

ABSTRACT
Predicting the future evolutionary state of a population is a primary goal of evolutionary biology. One can differentiate between forward and backward predictability, where forward predictability is the probability of the same adaptive outcome occurring in independent evolutionary trials, and backward predictability is the likelihood of a particular adaptive path given the knowledge of the starting and final states. Most studies of evolutionary predictability assume that alleles along an adaptive walk fix in succession with individual adaptive mutations occurring in monomorphic populations. However, in nature, adaptation generally occurs within polymorphic populations, and there are a number of mechanisms by which polymorphisms can be stably maintained by natural selection. Here we investigate the predictability of evolution in monomorphic and polymorphic situations by studying adaptive walks in diploid populations using Fisher’s geometric model, which has been previously found to generate balanced polymorphisms through overdominant mutations. We show that overdominant mutations cause a decrease in forward predictability and an increase in backward predictability relative to diploid walks lacking balanced states. We also show that in the presence of balanced polymorphisms, backward predictability analysis can lead to counterintuitive outcomes such as reaching different final adapted population states depending on the order in which mutations are introduced and cases where the true adaptive trajectory appears inviable. As stable polymorphisms can be generated in both haploid and diploid natural populations through a number of mechanisms, we argue that natural populations may contain complex evolutionary histories that may not be easily inferred without historical sampling.
INTRODUCTION
Predicting evolution is one of the fundamental challenges of evolutionary biology (reviewed in de Visser and Krug (2014)). This question became particularly prominent with Gould’s famous thought-experiment on “replaying the tape of life” (Gould, 1990). Gould wondered whether we would regenerate the observed evolutionary history of the world if we reset our evolutionary history to any point in the past and let evolution retake its course from there. More generally, we can ask whether it is possible to predict the path or the final destination of the evolutionary process from a given starting point. It is also possible, however, to ask whether we can reconstruct the true evolutionary trajectory given the final adapted state (Weinreichet al., 2006). This distinction between types of predictability is rarely made (however see Nourmohammadet al. (2013) and Szendro et al. (2013)), so we formalize the methods for studying predictability and utilize these distinctions to study the impact of polymorphism on the predictability of evolution.
Forward predictability of evolution
We define forward predictability as the probability of observing a particular future evolutionary outcome from a known starting state. Previous experimental evolution studies have generally (but not always) focused on the forward predictability of evolution. This type of analysis can be done at a number of levels, including the predictability of overall fitness changes, phenotypic shifts and different levels of genotypic changes (pathways, genes, and individual mutations).
For example, Fereaet al. (1999), Cooperet al. (2003) and Fong et al. (2005) evolved independent replicates of microbes and observed similar changes in gene expression and growth rate in the evolved clones. A large study of 145 parallel long-term experimental evolutions with Escherichia coli grown at elevated temperature showed that the same genes and pathways were repeatedly targeted for mutations in independent populations (Tenaillonet al., 2012) as did a study of 40 replicate Saccharomyces cerevisiae batch culture evolutions (Langet al., 2013) and a study that sequenced clones from 10 replicate evolutions for each of 13 different genetic backgrounds (Kryazhimskiyet al., 2014). Tenaillonet al. (2012) also observed a high degree of parallel evolution at the level of individual nucleotides, but nucleotide level parallelism was rarely observed by Langet al. (2013). Herron and Doebeli (2013) evolved E. coli under multiple carbon sources and repeatedly observed the evolution of two distinct ecotypes with differential ability to grow on each carbon source. By sequencing independent replicate clones of both ecotypes, they found the same genes, and sometimes the same exact mutations invading these replicate populations and differentiating the ecotypes. These studies suggest that evolution is indeed forward predictable to a surprising degree.
Repeated evolution has been observed at both the genetic and morphological levels in natural systems as well (reviewed in Stern (2013)). Kviteket al. (2008) showed that highly divergent yeast strains isolated from oak trees had similar growth rates across a panel of diverse growth conditions. Studies of Anolis lizards in the Caribbean show repeated independent adaptive radiations into similar niches across the islands (Losos, 1998). In addition, a study of the adaptive radiation of cichlid fish in Lake Tanganyika showed convergent morphological evolution when the skeletal morphology of the various species was compared to their phylogeny (Muschicket al., 2012).
