, Fig. S10). A subset of these conditions was then selected to measure growth rates of 182 strains covering SpA, SpB, SpC and SpC* in order to examine how SpC* strains compare to the parental SpC and SpB lineages. The 182 strains were assembled into two arrays of 1536 colonies on solid medium (12 replicates per strain per plate, omnitrays) following the procedures outlined in Rochette et al.44 on rich standard yeast YPD (yeast extract, peptone, dextrose) medium. Ten strains, including two SpC* strains of type SK, did not grow on these arrays and thus were removed from the downstream analyses. These arrays were then replicated on other YPD plates and incubated in a range of temperatures (10, 15, 20, 25, 20 and 35°C). In another experiment, strains were transferred from YPD plates to YP medium with alternative carbon sources (2% Maltose, 2% Galactose, 2% Mannose, 2% Fructose, 2% Methyl α-D-glucopyranoside, 2% Sucrose or 2% Glucose) or synthetic medium with various sources of nitrogen (5 g/L Isoleucine, 5 g/L Tyrosine, 5 g/L Proline, 5 g/L Glutamine, 5 g/L Asparagine, 5 g/L Lysine, 5 g/L Histidine or 5 g/L Glycine) and grown at 25°C. In each of the temperature and the carbon and nitrogen source experiments, strains were transferred for a second round of selection in selective conditions. All experiments were performed using a BMCBC S&P Robotic platform with 1536 pin tools (0.5 mm diameter). Colony size was measured by imaging plates and counting pixel intensities of colonies as described in Diss et al.45. Freezethaw survival was performed as described in Leducq et al.16. Data from Leducq et al. was used in addition to another set of 36 strains. Briefly, samples of cells were frozen at -80°C and a control sample was kept on ice. The two samples were then plated on YPD medium and the number of colony-forming units (CFUs) in the frozen sample/control sample was used as a measure of survival rate.
Measurement of reproductive isolation
The detailed procedures were described in Charron et al.17 The HO locus was inactivated by complete gene deletion using antibiotic resistance cassettes and homologous recombination, and strains were sporulated to isolate haploid strains. Mating types were tested by performing crosses with control strains. Heterothallic strains expressing an antibiotic resistance cassette at the HO locus were mated and diploid cells were selected using a pair of antibiotics. The resulting diploid strains were then sporulated and tetrads with 4 apparent spores were dissected and deposited on YPD plates. Spore survival was estimated as the number of colony-forming spores after 72h divided by the total number of spores dissected for a given cross.
Analysis of hybridization and identification of introgressed regions
Genomic islands of high diversity (He) and low differentiation (Fst) were identified from the 24 HC genomes in 500bp discrete windows along the genome. Expected heterozygosity (He) was estimated as He = 2p(1-p) and the fixation index Fst was computed using methods implemented in the R package HIERFSTAT46. Peaks of high Fst and low He values were identified by computing t-statistics (p-value < 0.0001) between He and Fst estimates in 10kb contiguous regions of 20x500bp regions. The phylogenetic approach used to identify introgressed regions was performed on discrete 2,000kb windows (Fig. S11) and the phylogeny of these regions were determined based on pairwise evolutionary distance (ED) using mutation probabilities (μp) estimated from Saccharomyces cerevisiae mutation accumulation experiments 28 (see supplementary material section 17 for details; Fig. S12). The extent of introgression in the SpC population was examined by identifying introgressed regions in the LC strains. We used the SpA reference genome (CBS432), 10 SpB and 6 SpC non introgressed HC genomes as reference and used sites with fixed alleles in each group as a diagnostic for introgression in the overall population (see supplementary material section 18). All strains were assigned to one group or the other based on these diagnostic markers across the entire genome using 5Kb discrete windows. Error rates were estimated using two sequencing libraries obtained from the same homozygous clone.