Backward predictability of evolution
In addition to Gould’s thought-experiment, one can study predictability in a historical manner. Given the current state, we can try to predict the ancestral state or the evolutionary path that resulted in the current state of the study system. We call this backward predictability, as it requires us to look backward in time. For example, one can try to predict exactly how corn or rice became domesticated from one or more wild ancestors (Matsuokaet al., 2002; Molinaet al., 2011), identify the ancestral species that gave rise to Darwin’s Finches (Darwin, 1872; Satoet al., 2001), or reconstruct the ancestral state of a particular protein (Ortlundet al., 2007).
Alternatively, if we already know the ancestral state, we can try to predict the particular order of mutations or phenotypic states that led to the evolution of the current state. Weinreichet al. (2006) conducted a seminal study of backward predictability in this sense, using a combinatorially complete reverse genetic study design pioneered by Malcolmet al. (1990). Weinreichet al. (2006) reconstructed every possible combination of five mutations in the beta-lactamase gene in E. coli which are known to lead to high levels of resistance to the drug rifampicin. They then assayed each genotype’s resistance to the drug, which they used as a proxy for fitness. Using this data, they determined the fitness changes involved in every step of each of the 5! = 120 possible mutational paths that converts the wild-type genotype to the resistant five-mutant genotype. A mutational path was deemed viable if fitness monotonically increased with every step, that is, there were no mutations along the path that decreased resistance to the drug.
Weinreichet al. (2006) found that only 18 of the 120 possible paths were viable, suggesting high backward predictability of evolution. In contrast, Khanet al. (2011) performed an analysis of five adaptive mutations from experimentally evolved bacterial lineages using identical methodology and found that a majority of the orders were viable. Finally, Frankeet al. (2011) studied backward predictability in all subsets of two to six mutations in an empirical eight-locus system and found that the number of viable paths varied widely for a given subset size. For example, they observed both zero and nine viable paths (out of 24 possible) in different four-locus subsets. The varying degrees of backward predictability found in these different systems does not yet allow us to draw general conclusions, and the laborious nature of the experiments makes it challenging to study more than a few mutations at a time. In addition, without knowing the true order in which the mutations arose in the population, it is unclear how accurate backward predictability analysis actually is.
Predictability in Fisher’s Geometric Model
Overall, there seems to be no consensus on whether evolution is backward predictable using the method of Weinreichet al. (2006). It is also unclear how forward and backward predictability are correlated with each other. In principle, one would want to conduct forward evolution and then conduct backward predictability analysis on the same system to understand their relationship. However such studies would be extremely laborious, and given the disparate answers coming out of different experimental systems, a large number of independent experiments in many systems would need to be conducted to give a convincing answer.
Another difficulty in experimental evolution studies of predictability are practical limitations in sampling adaptive mutations. As most studies can only afford to sample a few adapted individuals from a given experiment, mutations must be at high frequency to be observed and a common assumption is that each of these mutations fixed in the population in succession (Gillespie, 1983, 1984; Orr, 2002; Weinreichet al., 2006; Khanet al., 2011; Frankeet al., 2011). However, we know that mutations can be maintained in a polymorphic state by a number of mechanisms. These include negative frequency-dependent selection (Levinet al., 1988; Iserbytet al., 2013), spatial and temporal fluctuations in selection (Rainey and Travisano, 1998; Kasumovicet al., 2008; Saltz and Nuzhdin, 2014) and heterozygote advantage (also called overdominance, Takahata and Nei (1990)). Polymorphisms can also be present in an unstable form through clonal interference (Desai and Fisher, 2007; Herron and Doebeli, 2013; Kvitek and Sherlock, 2013; Langet al., 2013). The presence of functionally consequential polymorphisms in a population can in principle significantly alter predictability analysis as the selective effect of a new mutation may be dependent on other alleles segregating in the population (fitness epistasis). Many of these polymorphisms are either lost by the end of the experiment or are not observed in the sampled adapted individuals, leading to incorrect inferences of predictability. Additional complications can arise when estimating predictability as mutations can occur in multiple backgrounds in a given population, so the likelihood of each mutation occurring in a particular background also has to be taken into account, as well as any epistatic interactions the mutation has with the rest of that background.
Due to the challenges of isolating sufficient numbers of independent adaptive mutations from experimental populations to study predictability, we utilize a simulation-based approach to study the impact of polymorphisms on forward and backward predictability. We employ Fisher’s geometric model (FGM, Fisher (1930)), which is a well-studied (Orr, 1999, 2005) phenotypic model that treats individuals and alleles as a phenotype that is a vector in coordinate space with a fitness that is determined by the distance of the individual’s phenotype from a predefined optimal phenotype using a gaussian function (Figure 1a). Selliset al. (2011) showed that adaptive mutations in diploid FGM simulations are frequently overdominant if the mutations are sufficiently large in phenotypic space, resulting in balanced polymorphisms. Such overdominant mutations are stable but can be driven out of the population by subsequent adaptive mutations. As we are interested in the interaction between balanced polymorphic states and the predictability of evolution, we select the distribution of mutational effects such that some evolutionary trajectories contain overdominant mutations, generating stable polymorphisms, and others do not. We then compare both types of trajectories to understand how polymorphisms influence predictability. We conclude that the presence of polymorphic states has a substantial qualitative effect on the predictability of evolution, such that at least in this model, forward and backward predictability are inversely correlated.
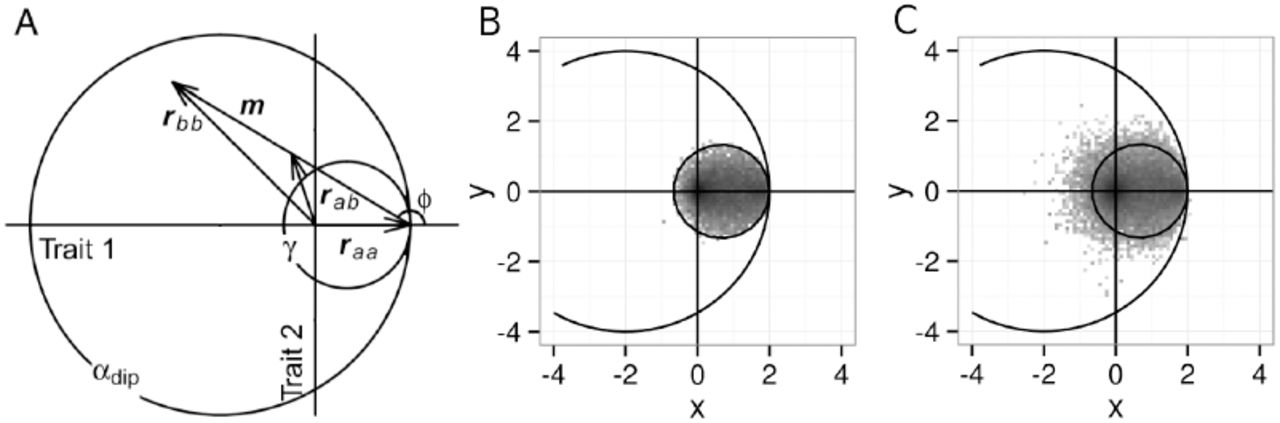
(A) Modified from Figure 2A Sellis et al 2011. Two orthogonal axes represent independent character traits. Fitness is determined by a symmetrical Gaussian function centered at the origin. Consider a population initially monomorphic for the wild-type allele  . A mutation m gives rise to a mutant phenotype vector
. A mutation m gives rise to a mutant phenotype vector  . The phenotype of the mutant heterozygote assuming phenotypic codominance (h = 1/2) is
. The phenotype of the mutant heterozygote assuming phenotypic codominance (h = 1/2) is  . The different circles specify the areas in which mutations are adaptive (i.e. successfully invade the population, αdip) and replacing (i.e. fix in the population, γ) in diploids. (B) Density plot of all phenotypes of homozygous individuals observed in the adaptive walks of FGM simulations that do not contain overdominant mutations. Note that all observed phenotypes lie within γ, as all mutations must be replacing and not balancing in this group of simulations. Circles denote αdip and γ as described in (A). (C) Homozygous phenotypes for simulations that do contain overdominant mutations. Note that a large number of phenotypes lie outside of γ, as expected for overdominant mutations, confirming that we are correctly separating walks with and without overdominant mutations. When comparing B and C, we observe that simulations with overdominant mutations are less forward predictable than those without such mutations.
. The different circles specify the areas in which mutations are adaptive (i.e. successfully invade the population, αdip) and replacing (i.e. fix in the population, γ) in diploids. (B) Density plot of all phenotypes of homozygous individuals observed in the adaptive walks of FGM simulations that do not contain overdominant mutations. Note that all observed phenotypes lie within γ, as all mutations must be replacing and not balancing in this group of simulations. Circles denote αdip and γ as described in (A). (C) Homozygous phenotypes for simulations that do contain overdominant mutations. Note that a large number of phenotypes lie outside of γ, as expected for overdominant mutations, confirming that we are correctly separating walks with and without overdominant mutations. When comparing B and C, we observe that simulations with overdominant mutations are less forward predictable than those without such mutations.