Chromosomal changes
Potential aneuploidies and chromosomal changes were analyzed by Pulse Field Gel Electrophoresis (PFGE). All samples were treated as described in Charron et al.17 and standard genomic DNA from Saccharomyces cerevisiae was used as control on all gels. Details on the mage analyses are provided in supplementary material (section 19). The genomes of the 24 HC trains were assembled using ABySS47. Several parameters were tested on a subset of strains and led to the selection of K=64 as a final parameter for the assembly (Fig. S13). Chromosomal rearrangements were detected using scaffolds aligned on the reference S288C genome with methods implemented in MAUVE48 and through the identification of chimeric scaffolds, i.e. scaffolds that align with two non-contiguous regions of the reference genome. Chromosomal fusions were confirmed by mapping reads on the chimeric scaffolds. One major chromosomal inversion (iVI) was confirmed by PCR in the collection of sequenced strains (Fig. S14, Table S11).
Analysis of segregation in F2 hybrids
We examined the segregation of the SpC and SpB-like genomic regions in the SpC × SpC* F2 hybrids by genotyping 25 pools of haploid strains (3–30 strains per pool) grouped by crosses and by number of spores that survived in a given tetrad (n =1–4). Pools were used to construct 25 Illumina Nextera XT libraries and were sequenced in one lane of Illumina HiSEQ2500. Reads can be retrieved from NCBI under the BioProject number PRJNA277692 (BioSamples SAMN03389818-SAMN03389842). Reads were mapped on the reference genome of S. paradoxus CBS43238 using Bowtie239. The depth of coverage (number of reads at a given position) was analyzed along the genome as a measure of ploidy to identify regions that segregate abnormally. We also controlled for coverage along the genome of parental strains to ensure that variations were not inherited by aneuploidy in natural populations, which was not the case (6% of strains; Figs. S15-S16). Variant calling was performed on the pooled libraries using Freebayes 41 with options –J and –p 2. Genotypes of the spores were compared directly with that of their parental strains. The inversion on chromosome iVI in the spores was genotyped in the spores from the same crosses by PCR using oligonucleotides flanking the inversion.
History reconstruction and timing of introgression
The time of divergence between SpC, SpB and SpC* was estimated using BEAST49 from the introgressed regions and in regions without traces of introgression using the set of HC genomes. Several groups were formed according to the overall phylogeny and the introgressed regions present: CBS432, SpA (5 strains), SpB (10 strains), SpC (5 strains), PE (1 SpC* strain), PP (2 SpC* strains) and IO2 (1 SpC* strains). As calibration time we assumed that SpC and SpB’s initial divergence was initiated at the onset of the last glaciation 110,000 years ago (see supplementary material section 28 for the justification).
Acknowledgments:
We thank A. K. Dubé, K. Lambert, R. Nuwal, S. Haughian, A.-E. Chrétien, M. Caouette, I. Kukavica-Ibrulj, R. Levesque and the IBIS sequencing platform (B. Boyle) for technical help; P. Sniegowski, M.-A. Lachance and J. Anderson for providing strains, I. Levade and C. Lemieux for discussion, and N. Aubin-Horth, A. Moses, L. Bernatchez, J. Shapiro, S. Pavey, F. Rousseau-Brochu, I. Gagnon-Arsenault, A. K. Dubé, A.-M. Dion-Côté, H. Vignaud, M. Nigg and three anonymous reviewers for comments on the manuscript. The data reported in this paper are provided in the supplementary materials and raw sequencing reads were deposited in NCBI (BioProject PRJNA277692).
Funding support: NSERC Discovery Grant and HFSP Grant (RGY0073/2010) to C.R.L. FRQS Fellowships to J.-B.L., NSERC USRA Summer scholarships to L.N.T., FRQNT and NSERC PhD Fellowships to G.C. Some of this material (yeast collection) is based upon work supported by the National Science Foundation under Grant No. DEB-1253634 (C.T.H.) and by the DOE Great Lakes Bioenergy Research Center (DOE Office of Science BER DE-FC02–07ER64494). C.T.H. is a Pew Scholar in the Biomedical Sciences, supported by the Pew Charitable Trusts. CRL is a FRQS Junior Investigator and a Canada Research Chair in Evolutionary Cell Biology.
The authors declare no conflict of interest. JBL, CRL, LNT and GC planned the experiments. GC, JBL and CE performed experiments. JBL, LNT and JPV performed bioinformatic analyses. PS, KS, CTH and GB provided strains and discussion in the early stages of this study. JBL and CRL drafted the manuscript with contributions from LNT, GC, CE, PS and GB.
Footnotes
↵† These authors contributed equally to this work.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}