METHODS
Simulations
We model adaptive walks in diploid populations with Wright-Fisher simulations using Fisher’s geometric model (FGM) as in Selliset al. (2011). In FGM, alleles are represented as a vector in n-dimensional phenotype space (Figure 1a). The simulations use code modified from Sellis et al. to allow for more than 2 dimensions. We perform 10,000 replicate simulations with population size N = 5, 000 for 10,000 generations. We explore two models, one with two dimensions and one with 25 dimensions. We partition our adaptive walks into those that do and those that do not contain overdominant mutations to study the impact of balanced states on predictability. For the remainder of our analysis, we identify the most frequent allele in each simulated population at the end of 10,000 generations of evolution and study the mutations present on that allele. We limit our analysis to studying the first five mutations of each adaptive walk and ignore simulations with fewer than 5 mutations in order to control for the length of the adaptive walk when studying predictability.
Forward Predictability Analysis
We calculate the forward predictability of the adaptive trajectory using two metrics. In both of these metrics, we only consider homozygous phenotypes. Our first metric, maximum pairwise distance, considers pairs of adaptive walks. We compute the maximum of the phenotypic distances between the observed single mutant phenotypes of the two adaptive walks, the double mutant phenotypes, the triple mutant phenotypes etc. Our second metric measures the maximal deviation from the optimal trajectory. For each adaptive walk, we compute the maximal phenotypic distance of any encountered (homozygous) phenotype from the line segment connecting the ancestral phenotype and the optimum.
Backward Predictability Analysis
We compute backward predictability on adaptive walks of exactly five mutations. We calculate the probability of all possible mutational orders for the given set of mutations in a manner similar to Weinreichet al. (2006), but generalized to allow balanced states as the experimental protocol of Weinreichet al. (2006) assumes that every mutation along each mutational order fixes in succession. We summarize the set of possible mutational orders for a given set of mutations through the effective number of trajectories statistic, which we define as
 where p is the probability of each mutational order possible for a given set of mutations. If no mutational order is viable (has nonzero probability), the effective number of trajectories is defined to be 0. Please see the Supplementary Methods for full methodological details.
where p is the probability of each mutational order possible for a given set of mutations. If no mutational order is viable (has nonzero probability), the effective number of trajectories is defined to be 0. Please see the Supplementary Methods for full methodological details.
RESULTS
We explore the predictability of evolution in the framework of Fisher’s geometric model (FGM) of adaptation. In FGM, alleles are represented as vectors in coordinate space, with individuals having a phenotype that is the average of the phenotypes of their constituent alleles. Mutations are vectors that modify the phenotype of an allele, and fitness is a guassian function of the distance of the individual’s phenotype from the optimal phenotype (which we define as the origin).
In order to focus on the effect of polymorphic states on the predictability of evolution, we choose a parameter regime that generates simulations both with and without overdominant mutations after a number of trial simulations with various parameter values. We perform 10,000 replicate simulations of adaptation under FGM in diploids with N = 5000 individuals. Mutational magnitudes are drawn from an exponential distribution with mean  and the population is initiated at two units from the optimum. The mutation rate is 5 ∗ 10−6, which results in a mutation-limited regime (significantly less than one mutation per generation as 2 ∗ N ∗ µ = 0.05), in order to minimize the generation of polymorphic states by clonal interference so that we can focus on only those polymorphic states generated by overdominant mutations.
and the population is initiated at two units from the optimum. The mutation rate is 5 ∗ 10−6, which results in a mutation-limited regime (significantly less than one mutation per generation as 2 ∗ N ∗ µ = 0.05), in order to minimize the generation of polymorphic states by clonal interference so that we can focus on only those polymorphic states generated by overdominant mutations.
We conduct our simulations using an FGM of two dimensions, and show that our qualitative results also hold at 25 dimensions. In the 25 dimension regime, we need to rescale our mutational magnitude mean to 5 in order to obtain a sufficient number of walks with five mutations over our 10,000 generation simulations for statistical analysis. For all of our statistical analyses, we consider only those mutations that are present on the most frequent allele at generation 10,000. Such mutations are typically the only ones available for analysis in a natural system. We additionally limit our analysis to studying the first five mutations of each adaptive walk, and ignore simulations with fewer than five mutations in order to compare adaptive walks of equal lengths. We partition the resulting five-mutation adaptive walks into those that do (n = 4975, 1548 in simulations with two and 25 dimensions, respectively) and do not (n = 1251, 10) contain overdominant mutations to study the impact of balanced polymorphisms on the predictability of evolution. The presence of overdominant mutations in an observed five-mutation adaptive trajectory is detected by the observation of a set of alleles during the FGM simulation that are capable of being maintained as a balanced polymorphism (Kimura, 1956). For details, please see the Supplementary Methods.
Predictability of Adaptive Walks
We first consider the forward predictability of phenotypic paths, which we define as the tendency of independent adaptive walks to explore similar portions of phenotypic space. The ability of adaptive walks with overdominant mutations to explore a larger phenotypic space compared to walks without overdominance (α-dip vs γ, Figure 1a) should lead to lower predictability of the phenotypic intermediates along the adaptive walk, which is confirmed by visual inspection of our simulations (Figure 1b,c) and is consistent with the findings of Selliset al. (2011).
We quantify forward predictability by measuring the distribution of maximal phenotypic distances between pairs of independent adaptive trajectories. Pairs of walks with overdominant states are, on average, 40% further apart than walks without overdominant mutations and are therefore less forward predictable (Figure 2, Kolmogorov-Smirnov test p ≪ 10−10). We also measure forward predictability as the maximal phenotypic distance of each observed trajectory from the optimal trajectory - the vector from the ancestral phenotype to the optimal phenotype. We observe that the presence of overdominant mutations in a walk increases the average distance from the optimal trajectory by 5% (Figure 3, Kolmogorov-Smirnov test p ≪ 10−10), again suggesting that overdominant mutations decrease forward predictability.

Shown are the cumulative distributions of the maximum phenotypic distance between independent pairs of adaptive walks, excluding the ancestral state. This is a measure of the phenotypic forward repeatability of independent walks on the same evolutionary landscape. The maximum phenotypic distance in simulations without overdominant states is significantly less than in simulations with such states (Kolmogorov-Smirnov test p ≪ 10−10).

Shown are the cumulative distributions of the maximum distance from the optimal trajectory of adaptive walks. This is a measure of the phenotypic forward predictability of walks. The maximum distance from the optimal trajectory in simulations without overdominant mutations is significantly less than those with such mutations (Kolmogorov-Smirnov test p ≪ 10-10).
We then study backward predictability in a manner similar to Weinreichet al. (2006). As before, we limit our analysis to adaptive walks of exactly five mutations, which is comparable to many recent experimental studies of backward predictability (Weinreichet al., 2006; Khanet al., 2011; Frankeet al., 2011). Backward predictability analysis requires knowledge of the five mutations that occurred during the FGM simulations and computes the likelihood of every possible order of those five mutations in generating the observed adapted five-mutation allele (e.g. see Weinreichet al. (2006) Figure 2). In order to conduct this analysis, we compute the probability of every possible path to the five-mutant state by successively introducing each of the five mutations into the population and assessing the probability of each of these mutations to successfully invade the population (see Supplementary Methods). Although we artificially constrain the available phenotypes to only those generated by combinations of the five mutations under consideration, this analysis is a model for studying predictability in situations where there are only a few possible adaptive mutations, such as the drug resistance mutations used by Weinreich et al. We compute the effective number of adaptive trajectories for each adaptive walk, with a higher number suggestive of a lower backward predictability.
The results of our backward predictability analysis are shown in Figure 4. We find that in contrast to forward predictability, overdominant states decrease the effective number of paths (and thus increase backward predictability) in a walk by 30%, on average (Kolmogorov-Smirnov test p ≪ 10−10). In other words, conditional on reaching a particular five-mutant state, it is more probable that independent trials of a walk that experienced at least one overdominant state will use the same mutational order in repeated trials relative to a walk without overdominant states. We also utilize the mean path divergence of Lobkovskyet al. (2011) to study backward predictability and find that overdominant states resulted in walks that were 10% less divergent (and thus more backward predictable), on average (Kolmogorov-Smirnov test p ≪ 10−10).

Shown are the cumulative distributions of the effective number of paths for adaptive walks with five mutations. This is a metric of backward predictability of evolution. Each mutation is introduced into the ancestral background in every possible order, and the number of viable mutational orders, weighted by their probabilities, determines the effective number of paths. The effective number of paths in simulations without overdominant mutations is significantly greater than in simulations with such mutations (Kolmogorov-Smirnov test p ≪ 10-10).
Multiple End States
In addition to studying the probability of a given mutational order in our backward predictability analysis, we also study the adapted population state that results from each viable mutation order. In particular, we observe that when mutations are introduced in different orders, the population encounters different intermediate alleles, resulting in instances where the final adapted five-mutant allele can balance against different intermediate alleles depending on the order in which the mutations were introduced into the population. We also observe instances where walks that did not experience balanced states in the FGM simulations generate balanced states when introduced in a different order.
We find that 53% of all walks have at least two different end population states containing the final adapted allele, with a maximum of 19 different population states for a single set of five mutations. We also find that the presence of overdominant mutations in the FGM simulation has a significant effect on whether there are multiple end states observed. The presence of an overdominant mutation in the observed walk increases the frequency of multiple end states from 30% to 60%. Our results suggest that adaptation occurring in the same genetic background, in response to the same selection pressure and using the same mutations, can result in significantly different final population states depending on the historical order in which the adaptive mutations occurred.
Qualitative categorization with regard to backward predictability
We analyze our backward predictability results to discern qualitative categorizations of our simulations. We find four broad categorizations of simulations: 1) simulations whose backward predictability reconstructions of the five-mutant allele by introducing the mutations in the order observed in the FGM simulation generate no balanced states, 2) those reconstructions that do generate balanced states, 3) reconstructions where the order of mutations that was observed in the simulation was impossible to reconstruct due to deleterious intermediate states during the reconstructions and 4) reconstructions where every possible order of mutations was impossible due to deleterious intermediate states (which is a subset of category 3).
We observe 2326, 3898, 89 and 5 simulations in each of these four categories, respectively. We can further separate these categories by conditioning on our original definitions of whether or not a simulation contained an overdominant intermediate state (i.e. whether there was a set of alleles that could be maintained in a stable balanced state at any point during the FGM simulation before the 5-mutant state reached 5% frequency). We find 1187, 62, 2 and 0 simulations in each of these four categories, respectively, among the simulations that we had previously identified as not containing overdominant intermediate states while we observe 1139, 3836, 87 and 5 simulations in each of these four categories, respectively, among simulations that we had previously identified as containing overdominant intermediate states.
The presence of backward predictability reconstructions where the observed order (and in a few cases, every order) of mutations is impossible is surprising. We hypothesize that this is due to the presence of adaptive alleles that are generated and stably maintained during a walk that are transient and do not survive until the end of the simulation. We call these “hidden alleles”, as they are hidden from almost all modern experimental studies of adaptation. Lack of knowledge of hidden alleles appear to decrease the computed probability of the true adaptive path observed in the FGM simulations, and in extreme cases, can make the true path impossible to reconstruct. Visual inspection of adaptive trajectories that are unable to be successfully reconstructed confirms this intuition (Figure 5). Backward predictability reconstructions that incorporate all mutations present at ≥ 1% frequency at any point in the simulation, regardless of whether the mutation was present on the allele sampled at the end of the simulation, can successfully reconstruct the observed adaptive trajectory of this previously impossible evolutionary outcome, confirming the necessity of hidden alleles for the viability of the observed adaptive trajectory in these instances.

The frequency of the two mutational lineages that reached at least 1% frequency in the population are shown throughout the 10,000 generations of the simulation. The main lineage, ending with allele ABCDGH, is at high frequency at the end of the simulation, while the minor lineage, ending with allele ABCDEF (a “hidden allele”) is at low frequency at the end of the simulation.
In the simulation, four mutations initially fix in quick succession, resulting in allele ABCD fixed in the population. At this point, mutations causing balanced polymorphisms result in branched mutational lineages. Mutation E is the first mutation to occur on allele ABCD, generating a balanced polymorphism between alleles ABCD and ABCDE and allowing both alleles to be stably maintained in the population at intermediate frequency. Mutation F then quickly occurs on the background of allele ABCDE, generating allele ABCDEF which also balances with allele ABCD. Mutation G then occurs on the background of allele ABCD generating allele ABCDG soon afterwards, which balances with allele ABCDEF. Finally, mutation H occurs on allele ABCDG generating allele ABCDGH, which outcompetes all other alleles and is nearly fixed by the end of the simulation.
In our backward predictability reconstructions, we consider only the first five mutations of the most frequent allele at the end of the simulation, that is, we consider only mutations A, B, C, D and G as these were the first five mutations on allele ABCDGH. In attempting to reconstruct this observed order of mutations, we find that we can successfully introduce mutations A, B, C and D in order, but mutation G, which results in allele ABCDG, is not beneficial if allele ABCD is the only other allele in the population (data not shown). Therefore, the true order of mutations is impossible to reconstruct in this case when only sampling allele ABCDGH at the end of the simulation. However, if we also consider mutations E and F, we are able to successfully reconstruct the intermediate steps of the observed adaptive trajectory, suggesting that the presence of allele ABCDEF is necessary for allele ABCDG to be beneficial (data not shown).
We then compare the forward and backward predictability metrics described above on the different categories of simulations. In particular, we compare the simulations that were initially defined as not containing overdominant states at any point to those that did not have balanced states in the backward predictability analysis but did have balanced states during the FGM simulation. We find no significant difference between these sets of simulations by any of our predictability metrics (maximum pairwise distance, maximum distance from optimal trajectory and effective number of paths Kolmogorov-Smirnov test p > 0.05). This result suggests that the signal in our predictability metrics is being driven by the presence of balanced states between intermediate alleles along the adaptive trajectory to the five-mutant allele rather than a general feature of observing balanced states in our simulations as a whole.
High Dimensionality
In our implementation of Fisher’s Model, balanced states arise when mutations are overdominant. The presence of additional phenotypic dimensions, which seems realistically plausible from observed rates of pleiotropy (Dudleyet al., 2005; Albertet al., 2008), increases the frequency of overdominant mutations (Selliset al., 2011). However, this concordantly decreases the fitness advantage of the average new beneficial mutation, decreasing the number of adaptive mutations that successfully invade the population over our 10,000 generation FGM simulations. To study the impact of high dimensional landscapes on predictability, we conducted simulations using 25 dimensions with a mean mutation size of 5. The increase in mean mutation size relative to our original two dimensional simulations is necessary to generate a sufficient number of walks containing at least 5 mutations within 10,000 generations. We again partitioned the simulations into those with (n = 1548) and without (n = 10) overdominant mutations at any point of the FGM simulation before the time when the five-mutant allele reached 5% frequency.
We observe the same qualitative results in 25 dimensions as in 2 dimensions (see Supplementary Figures 1-4). In general, it appears that our conclusions about predictability of adaptive walks do not depend on the dimensionality of the system, and only on the presence of overdominant mutations in the adaptive walk.
DISCUSSION
In this study, we explored the predictability of evolution using Fisher’s geometric model. We distinguished between forward and backward predictability, where forward predictability measures the likelihood of the same or a similar adaptive trajectory occurring in independent evolutions, while backward predictability measures the likelihood of a particular order of adaptive mutations given the ultimate adapted state. We knew from prior work that diploids frequently generate overdominant mutations under Fisher’s geometric model (Selliset al., 2011), so we studied predictability using walks with and without overdominant mutations to understand the impact of balanced polymorphisms on predictability.
We found that simulations without overdominant mutations are more forward predictable than simulations with overdominance, while the reverse is true for backward predictability. The anti-correlation between forward and backward predictability can be intuitively understood by considering the the nature of adaptation in Fisher’s geometric model. In walks without overdominant mutations, mutations are confined to within γ (Figure 1a), leading to high forward predictability. There is minimal opportunity for deviation from the optimal trajectory, and most of the adaptive mutations that occur during these walks have similar direction vectors to the optimal trajectory. Therefore, regardless of the order of mutations, each step will move the population closer to the optimum, making most of the trajectories viable, and resulting in low backward predictability. The reverse is true in walks with overdominant mutations, which explore a much larger portion of phenotypic space (αdip). Overdominant mutations tend to overshoot the optimum and are frequently followed by compensatory mutations. The larger amount of phenotypic space explored generates lower forward predictability, while the high frequency of compensatory mutations, and thus the importance of the order in which the mutations are introduced, results in high backward predictability. While Fisher’s geometric model is a useful tool to consider adaptation under phenotypic stabilizing selection, further work is required to determine the extent to which this anti-correlation is generalizable to biological systems. Nevertheless, the anti-correlation we observe between forward and backward predictability highlights the importance of distinguishing between types of predictability in future studies.
In natural populations, stable polymorphisms can be due to overdominance or other types of balancing selection, such as negative frequency dependent selection (Levinet al., 1988; Iserbytet al., 2013), and spatially or temporally variable selection (Rainey and Travisano, 1998; Kasumovicet al., 2008; Saltz and Nuzhdin, 2014). Transient functional polymorphisms at intermediate frequencies can also be generated via clonal interference (Desai and Fisher, 2007; Herron and Doebeli, 2013; Kvitek and SHERLOCK, 2013; Langet al., 2013). Both frequency dependent selection and clonal interference can occur in both haploid and diploid populations. Our work shows that the presence of polymorphisms in the population, regardless of source, significantly complicates analysis of adaptive trajectories, and these complications must be considered in all natural systems.
One such complication is the existence of simultaneous mutational lineages, which can result in hidden alleles (i.e. alleles that are not present at the end of the evolution) and transient population states that nevertheless significantly impact the future course of evolution. Ignoring hidden alleles can significantly modify the inferred backward predictability, and in extreme cases, can incorrectly suggest that the true order of mutations is impossible. Different orders of mutations can also generate different sets of heterozygous genotypes and different end population states, requiring the consideration of the state of the entire adapted population rather than the presence of a particular adapted allele.
Polymorphic states also drastically increase the number of possible adaptive paths. In systems where adaptation proceeds through sequential fixation, one only needs to consider the fitness of the 2n possible genotypes relative to the ancestral background for an n-mutation system. This is the methodology used in the experimental backward predictability studies of Weinreichet al. (2006), Khanet al. (2011) and Franke et al. (2011). However, in regimes where polymorphic states are frequently generated, the fitness of an invading mutation can vary depending on the alleles already present in the population. Within each adaptive trajectory, every mutation along the trajectory needs to be introduced into the prior population at low frequency on every available allele and tracked until the frequency of the new mutation has been stabilized in order to establish that the mutation is truly beneficial. Such a study would be extremely laborious, and to our knowledge, has never been conducted in any system.
Experimental Implications
In an experimental setting, high forward predictability means it is likely that the same set of mutations will be generated in independent adaptive walks, which make the probabilities generated through backward predictability analysis meaningful for predicting future events. This can occur by either a small mutational target size such as mutations that cause resistance to drugs, or a large mutational input into the population which makes rare but extremely beneficial mutations dominate the adaptive process (e.g. Desai and Fisher (2007); Kvitek and Sherlock (2011); Gersteinet al. (2012); Pennings (2012)). A study in FGM also suggests that a multi-locus FGM where each locus only influences a subset of the independent phenotypic dimensions (restricted pleiotropy) also promotes forward predictability, which the authors call parallel evolution (Chevinet al., 2010). Despite the large number of replicates required to achieve statistical significance, experimentally determining forward predictability has been shown to be feasible.
On the other hand, the possibility of hidden alleles makes accurate estimates of backward predictability impossible in both natural and artificial experimental systems. Since we do not have access to hidden alleles from natural populations, it is impossible to accurately compute the backward predictability of the adaptive walk leading to the current population state. Studying backward predictability using forward evolutions and constant sampling is equally infeasible. Even if we could sample every mutation that rises to reasonable frequency in a population, almost all of these mutations will be lost, and there may be far too many to determine the subset which are non-neutral. As mentioned above, there is also the problem of combinatorially many adaptive walks possible for even a few mutations, making complete experimental analysis of even a five mutation system extremely challenging. As others have mentioned, sampling a few high-fitness mutations and conducting backward predictability experiments may not generate a correct representation of the probability of any particular adaptive walk, as there may be alternative adaptive peaks (Weinreichet al., 2006). Additionally, there is the possibility of adaptation and potential epistatic interactions at sites not under consideration, and spatial or temporal fluctuations in selection pressures can further complicate accurate assessments of backward predictability in natural systems, and calls into question the accuracy of reconstructed ancestral states.
Finally, the impact of hidden alleles on evolutionary trajectories depends on the rate at which stable polymorphic states are generated. Rainey and Travisano (1998), for example, observed adaptive radiation by niche construction in every replicate evolution experiment they conducted. Under these conditions, we may expect hidden alleles to be frequent in a large evolving population. The adapted state of natural populations may thus experience a strong historical dependence on transient mutations that are eventually lost and impossible to sample, decreasing the forward predictability of evolution and making the inference of backward predictability impossible. The rate at which polymorphic states are generated in natural systems and potential differences between types of polymorphic states and their impact on forward and backward predictability should be further explored to improve our understanding of the predictability of evolution.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}